


Abstract
Lymphocyte trafficking out of secondary lymphoid organs is regulated by concentration gradient–dependent interactions between the membrane-derived lysophospholipid signaling molecule sphingosine 1-phosphate (S1P) and the G-protein–coupled receptor, S1P1. Etrasimod is a novel, next-generation, small-molecule, oral S1P receptor modulator in clinical development for the treatment of immune-mediated inflammatory disorders, including ulcerative colitis. In preclinical pharmacology studies, etrasimod was a full agonist of recombinant human (6.1 nM EC50), mouse (3.65 nM EC50), dog (4.19 nM EC50), and monkey (8.7 nM EC50) S1P1 receptors, and a partial agonist of human S1P4 (147 nM EC50) and S1P5 (24.4 nM EC50), with relative efficacies of 63% and 73% of S1P response, respectively; whereas neither agonist nor antagonist activity was observed for human S1P2 or S1P3. A dose-dependent relationship was observed for etrasimod plasma concentration and lymphocyte count in mice, and chronic treatment with etrasimod resulted in attenuation of inflammation in a CD4+CD45RBhigh T-cell transfer mouse model of colitis.
Introduction
Ulcerative colitis and Crohn’s disease are debilitating inflammatory bowel diseases characterized by inappropriate and sustained immune responses (Torres et al., 2017; Ungaro et al., 2017). As a greater understanding of the underlying immune pathology of these diseases developed, treatment evolved toward a more targeted approach, with therapies aimed at proinflammatory molecules such as cytokines [e.g., tumor necrosis factor (TNF), interleukin (IL)-12/IL-23] or other biologic processes known to contribute to disease pathology, such as leukocyte trafficking (Argollo et al., 2017). Although these biologically relevant therapies have added considerable value to the treatment of the inflammatory bowel diseases, they are not equally effective in all patients, and many patients lose response to treatment over time (Billioud et al., 2011). To date, nearly all biologic therapies explored and/or approved for the treatment of inflammatory bowel disease have been developed as antibodies against potential disease targets. Paradoxically, these large molecules are themselves immunogenic, which frequently leads to altered pharmacokinetics, reduced effectiveness, and/or safety and tolerability concerns (Hindryckx et al., 2017). Moreover, the size of these molecules precludes an oral route for administration, and the costs associated with their synthesis and use are considerable. Novel, orally administered, small-molecule therapies are therefore needed (Olivera et al., 2017).
Therapies targeting molecules and pathways involved in immune cell trafficking have demonstrated efficacy in their treatment (Danese and Panés, 2014; Zundler and Neurath, 2017). A crucial component of lymphocyte circulation involves the egress of these cells out of secondary lymphoid organs and into blood and lymph (Schwab and Cyster, 2007). This process is regulated by concentration gradient–dependent interactions between the membrane-derived lysophospholipid signaling molecule sphingosine 1-phosphate (S1P) and the G-protein–coupled receptor S1P1 [one of five known S1P receptors (S1P1–5)] (Peyrin-Biroulet et al., 2017). Antigen-activated T cells within peripheral lymphoid organs transiently downregulate S1P1 and are unresponsive to S1P egress signals, resulting in lymphocyte sequestration and inhibition of trafficking (Shiow et al., 2006).
Etrasimod (Fig. 1) is a synthetic next-generation S1P receptor modulator in clinical development for the treatment of immune-mediated inflammatory disorders, including ulcerative colitis. This report describes the characterization of the preclinical in vitro pharmacology of etrasimod S1P receptor binding, as well as the pharmacodynamic and in vivo effects of etrasimod in a mouse model of experimental colitis.

Chemical structure of etrasimod: (R)-2-(7-((4-cyclo-pentyl-3-(trifluoromethyl)benzyl)oxy)-1,2,3,4-tetrahydro-cyclopenta[b]indol-3-yl)acetic acid).
Materials and Methods
Etrasimod (99.6% purity as determined by chiral high-performance liquid chromatography) tested in the studies detailed herein was prepared in the laboratories of Arena Pharmaceuticals (San Diego, CA) as previously described (Buzard et al., 2014).
In Vitro Pharmacological Characterization of Etrasimod S1P Receptor Binding.
Human recombinant S1P1–5 and mouse, dog, and monkey S1P1 receptors were stably expressed in PathHunter HEK293 (DiscoveRx, Freemont, CA) parental cell lines after confirmation of cDNA accuracy against current National Center for Biotechnology Information reference sequences. Agonist (S1P1–5) and antagonist (S1P2 and S1P3) β-arrestin recruitment assays were performed according to the manufacturer (DiscoveRx) instructions. Briefly, PathHunter (DiscoveRx) HEK293 cells stably expressing recombinant receptors were seeded onto 384-well microtiter plates (2000–5000 cells/well depending on receptor) in OptiMem (Thermo Fisher Scientific, Waltham, MA) containing 0.05% fatty acid–free bovine serum albumin and incubated overnight in a humidified chamber. Plates were then equilibrated to room temperature (RT) for 1 hour followed by the addition of 5 μl of at least 10 different etrasimod concentrations (<10 μM) and incubation at RT for 2–3 hours. Lysis/detection reagents (12 μl total) were then added, and the plates were sealed and incubated for an additional 2 hours at RT. Plates were read on an EnVision (Perkin Elmer, San Jose, CA) or PheraStar (BMG, Cary, NC) plate reader. All etrasimod EC50 values were determined with a minimum of 10 different concentrations in triplicate. An EC90 of 200 and 300 nM S1P was added to plates 10 minutes after the addition of etrasimod for S1P2 and S1P3 antagonist assays, respectively.
In Vivo Regulation of Lymphocyte Counts in Mice.
Adult male Balb/c mice (Harlan, Indianapolis, IN) (N = 252) were used in this study and housed in climate-controlled conditions with a 12-hour automatic light/dark cycle with free access to certified rodent diet and water. All of the in vivo study protocols were reviewed and approved by the Arena Pharmaceuticals Institutional Animal Care and Use Committee.
Mice weighing 24.3 ± 2.1 g (mean ± S.D.) were randomly assigned to vehicle (n = 48), or 0.03, 0.10, 0.30, or 1.0 mg/kg etrasimod dose groups (n = 51 per dose) and received a single oral gavage dose (in the fed state) of either vehicle (0.5% methylcellulose) or etrasimod at a volume of 10 ml/kg. Whole-blood samples were collected by cardiac puncture and treated with potassium-EDTA. Plasma was prepared by centrifugation and stored frozen (−80°C) until analysis. The total lymphocyte count in whole blood was analyzed within 30 minutes of collection at 1, 3, 5, 8, 16, 24, and 32 hours postdose using an automated laser hematology analyzer (CELL-DYN 3700; Abbott Laboratories, Abbott Park, IL) designed for in vitro diagnostic use in clinical laboratories.
Plasma etrasimod concentrations at 0.25, 0.5, 1, 2, 3, 5, 8, 16, 24, and 32 hours postdose were analyzed using a selective liquid chromatography-mass spectrometry method. Plasma proteins were extracted with acetonitrile in the presence of an internal standard followed by centrifugation at 3700 rpm for 20 minutes. The supernatant from the processed plasma samples was injected into a high-performance liquid chromatography system equipped with either an Applied Biosystems API 4000 or API 5000 mass spectrometer (Thermo Fisher Scientific). Peak areas for the transitions of mass/charge ratio (m/z) 458.2 → 159.3 product ion of etrasimod were measured against the m/z 388.9 → 278.1 product ion of the internal standard, in positive-ion multiple reaction–monitoring mode.
Experimental Colitis Model.
Female, 9-week-old CB17/Icr-Prkdcscid/IcrCr mice (N = 45; strain 561; Charles River Laboratories, San Diego, CA) were housed in climate- and lighting-controlled conditions (reverse light/dark cycle, lights off 11:30–19:30) and allowed 1 week of habituation to this environment prior to study initiation. Animals had free access to sterilized rodent chow (2018 Teklad; Harlan) and water.
Spleens were dissected from BALB/c female donor mice (Charles River Laboratories) and placed in a staining buffer [PBS Ca/Mg2+ free; 1 mM EDTA, 25 mM HEPES (pH 7), 4% FBS (heat inactivated); 10 U/ml DNase II]. Single-cell suspension was generated by mashing spleens through 70-μm nylon mesh strainers (Falcon; Thermo Fisher Scientific). Cells were pelleted and suspended 1:3 in Life Technologies ACK (ammonium-chloride-potassium) lysis buffer (Thermo Fisher Scientific) for 3–5 minutes at RT followed by the addition of cold staining buffer. Cells were pelleted and suspended in MACS buffer (PBS, 0.5% BSA, 2 mM EDTA, pH 7.2; Miltenyi Biotec, San Diego, CA). CD4+ T cells were enriched by negative selection using a CD4+ T-cell isolation kit (Miltenyi Biotec) and LS magnetic columns (Miltenyi Biotech). Briefly, 107 cells were suspended in 40 μl of ice-cold MACS buffer to which 10 μl of biotin-antibody cocktail was added, mixed, and incubated on ice for 5 minutes. An additional 30 μl of ice-cold MACS buffer was added, as well as 20 μl of anti-biotin microbeads, followed by incubation on ice for 10 minutes. Cells, antibody, and bead suspensions were then added to LS columns (Miltenyi Biotech) previously rinsed with 3 ml of MACs buffer. The flow through was collected, and cells were centrifuged and resuspended in 100 μl of basic sorting buffer [PBS Ca/Mg2+ free; 1 mM EDTA, 25 mM HEPES (pH 7), 4% heat-inactivated FBS; 10 U/ml DNase II; 0.2 μM filter sterilized] per 106 cells. Monoclonal antibodies (CD45RB fluorescein isothiocyanate; CD4 APC; CD25 PE; all from BD Biosciences) were added and incubated on ice for 30 minutes in the dark.
Cells were washed with ice-cold staining buffer and stained with 1 μM 4′,6-diamidino-2-phenylindole. The 4′,6-diamidino-2-phenylindole–negative, CD4+, and CD45RBhigh cells were sorted on an Astrios (100 μM, 25 psi; Beckman Coulter Life Sciences, Indianapolis, IN) into 50% FBS, centrifuged at 3000 rpm for 15 minutes and suspended in ice-cold PBS (Ca, Mg2+ free) at 1 × 106 cells/ml. A total of 5 × 105 cells were intraperitoneally injected into female severe combined immunodeficiency (SCID) mice. SCID mice injected with 5 × 105 unsorted enriched T cells served as baseline controls. SCID mice injected with colitogenic T cells were orally dosed daily beginning on the day following transfer until the day before tissue harvest (Day 32) with either vehicle (negative control; n = 11) 1 (n = 12) or 3 mg/kg (n = 12) etrasimod or 1 mg/kg fingolimod (positive control; n = 12) in a volume of 4 ml/kg. All mice were weighed daily.
Prior to tissue harvest, blood was collected and mice were anesthetized with isoflurane and then euthanized. Colon segments were dissected, measured, and flushed with saline prior to weighing. Approximately 4 cm of the distal colon was fixed in formalin for 48 hours at RT, dehydrated, paraffin embedded, and sectioned (10 μm thickness), followed by staining with H&E. The remaining colon tissues was frozen in liquid nitrogen and stored at −80°C for RNA isolation.
Frozen tissue was subsequently homogenized in tubes containing lysing matrix A (MP Biomedicals, Santa Ana, CA) with 1 ml of TRIzol (Thermo Fisher Scientific) using an MP FastPrep homogenizing machine (MP Biomedicals). Ultrapure phenol:chloroform:isoamyl alcohol (200 ml; Thermo Fisher Scientific) was immediately added. Samples were incubated at RT for 10 minutes, followed by centrifugation at 15,000 rpm for 15 minutes at 4°C. The upper clear phase was removed and placed in a new tube containing 500 μl of isopropanol, followed by incubation for 10 minutes at RT and centrifugation at 15,000 rpm for 15 minutes. The RNA pellet was washed once with 70% ethanol, and centrifuged for 15 minutes at 15,000 rpm before reconstitution in RNase/DNase-free water. Quantitation of RNA was performed with NanoDrop Lite (Thermo Fisher Scientific) prior to treatment with DNase (Thermo Fisher Scientific) and reverse transcription using iScript cDNA Synthesis Kit (Bio-Rad, Hercules CA). Quantitative polymerase chain reaction (PCR) was performed using QuantStudio 6 (Thermo Fisher Scientific) for CD4 (Mm00442754_m1), CD3γ (Mm00438095_m1), CD11b (Mm00434455_m1), IL-17A (Mm00439618_m1), IL-13 (Mm00434204_m1), interferon-γ (Mm01168134_m1), TNF-α (Mm00443258_m1), IL-1β (Mm00434228_m1), IL-6 (Mm00446190_m1), and IL-10 (Mm00439616_m1; all Thermo Fisher Scientific); and Ly6G (Beacon Discovery, San Diego, CA).
For the assessment of histologic disease severity, blinded histopathological assessment was performed using a modification of previously described methods (Ostanin et al., 2006; Pavlick et al., 2006). Briefly, the following scoring criteria were assessed in a blinded fashion by a single pathologist: 1) crypt score based on abnormal crypt architecture including distortion, branching, atrophy, and crypt loss (range, 0–3); 2) goblet cell loss (range, 0–2); and 3) mucosal erosion and ulceration (range, 0–1), and 4) immune cell infiltration (range, 0–2). A total histopathological score is calculated by combining the scores for each of the four parameters for a maximum score of 8.
Data Analysis.
For receptor assays, efficacies were calculated as a percentage of the S1P receptor dose-response curve height, which was defined as 100% in each experiment. Mean EC50 values were calculated from mean pEC50.
Composite sampling was used to determine the blood lymphocyte and plasma concentration versus time profiles. The relationship between etrasimod plasma concentration and lymphocyte count was determined using an indirect pharmacokinetic (PK)/pharmacodynamic (PD) model. Initial observations indicated that the blood lymphocyte count was associated with a circadian rhythm. To discern between the lymphocyte baseline diurnal effect and the effect of etrasimod on lymphocyte egress, a circadian equation was built into the model. The relationship between drug concentration and pharmacological effect were expressed by model estimates IC50 and Imax. These values were determined by simultaneously modeling the blood lymphocyte counts over time for all doses (including vehicle control) while holding the PK parameters constant. This PK/PD model was also used to calculate plasma concentrations and associated lymphocyte counts for a 3 mg/kg dose to provide estimates for the dose used in the adoptive transfer study.
Body weight was expressed as a percentage of initial body weight at the day of T-cell transfer and subjected to ANOVA followed by Dunnett’s multiple-comparison test with all groups compared with vehicle control. For quantitative PCR, mRNA level was first normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) level within each sample to obtain a ratio, all values were then normalized to the average of values from naive SCID mice, and expressed as a fold value over control (naive). One-way ANOVA followed by Dunnett’s test was used to compare multiple groups. All analyses were done with GraphPad PRISM 7 (2016).
Results
S1P Receptor Binding.
Etrasimod has previously been shown to be a full agonist of human S1P1 and a partial agonist of S1P4 and S1P5 in a β-arrestin assay; and a full agonist of human, mouse, rat, dog and monkey S1P1 demonstrated in a cAMP assay (Buzard et al., 2014). Here, we further demonstrate that etrasimod is a full agonist of human, dog, mouse, and monkey recombinant S1P1 receptors in β-arrestin recruitment assays (Table 1). Additionally, neither agonist nor antagonist activity was observed for etrasimod on either the human recombinant S1P2 or S1P3 receptors. The mean EC50 values of etrasimod for S1P1 were similar among the species tested and ranged from 3.65 to 8.70 nM. Selectivity for human S1P1 was 24-fold and 4-fold compared with S1P4 and S1P5, respectively, and ≥1000-fold compared with S1P2 and S1P3.
Etrasimod mean effective concentrations and efficacy on recombinant S1P receptors by β-arrestin assay
Effect of Etrasimod on Lymphocyte Counts.
Etrasimod was rapidly absorbed after oral administration, with quantifiable plasma concentrations detected at the first time point measured (0.25 hour), and subsequent dose-proportional increases were observed for all doses tested.
Etrasimod produced dose-dependent blood lymphopenia in mice, and plasma etrasimod concentrations and lymphocyte counts were inversely related (Fig. 2). The calculated IC50 value was 46.3 ng/ml (planar 95% confidence interval, −13.7 to 106), which is equivalent to a median effective dose of approximately 0.2 mg/kg. The Imax of lymphocyte egress based on model estimates was approximately 1.03 (planar 95% confidence interval, 0.686–1.36) or 100%. Additional model estimates suggest a further blunting of lymphocyte egress after a 3 mg/kg dose. Also of note is a clear dose-dependent blunting of the circadian rhythm associated with lymphocyte trafficking.
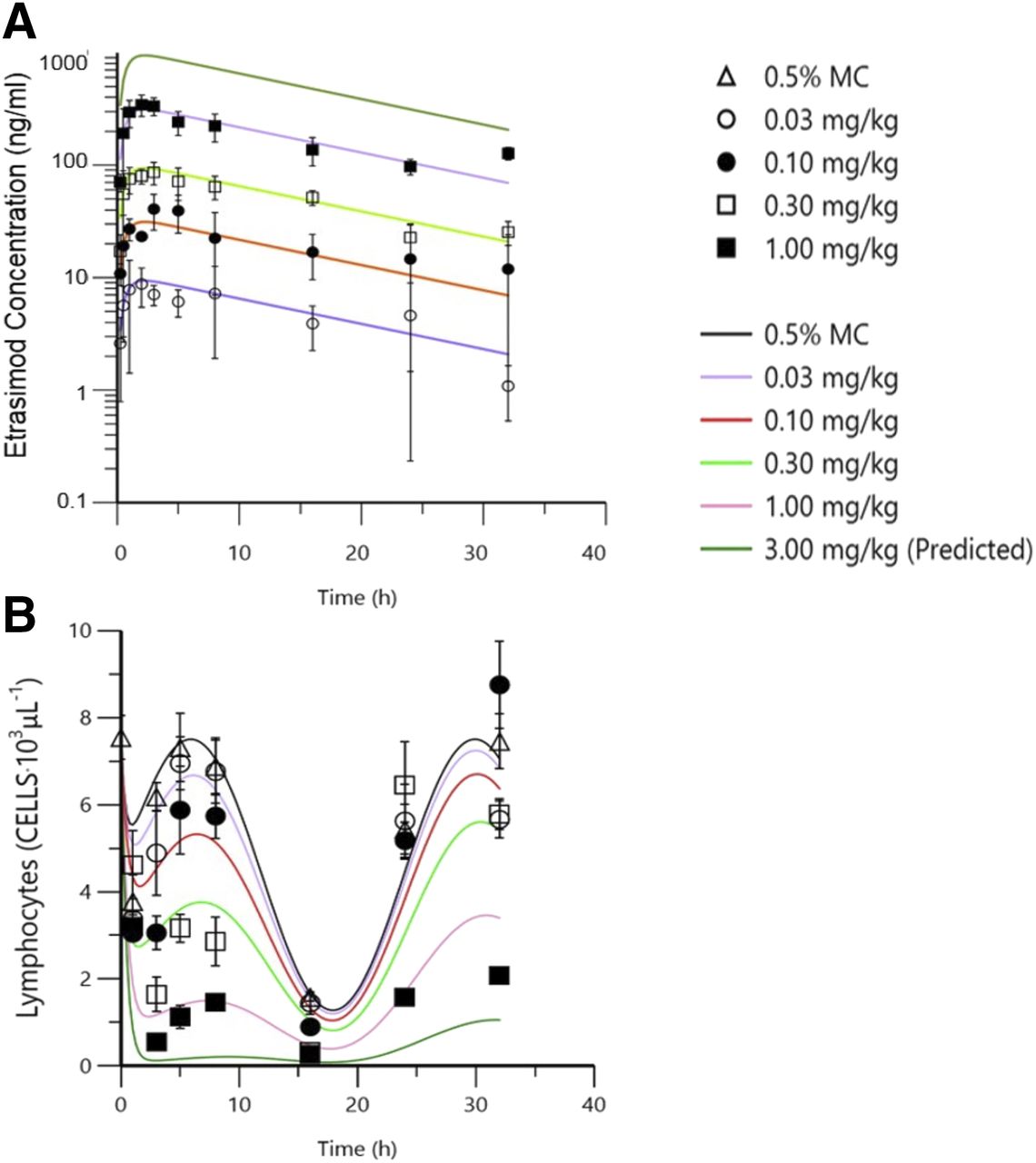
Comparison of mean etrasimod plasma concentration and blood lymphocyte count vs. time profiles. Dose-dependent increases in plasma concentration plotted over time (A) are inversely related to peripheral blood lymphocyte counts over the same time period (B). Note: collected data were used to build a PK/PD model that was then used to derive the predicted exposure and associated lymphocyte counts represented by the continuous line plots. MC; methylcellulose.
Attenuation of Disease in Mouse Colitis Model.
Onset of progressive colitis-like symptoms (loose, mucinous stool and loss of body weight) began between 2 and 3 weeks after adoptive transfer of CD4+ CD45RBhigh T cells. Mice that received colitogenic T cells showed significant weight loss over the course of the study compared with controls treated with unsorted T cells. Etrasimod treatment dose-dependently attenuated this loss, with a statistically significant effect observed at 3 mg/kg. Similarly, fingolimod treatment significantly inhibited weight loss compared with vehicle-treated controls (Fig. 3A).

Treatment with etrasimod or fingolimod inhibits weight loss associated with disease onset in SCID mice receiving CD4+ colitogenic T cells. Etrasimod attenuates weight loss (A) and reduces colon weight-to-length ratio (B) dose-dependently compared with vehicle-treated controls (vs. vehicle: **P ≤ 0.01; ***P ≤ 0.001 Dunnett’s test).
Compared with naive mice and mice who received unsorted CD4 T cells, CD4+CD45RBhigh SCID mice treated with vehicle had an increase in colon weight:length ratio. Treatment with 1 and 3 mg/kg etrasimod and 1 mg/kg fingolimod significantly inhibited the increase in colon weight:length ratio in CD4+CD45RBhigh SCID mice (Fig. 3B).
Consistent with the increased colon weight:length ratio, histopathologic examination suggested that the colonic mucosae of CD4+CD45RBhigh SCID mice were thicker than those of naive SCID mice and mice who had received unsorted T cells. Treatment with etrasimod (3 mg/kg) or fingolimod (1 mg/kg) resulted in significant reductions in mucosal thickness (Fig. 4A), and lower histopathology scores (Fig. 4B). These differences are readily observable in photomicrographs of colon cross sections from the respective groups (Fig. 4C) wherein etrasimod and fingolimod reduce the number of inflammatory cell infiltrates compared with vehicle-treated mice transferred with colitogenic T cells.

Etrasimod inhibits the increase in histologic (Histo) markers of disease. (A) Etrasimod inhibits increases in mucosal thickening. (B) Etrasimod reduces the increase in disease index scores. (C) Photomicrographs of H&E-stained colon cross sections suggest that etrasimod decreases inflammatory cell infiltration (arrows) in the colons of mice transferred with colitogenic T cells (vs. vehicle: *P ≤ 0.05; **P ≤ 0.01 Dunnett’s test). Max, maximum.
Reduction in Inflammatory Responses in Mouse Colitis Model.
Increased expression of immune cell markers and cytokines as estimated by quantitative PCR was observed in the colonic tissue isolated from CD4+CD45RBhigh SCID mice and mice that had received unsorted T cells compared with naive mice, with comparably higher levels of CD4 (T cells, natural killer cells, and dendritic cells), CD3γ (T lymphocytes) and CD11b (monocytes, natural killer cells, and dendritic cells), and unchanged levels of Ly6G (neutrophils, and granulocytes) observed in CD4+CD45RBhigh SCID mice (Fig. 5). Treatment with etrasimod (3 mg/kg) or fingolimod (1 mg/kg) significantly reduced the expression of the T-cell and monocyte markers, suggesting that both agents decreased infiltration and expansion of these cell populations in the colons of CD4+CD45RBhigh SCID mice.

Etrasimod reduces infiltration and expansion of T cell and macrophage populations. QPCR of colon homogenates shows statistically significant decreases in (A) CD4, (B) CD3γ and (C) CD11b, but not (D) Ly6G in mice treated with etrasimod. Data shown as relative expression to GAPDH. (vs. vehicle *P < 0.05; **P < 0.01. Dunnet’s).
In support of these data, T-cell and/or monocyte-derived proinflammatory cytokines TNF-α, IL-1β, IL-6, and IL-17A were also significantly lower in CD4+CD45RBhigh SCID mice treated with etrasimod (3 mg/kg) and fingolimod (1 mg/kg) compared with mice treated with vehicle (Fig. 6). Etrasimod induced dose-dependent increases in the anti-inflammatory cytokine IL-10. Expression levels of this cytokine were comparable between animals treated with the high dose of etrasimod and with fingolimod, and both were significantly higher than those in the vehicle-treated control group (P < 0.05 Dunnett’s test) (Fig. 6). Last, no clear differences between the treatment groups were observed in both IL-13 and interferon-γ expression (data not shown).

Etrasimod reduces elevations in pro-inflammatory cytokines and increases anti-inflammatory cytokine. QPCR analysis of colon tissue homogenates show statistically significant decreases in macrophage and/or T-cell derived pro-inflammatory cytokines (A) TNF-α, (B) IL-1β, (C) IL-6, and (D) IL-17A, and increases anti-inflammatory (E) IL-10 in response to treatment with etrasimod compared to vehicle control. Data shown as relative expression to GAPDH. (vs. vehicle *P < 0.05; **P < 0.01. Dunnet’s).
Discussion
In this report, we further characterize the preclinical in vitro pharmacology of etrasimod S1P receptor binding, as well as the pharmacodynamic and in vivo effects of etrasimod in a mouse model of experimental colitis. The data demonstrate that etrasimod is a full agonist of S1P1 across multiple species with greater than 1000-fold selectivity versus human S1P2 and S1P3, with neither agonist nor antagonist activity observed for these latter receptors. Etrasimod was rapidly absorbed after oral administration and produced dose-dependent reductions in lymphocyte counts in mice, which were inversely related to etrasimod plasma concentrations. Consistent with previous reports, there was a clear circadian pattern in lymphocyte egress as measured with lymphocyte counts, which also showed a dose-dependent response to increasing concentrations of circulating etrasimod (Druzd et al., 2017). These circadian fluctuations were almost completely blunted at higher doses of etrasimod. Importantly, oral administration of etrasimod at a dose of 3 mg/kg was efficacious in a mouse model of colitis, demonstrating attenuation of colitis-like symptoms and reductions in mucosal thickness and histologic disease activity scores. These outcomes were likely a result of the inhibitory effects of etrasimod observed on the expression of proinflammatory cytokines (TNF-α, IL-1β, IL-6, IL-17A) and increased anti-inflammatory IL-10 expression. It is important to note that etrasimod treatment in the mouse colitis model did not affect the expression of the neutrophil cell surface marker, Ly6G, suggesting that innate immune surveillance by these cells is likely protected with etrasimod treatment.
Proof-of-concept support for S1P receptor targeting in chronic inflammatory diseases was first demonstrated in clinical trials of fingolimod, a nonselective S1P modulator approved for the treatment of relapsing multiple sclerosis (Calabresi et al., 2014). Adverse effects of fingolimod, such as macular edema, dysregulated pulmonary function, and hypertension, however, are presumed to stem from the nonspecific S1P1–5 activity (Forrest et al., 2004; Camm et al., 2014). S1P2 and S1P3 signaling are implicated in a myriad of biologic activities (Sanna et al., 2004; Blaho and Hla, 2014). Although a complete understanding of the specific biologic functions mediated by the individual receptors is lacking, in general, S1P1, S1P4, and S1P5 appear to be fundamentally involved in the regulation of the immune system (Blaho and Hla, 2014), whereas S1P2 and S1P3 have been implicated in processes such as vasoconstriction and fibrosis, mechanisms that may play a role in the renal ischemia-reperfusion injury potentially observed with S1P2 (Park et al., 2012) and the hypertension observed with fingolimod (Fryer et al., 2012). More selective S1P receptor modulators (ozanimod, siponimod, ponesimod) in clinical development have demonstrated improved clinical safety profiles compared with fingolimod (Piali et al., 2011; Gergely et al., 2012; Tran et al., 2018). In this regard, the receptor-binding profile of etrasimod should theoretically avoid some of the adverse effects associated with modulation of these receptors, while simultaneously decreasing intestinal inflammation.
The selectivity of etrasimod for S1P1 has important and specific disease-related implications for the treatment of inflammatory bowel disease. In a study designed to investigate the role of the S1P pathway on inflammatory bowel disease pathogenesis, Karuppuchamy et al. (2017) found S1P1 expressed on naive, central memory cells and subsets of gut-homing effector T cells, activated dendritic cells, and endothelial cells. Furthermore, chronic inflammatory signals were shown to upregulate S1P1 on both T cells and the endothelium, and a similar pattern of dysregulation was observed for enzymes that control tissue S1P levels in inflamed mouse and human intestines (i.e., induction of S1P synthesis and suppression of degradation). Karuppuchamy et al. (2017) hypothesized that S1P1 inhibitors could have multiple relevant anti-inflammatory mechanisms of action in the treatment of inflammatory bowel disease apart from their lymphopenic effects, including potential effects on dendritic cell migration and vascular barrier function (Karuppuchamy et al., 2017). Initial clinical results for selective S1P receptor modulators in the treatment of inflammatory bowel disease have been encouraging. The S1P receptor modulator ozanimod, which targets both S1P1 and S1P5, has demonstrated efficacy and tolerability in phase 2 clinical trials of ulcerative colitis (Sandborn et al., 2016) and in phase 3 clinical trials of multiple sclerosis (Cohen et al., 2017; Comi et al., 2017). However, longer-term data and larger trials are required to fully characterize the efficacy and safety profile of this agent in patients with inflammatory bowel disease.
In conclusion, etrasimod is a second-generation, oral, synthetic small-molecule S1P receptor modulator that may address some of the limitations associated with currently approved monoclonal antibodies (i.e., route of administration, immunogenicity, prolonged half-lives, and manufacturing costs). Etrasimod receptor selectivity (S1P1,4,5) may avoid off-target effects related to broader receptor binding, such as those observed with fingolimod, and provide relevant mechanistic effects specifically related to the role of S1P1 in the pathology of inflammatory bowel disease. Furthermore, an oral route of administration may provide additional convenience and options to patients and their treating gastroenterologists. Future clinical trials in humans will provide more efficacy and safety data on etrasimod and its potential placement in the treatment armamentarium for ulcerative colitis.
Authorship Contributions
Participated in research design: Al-Shamma, Lehmann-Bruinsma, Carroll, Solomon, and Adams.
Conducted experiments: Al-Shamma, Lehmann-Bruinsma, Carroll, and Solomon.
Performed data analysis: Al-Shamma, Lehmann-Bruinsma, Carroll, and Solomon.
Wrote or contributed to the writing of the manuscript: Al-Shamma, Komori, Peyrin-Biroulet, and Adams.
Footnotes
- Received October 9, 2018.
- Accepted March 5, 2019.
This work was funded by Arena Pharmaceuticals, Inc. HA-S is an employee of Beacon Discovery Inc., which is the originating laboratory for the studies; KL-B is an employee of Beacon Discovery Inc.; CC is an employee of Beacon Discovery Inc.; MS reports; HKK is an employee of Arena Pharmaceuticals, Inc., the company that funded the studies; LP-B reports receiving honoraria from Merck, Abbvie, Janssen, Genentech, Ferring, Tillots, Vifor, Pharmacosmos, Celltrion, Takeda, Biogaran, Boerhinger-Ingelheim, Lilly, Pfizer, Index Pharmaceuticals, Amgen, Sandoz, Celgene, Biogen, Samsung Bioepis, Alma, Sterna, Nestlé and Enterome; JA is an employee of Arena Pharmaceuticals, Inc.
Abbreviations
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IL
- interleukin
- PCR
- polymerase chain reaction
- PD
- pharmacodynamic
- PK
- pharmacokinetic
- RT
- room temperature
- S1P
- sphingosine 1-phosphate
- SCID
- severe combined immunodeficiency
- TNF-α
- tumor necrosis factor-α
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}