


Abstract
Class II antiarrhythmics or β-blockers are antisympathetic nervous system agents that act by blocking β-adrenoceptors. Despite their common clinical use, little is known about the effects of β-blockers on free intracellular calcium (Ca2+i), an important cytosolic second messenger and a key regulator of cell function. We investigated the role of four chemical analogs, commonly prescribed β-blockers (atenolol, metoprolol, propranolol, and sotalol), on Ca2+i release and whole-cell currents in mammalian cancer cells (PC3 prostate cancer and MCF7 breast cancer cell lines). We discovered that only propranolol activated free Ca2+i release with distinct kinetics, whereas atenolol, metoprolol, and sotalol did not. The propranolol-induced Ca2+i release was significantly inhibited by the chelation of extracellular calcium with ethylene glycol tetraacetic acid (EGTA) and by dantrolene, an inhibitor of the endoplasmic reticulum (ER) ryanodine receptor channels, and it was completely abolished by 2-aminoethoxydiphenyl borate, an inhibitor of the ER inositol-1,4,5-trisphosphate (IP3) receptor channels. Exhaustion of ER stores with 4-chloro-m-cresol, a ryanodine receptor activator, or thapsigargin, a sarco/ER Ca2+ ATPase inhibitor, precluded the propranolol-induced Ca2+i release. Finally, preincubation of cells with sotalol or timolol, nonselective blockers of β-adrenoceptors, also reduced the Ca2+i release activated by propranolol. Our results show that different β-blockers have differential effects on whole-cell currents and free Ca2+i release and that propranolol activates store-operated Ca2+i release via a mechanism that involves calcium-induced calcium release and putative downstream transducers such as IP3. The differential action of class II antiarrhythmics on Ca2+i release may have implications on the pharmacology of these drugs.
Introduction
Class II antiarrhythmics or β-blockers have been in clinical use for the treatment of cardiovascular conditions such as angina and hypertension for more than five decades (Black et al., 1964; Chobanian et al., 2003). The cardioprotective effects of class II antiarrhythmics are linked to the inhibition of β-adrenoceptor (β-AR) signaling. There are three types of β-ARs: β1-AR, found mainly in cardiac cells; β2-AR, present in bronchial and vascular tissue; and β3-AR, largely expressed in adipose tissue. β-ARs are also expressed in many primary and metastasized tumors (Daly and McGrath, 2011; Cole and Sood, 2012).
Upon stimulation by catecholamines, β-AR signaling results in elevated cAMP levels and activation of the cAMP-dependent protein kinase A (Naga Prasad et al., 2001; Cole and Sood, 2012), which targets L-type Ca2+ channels (CaV1.2), activating a calcium influx (Weiss et al., 2013). Few reports have analyzed the direct effects of β-blockers on free intracellular calcium (Ca2+i) although previous studies have suggested that there is a reduction in the levels of free Ca2+i in platelets and erythrocytes of hypertensive patients treated with β-blockers (Erne et al., 1984; Baumgart et al., 1986).
Free Ca2+i is a potent second messenger that regulates many different cellular processes, including cell proliferation, cell differentiation, gene transcription, and apoptosis (Berridge et al., 2000; Carafoli et al., 2001). The level of cytosolic-free calcium (∼100 nM) is tightly regulated, and most Ca2+i resides in intracellular stores, which in nonmuscle cells are principally located in the endoplasmic reticulum (ER) (Berridge et al., 2003). Ca2+i can be released from the ER through ryanodine receptor (RyR) (Zorzato et al., 1990) and inositol-1,4,5-trisphosphate (IP3) receptor channels (Nixon et al., 1994). Calcium transients resulting from Ca2+ influx or Ca2+i release give rise to fast Ca2+ spikes or slower oscillatory waves that, depending on their kinetics and amplitude, translate to different cellular funtions (Berridge et al., 2003).
Despite the critical role of free Ca2+i as a regulator of cell function, the effects of β-blockers on the mobilization and kinetics of Ca2+i have received limited attention. Some reports regarding the regulation of calcium by β-blockers exist and include the investigations of Ca2+i levels in different disease models of heart failure or hypertension (Doi et al., 2002; Reiken et al., 2003; Tuncay et al., 2013; Cseplo et al., 2016). Other studies have examined the effects of β-blockers on β-adrenergic mediated calcium entry (e.g., after activation with β-AR agonists, such as isoproterenol or albuterol) and their role on cell contraction and vasorelaxation in different tissues and cell types (Sakanashi and Takeo, 1983; Yao et al., 2003; Priviero et al., 2006; Shahbaz et al., 2011; Cekic et al., 2013; Keller et al., 2014) (see Supplemental Table 1 for details). Surprisingly, the direct activation of free Ca2+i release by β-blockers has not been examined before in excitable or nonexcitable cells.
We have previously shown that several membrane potential regulating compounds (MPRCs), including the antiarrhythmics amiodarone and dofetilide, activate store-operated Ca2+i release in mammalian cancer cells (Petrou et al., 2017). Several epidemiologic studies have reported that the use of β-blockers correlates with a lower incidence of cancer progression and mortality for prostate (Perron et al., 2004; Grytli et al., 2013), breast (Powe et al., 2010; Barron et al., 2011; Melhem-Bertrandt et al., 2011), and skin cancers (De Giorgi et al., 2011; Lemeshow et al., 2011; De Giorgi et al., 2017).
We asked whether β-blockers could activate Ca2+i release in cancer cells. We concentrated on four commonly used β-blockers, including two β1-selective β-blockers—atenolol and metoprolol—and two nonselective β-blockers—propranolol and sotalol. We used PC3 and MCF7, prostate and breast cancer cell lines, respectively, to measure Ca2+i release by ratiometric live calcium imaging. In addition, we used a medium throughput patch-clamp system to measure ionic currents in the cells. The results show that 1) only propranolol activated a Ca2+i release, with distinct kinetics and amplitude; 2) the propranolol activation of Ca2+i stores was mediated by calcium-induced calcium release (CICR); and 3) the four β-blockers regulate endogenous whole-cell currents in cancer cell lines. Our results show differential activation of calcium stores and free Ca2+i release by several class II antiarrhythmics in nonexcitable, cancer cells and may have important implications for the mechanism of action and pharmacology of these β-blockers.
Materials and Methods
Compounds.
All β-blockers were purchased from Sigma-Aldrich (Gillingham, UK). Stock solutions were prepared in dimethyl sulfoxide (DMSO; Sigma-Aldrich) for atenolol and propranolol or in phosphate buffer saline (PBS), pH 7.4, without Ca2+ or Mg2+ (cat. no. 10010; Gibco ThermoFisher, Loughborough, UK) for metoprolol and sotalol according to the manufacturer’s instructions. The following are the systematic names for the β-blockers: atenolol (cat. no. A7655), (±)-4-[2-hydroxy-3-[(1-methylethyl)amino]propoxy]benzeneacetamide; metoprolol tartrate (cat. no. M5391), (±)1-(isopropylamino)-3-[p-(β-methoxyethyl)phenoxy]-2-propanol (+)-tartrate salt; propranolol hydrochloride (cat. no. P8688): (S)-1-isopropylamino-3-(1-naphthyloxy)-2-propanol hydrochloride; and sotalol hydrochloride (cat. no. S0278), N-[4-[1-hydroxy-2-(isopropylamino)ethyl]phenyl]methanesulfonamide hydrochloride (see Supplemental Fig. 1 for chemical structures). For all β-blockers, two different lots were purchased and tested in our experiments. Loxapine (cat. no. L106; Sigma-Aldrich), previously shown to activate store-operated free Ca2+i release via CICR in cancer cells (Petrou et al., 2017), was used as a positive control for some live Ca2+i imaging experiments. Timolol (cat. no. T6394; Sigma-Aldrich) was used as an additional nonselective β-blocker to investigate the contribution of β-ARs to Ca2+i release. Ethylene glycol tetraacetic acid (EGTA), dantrolene, 4-chloro-m-cresol (4-CmC), thapsigargin (Sigma-Aldrich), and 2-aminoethoxydiphenyl borate (2-APB; Tocris Bioscience, Abingdon, UK) were also used in some experiments (see later). Stock solutions were prepared in DMSO (dantrolene, 2-APB, and thapsigargin), ethanol (4-CmC), or PBS (EGTA).
Cell Culturing.
PC3 prostate cancer and MCF7 breast cancer cell lines were obtained from the American Type Culture Collection (Teddington, UK). Details of cell culture procedures have been described elsewhere (Thrasivoulou et al., 2013; Petrou et al., 2017). Briefly, cells were maintained in RPMI 1640 medium (Gibco ThermoFisher) supplemented with 10% fetal bovine serum and 5 mM l-glutamine and cultured at 37°C in a humidified incubator with 5% CO2 and 21% O2 atmosphere. For live calcium imaging experiments, 105 cells were seeded in 35-mm FluoroDishes (World Precision Instruments, Hitchin, UK), and experiments were performed in at least four to eight different passages (passage numbers 23–38 for PC3 cells and 35–44 for MCF7 cells).
Intracellular Live Calcium Imaging.
Free Ca2+i release was measured as a change in the ratio of Fluo-4/FuraRed (free calcium/bound calcium) over time. The two indicators have reciprocal shifts in intensity owing to calcium binding and are used together in a ratiometric probe strategy described previously (Wang et al., 2010b; Thrasivoulou et al., 2013; Petrou et al., 2017). Briefly, cells were grown as a monolayer in 35 mm FluoroDishes for 3 to 4 days. Before imaging, the cells were incubated for 30–40 minutes at 37°C with the acetoxymethyl ester derivatives of the calcium indicators Fluo-4 and FuraRed (ThermoFisher Scientific) at 1.1 and 1.4 µg/ml, respectively. Cells were washed (3×) with and replaced in 1 ml PBS without Ca2+ or Mg2+ (Gibco ThermoFisher, as described) for live calcium imaging, performed using an Olympus FluoView FV100 confocal microscope (Olympus, UK) equipped with a 20×/0.75 NA objective and a temperature-controlled chamber at 37°C. Calcium indicators were excited with an argon laser at 488 nm, and fluorescence was recorded every 2.2 seconds in the green channel for Fluo-4 (500–580 nm) and in the red channel for FuraRed (630–730 nm). Confocal imaging was started and, after a baseline was achieved, Ca2+i release was measured by applying β-blockers to the FluoroDish as a bolus, at a volume of 0.5–5 µl, to achieve a final β-blocker concentration of 25, 50, 100, 150, or 250 µM; vehicle controls were performed using a similar protocol. Data acquisition was performed using Olympus FV10-ASW 4.2 software.
Although the PBS used here is nominally Ca2+ free, the residual Ca2+ concentration in PBS was measured to be >60 µM in this solution (Petrou et al., 2017). In some experiments, 5 mM EGTA was added to the PBS used as imaging media to chelate free residual Ca2+; cells were preincubated for 5 minutes before imaging was started. We have previously shown that using 5 mM EGTA in PBS reduces the residual free Ca2+ to <10 nM (Petrou et al., 2017). In other experiments, for the pharmacologic characterization of the mechanisms of Ca2+i store activation, cells were preincubated with the following: 1) 10 µM dantrolene for 5 minutes at 37°C to inhibit the ER RyR channels (Zhao et al., 2001); 2) 1 mM 4-CmC for 12 minutes at 37°C to activate RyR channels and exhaust the Ca2+i ER stores (Zorzato et al., 1993); 3) varying concentrations (1, 25, 50, or 100 µM) of 2-APB for 10 minutes at 37°C to inhibit the ER IP3 receptor channels (Maruyama et al., 1997); 4) 5 µM thapsigargin for 15–20 minutes at 37°C to discharge Ca2+i from the ER (Thastrup et al., 1990); or 5) 250 µM sotalol or timolol (nonselective β-blockers) for 2 minutes at 37°C for blockade of β-ARs (Baker, 2005).
Data Analysis and Statistics.
Data from live calcium imaging experiments were analyzed for time kinetics and amplitude as described previously (Thrasivoulou et al., 2013) using a Mathematica script (Wolfram, Hanborough, UK) (Petrou et al., 2017). The kinetics of the calcium waveform were characterized by different time constants: rise time (time from baseline to peak), dwell time (duration of the plateau phase), and fall time (time to return to baseline). The amplitude of the response was calculated as a fold increase in fluorescence intensity from baseline to peak (ΔF/F0, where ΔF = F – F0 and F is the maximum fluorescence intensity over basal level, F0). Data were analyzed using the D’Agostino-Pearson test for normal distribution and the Mann-Whitney U test for statistical significance using MedCalc (Ostend, Belgium), and plotted using OriginPro 2016 (OriginLab, Northampton, MA).
Automated Medium-Throughput Electrophysiology.
Endogenous whole-cell currents of PC3 cells in response to the application of β-blockers were measured using the QPatch automated cell patch-clamp system (Sophion Bioscience, Ballerup, Denmark), as described previously (Petrou et al., 2017). PC3 cells were cultured and harvested using Detachin (Genlantis, San Diego, CA) and kept in the QStirrer of the QPatch for up to 4 hours before the automatic preparation. The cells were transferred to the QFuge, centrifuged, and washed 2× in extracellular solution (see following) before being applied to the measuring site in the QPlate of the QPatch. A pressure of −70 mBar was applied to obtain positioning and sealing of the cells, and a whole-cell protocol with pressure pulses at −150 mBar was used to obtain whole-cell formation. Gigaseals were formed upon execution of a combined suction/voltage protocol. The intracellular solutions and compounds were applied by eight pipettes. The intracellular solution contained (in millimolar concentrations): 5.3 CaCl2, 1.7 MgCl2, 10 EGTA, 10 HEPES, 120 KCl, and 4 Na2-ATP; with pH 7.2, osmolarity of 295 mOsm, and a calculated free calcium concentration of 680 nM. The extracellular solution contained (in millimolars): 2 CaCl2, 1 MgCl2, 10 HEPES, 4 KCl, and 145 NaCl, with pH 7.4, and osmolarity adjusted to 285–295 mOsm. Currents were recorded using a command ramp from −120 to +120 mV at 0.5 mV/ms every 3 seconds, with a holding voltage of −10 mV between executions of the ramps. Data were sampled at 5 kHz and filtered using a fourth-order Bessel filter.
Six different concentrations of individual drugs were used: increasing concentrations of the β-blocker from 0 to 500 µM were applied sequentially on the same cell. The QPatch system implements fine microfluidic control of the drug delivery time, and complete solution changes were made within 500 milliseconds. Average currents at +100 and −100 mV were analyzed to explore the concentration dependence of the modulation of endogenous whole-cell currents by β-blockers; statistical significance was calculated with the Wilcoxon test (MedCalc). Current-voltage (I–V) curves of control versus β-blocker were also constructed. Additional analysis of medium-throughput data from QPatch was carried out using Matlab (Mathworks, Natick, MA).
Real-Time Polymerase Chain Reaction .
Total RNA was purified from PC3 cells by using the RNeasy Plus kit (Qiagen, Manchester, UK) and reverse-transcribed with the Omniscript RT kit (Qiagen), according to the manufacturer’s instructions. Polymerase chain reaction was performed with the SYBR-Green PCR master mix (Applied Biosystems, Foster City, CA) using a Bio-Rad (Watford, UK) CFX384 thermocycler. Expression levels of the genes of interest were normalized to GAPDH or β-actin, and melting curves were analyzed using Bio-Rad CFX Manager to verify the products. The following primers were purchased from Sigma-Aldrich: β1-AR (ADRB1) sense 5′-TACGGCTCCTTCTTCTGCGA-3′ and antisense 5′-CAGGTACACGAAGGCCATGAT-3′; β2-AR (ADRB2) sense 5′-CATTGAGACCCTGTGCGTGA-3′ and antisense 5′-AGGGCTTTGTGCTCCTTCAA-3′; β3-AR (ADRB3) sense 5′-GTTTTCGTGGTGGCTACGC-3′ and antisense 5′-CCTAGCCAGTTCAGGGCAAG-3′; glyceraldehyde 3-phosphate dehydrogenase (GAPDH) sense 5′-CGGATTTGGTCGTATTGGGC-3′ and antisense 5′-TGGTCATGAGTCCTTCCACG-3′; β-actin (ACTB) sense 5′-CTGTGCTATCCCTGTACGCC-3′ and antisense 5′-ATCTTCATTGTGCTGGGTGCC-3′ (annealing at 60°C for 40 cycles; 70 melting curve reads off, from 60 to 95°C).
Results
Propranolol Activates Ca2+i Release in Cancer Cells with Distinct Kinetics but Other β-Blockers Do Not.
The addition of 50 µM propranolol to the extracellular solution caused a change in the ratio of Fluo-4/FuraRed in PC3 cells (Fig. 1), indicating release of free Ca2+i; there was no observable Ca2+i release in response to atenolol, metoprolol, and sotalol (Fig. 2A). Similar results were observed in MCF7 breast cancer cells, indicating that this phenomenon is not cell line specific (Fig. 2B). We also tested various concentrations of β-blockers (Fig. 3) within the pharmacologic range (Joint Formulary Committee, 2017).

Intracellular calcium (Ca2+i) release in PC3 cells in response to 50 µM propranolol. PC3 cells were loaded with the calcium indicators Fluo-4 (green fluorescence, an indicator of free calcium) and FuraRed (red fluorescence, an indicator of bound calcium) and monitored by time-lapse video microscopy every 2.2 seconds using an Olympus FluoView FV100 confocal microscope. Representative snapshots of free Ca2+i release at four time points (A–D) are shown: (A) Baseline before the addition of propranolol; (B) approximately 30% of cells in the observable frame undergoing Ca2+i release at t = 338 seconds, as observed by the simultaneous increase in Fluo-4 fluorescence and decrease in FuraRed signal; (C) peak of the Ca2+i release response (t = 374 seconds) with almost all cells showing a response (green); and (D) cells returning to baseline after the propranolol-induced Ca2+i release. Scale bar, 50 µm, original magnification, 20×.

Representative traces of Ca2+i release in (A) PC3 prostate cancer and (B) MCF7 breast cancer cells in response to the addition of 50 µM β-blockers: propranolol (green), atenolol (magenta), metoprolol (yellow), and sotalol (blue). The x-axis represents time (s) and the y-axis represents the Fluo-4/FuraRed ratio (free calcium/bound calcium, respectively) in arbitrary units (AU). The waveform was used to calculate the kinetics (rise, dwell, and fall times) of the Ca2+i release (see Fig. 5). The mean amplitude and time constants were used to select a representative trace for each compound (n = 5–8 experiments, n = 151–448 single-cell measurements per β-blocker). Of the four β-blockers tested, propranolol was the only one to induce Ca2+i release in both PC3 and MCF7 cell lines.

Characterization of free Ca2+i release in response to various concentrations of β-blockers in PC3 cells. PC3 prostate cancer cells loaded with the calcium indicators Fluo-4 and FuraRed were treated with β-blockers, added as a bolus into the FluoroDish, and monitored over time using time-lapse confocal microscopy. Ca2+i release was measured as changes in the Fluo-4/FuraRed ratio; the amplitude of the Ca2+i release was calculated as fold increase in fluorescence intensity from baseline to peak (ΔF/F0) of the Fluo-4/FuraRed waveform and is presented as box plots. At a range of pharmacologic concentrations (see Supplemental Fig. 1 for details), only propranolol activated the release of Ca2+i. Other β-blockers, atenolol, metoprolol, and sotalol, did not mobilize Ca2+i (n = 107 imaging experiments and n = 7256 individual cells analyzed; with at least n ≥ 2 experiments and n > 130 cells per concentration for each β-blocker).
Three of the β-blockers used did not induce free Ca2+i release in either PC3 or MCF7 cancer cell lines (Fig. 2). We performed additional validation experiments to exclude the possibility that the lack of Ca2+i release by atenolol, metoprolol, and sotalol may have been due to cellular or technical factors. First, to confirm that the cells remained responsive to free Ca2+i release, loxapine, a compound previously shown to induce Ca2+i release in cancer cells (Petrou et al., 2017), was used as a positive control. In these experiments, atenolol, metoprolol, or sotalol were first added to the cells for 10 minutes, followed by loxapine in live calcium imaging experiments. There was no Ca2+i release in response to the three β-blockers (n = 3); however, the addition of loxapine to the same cells activated Ca2+i release (Fig. 4). Second, we procured different lots for the three drugs from the vendor (Sigma-Aldrich) and tested these in live Ca2+i imaging experiments. No Ca2+i release occurred in response to addition of any of the different lots of atenolol, metoprolol, or sotalol. The effect of the solvent vehicle used for drug suspension (either DMSO or ethanol) was tested and was found to have no effect on any of the responses (i.e., only propranolol, but not atenolol, metoprolol, or sotalol, induced Ca2+i release) (Supplemental Fig. 2).

Atenolol, metoprolol, and sotalol did not induce the release of Ca2+i in PC3 prostate cancer or MCF7 breast cancer cells. To confirm this observation, PC3 cells loaded with the calcium indicators Fluo-4 and FuraRed were treated with 50 µM β-blocker (atenolol, metoprolol, or sotalol, added as a bolus) and after 10 minutes of imaging, the cells were treated with 50 µM loxapine, a dibenzoxazepine antipsychotic drug known to activate the release of free Ca2+i in human cancer cells (see Fig. 4 from Petrou et al., 2017 for details). Representative traces of Ca2+i release (amplitude of the Fluo-4/FuraRed ratio over time) in response to the addition of (A) atenolol (magenta), (B) metoprolol (yellow), or (C) sotalol (blue), followed by the addition of loxapine; arrows indicate the time of addition (n = 3 per β-blocker, with n = 161–272 individual cells). A representative trace of loxapine-induced Ca2+i release in control cells (no previous treatments) is shown in the insert (scale: y-axis 1 AU and x-axis 100 seconds). Note that there was no Ca2+i release in response to any of the three β-blockers whereas loxapine caused an immediate release of Ca2+i in the cells.
The waveform of Ca2+i release was used to measure the amplitude and the time kinetics (rise, dwell, and fall times) as described elsewhere (Thrasivoulou et al., 2013). Figure 5 shows the distinct time constants of the Ca2+i release in response to 50 µM propranolol in PC3 and MCF7 cells; similar kinetics were observed in both cell lines.

Characterization of the propranolol-induced Ca2+i release kinetics in PC3 prostate cancer and MCF7 breast cancer cells. (A) The Ca2+i waveform generated by the addition of 50 µM propranolol to PC3 or MCF7 cells was defined by three time constants: rise, dwell, and fall times, represented as box plots (n = 5–8 imaging experiments per cell line, with n = 151–448 single cell measurements). The Ca2+i release activated by propranolol had comparable time constants in both cancer cell lines. (B) Superimposed traces for individual cells of the propranolol-induced Ca2+i release in PC3 and MCF7 cell lines; arrowheads indicate the time of propranolol addition to the cells.
Role of Calcium-Induced Calcium Release Mechanism in the Propranolol-Induced Ca2+i Release.
We tested the hypothesis that CICR is the mechanism by which propranolol-induced Ca2+i release occurs in cancer cells. Four sets of experiments were performed. First, we added 5 mM EGTA to chelate the residual Ca2+ (>60 µM in the PBS used for calcium imaging experiments), as it is known that even micromolar levels of extracellular Ca2+ can activate CICR pathways (Berridge et al., 2003; Endo, 2009). The addition of 5 mM EGTA, which chelates extracellular Ca2+ concentration to <10 nM (Petrou et al., 2017), significantly inhibited the propranolol-induced Ca2+i release (Fig. 6).

Calcium-induced calcium release (CICR) as a putative mechanism for the propranolol-induced Ca2+i release. PC3 cells were treated with 50 µM propranolol (Pro; added as a bolus) and different chelators or inhibitors of CICR pathways; the mobilization of free Ca2+i release was monitored by time-lapse confocal microscopy. (A) Amplitude of the propranolol-induced Ca2+i release, calculated as fold increase in the fluorescence intensity of the calcium waveform (ΔF/F0). Box plots (L–R): 1) control cells treated with propranolol only, 2) EGTA 5 mM (chelation of extracellular calcium), 3) dantrolene 10 µM (inhibition of the ER RyR channels), 4) 4-CmC 1 mM (depletion of ER stores via activation of RyR channels), 5) 2-APB 50 µM (inhibition of the ER IP3 receptor channels), and 6) thapsigargin 5 µM (exhaustion of ER stores via inhibition of SERCA pump). Blocking CICR pathways significantly inhibited the propranolol-induced Ca2+i release (Mann-Whitney U Test, ***P < 0.001; n = 3–8 imaging experiments and n = 186–575 single-cell measurements per condition). (B) Superimposed traces for individual cells of the propranolol-induced Ca2+i release after EGTA, dantrolene, 4-CmC, 2-APB, or thapsigargin; the time of propranolol addition is indicated by arrowheads. A representative trace for propranolol-induced Ca2+i release can be found in Fig. 2A.
Second, we used dantrolene and 4-CmC, modulators of RyR channels found in the ER (Zorzato et al., 1993; Zhao et al., 2001), to determine whether the ER calcium stores were activated in response to extracellular addition of propranolol. Incubation with 10 µM dantrolene, an inhibitor of RyRs, significantly inhibited the Ca2+i release induced by propranolol (Fig. 6). Incubation of cells with 1 mM 4-CmC, an activator of RyRs that is known to induce Ca2+i stores (Supplemental Fig. 3A), also inhibited the propranolol-induced Ca2+i release (Fig. 6).
Third, we used 2-APB, an inhibitor of IP3-induced Ca2+i release from the IP3 receptor channels located in the ER (Maruyama et al., 1997) and also known to inhibit various TRP ion channels (Xu et al., 2005; Togashi et al., 2008). Incubation with 50 µM 2-APB, a concentration within the range of IC50 values for the inhibition of IP3-induced Ca2+i release (Maruyama et al., 1997; Bootman et al., 2002; Saleem et al., 2014), also abolished the propranolol-induced Ca2+i release (Fig. 6). Similar results were observed when cells were incubated with 25 and 100 µM 2-APB (Supplemental Fig. 4).
Fourth, incubation with 5 µM thapsigargin, an inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase or SERCA (Thastrup et al., 1990), completely abolished the propranolol-induced Ca2+i release (Fig. 6). Thapsigargin, like 4-CmC, discharges the Ca2+i from thapsigargin-sensitive stores (e.g., the ER; Supplemental Fig. 3B) and has been previously used to investigate ligand-induced Ca2+i release (Thrasivoulou et al., 2013). In summary, these results indicate that the propranolol-induced Ca2+i release is likely to be a CICR-facilitated mechanism in which extracellular calcium contributes to the activation of Ca2+i release from the ER through RyR channels and IP3 receptor channels.
β-ARs and Propranolol-Induced Ca2+i Release.
The involvement of β-ARs in general for the activation of Ca2+i release by propranolol was investigated by using two different nonselective β-blockers: sotalol and timolol. We tested the hypothesis that blocking β-ARs using the known nonselective β-blockers would interfere with the propranolol-induced Ca2+i release. Neither sotalol nor timolol activated Ca2+i release in PC3 cells, and this was further confirmed by treating the same cells with loxapine (Fig. 4C; Supplemental Fig. 5); however, incubating the cells with either sotalol or timolol significantly inhibited the propranolol-induced Ca2+i release (Fig. 7A), suggesting that propranolol exerts its function via the β-ARs. The Ca2+i release induced by loxapine (a dibenzoxazepine) remained unaffected by preincubation with sotalol or timolol (Fig. 7B).

Preincubation with nonselective β-blockers precludes normal propranolol-induced Ca2+i release. (A) PC3 cells were loaded with the calcium indicators Fluo-4 and FuraRed, as in previous experiments. Cells were pretreated with 250 µM of either sotalol or timolol (added as a bolus), nonselective β-blockers used to block the β-ARs. After 2 minutes of incubation, 50 µM propranolol was added to the cells, and intracellular calcium levels were monitored over time. Sotalol and timolol significantly inhibited the propranolol-induced Ca2+i release. (B) Control experiments were performed likewise using loxapine, a drug from a different pharmacologic class (i.e., dibenzoxazepine) that is known to activate Ca2+i release in these cells (Petrou et al., 2017). The loxapine-induced Ca2+i release was not affected by the blockade of β-ARs with sotalol or timolol (n = 3–5 experiments and n = 191–441 individual cells per condition; Mann-Whitney U Test, ***P < 0.001, n.s., nonsignificant). (C) Superimposed traces for individual cells of the propranolol- and loxapine-induced Ca2+i release after sotalol and timolol incubation; the time of propranolol or loxapine addition is indicated by arrowheads. Representative Ca2+i release traces are shown in Fig. 2A for propranolol and Fig. 4 insert for loxapine.
Electrophysiological Characteristics of β-Blockers on Medium-Throughput Whole-Cell Currents in PC3 Cells.
The electrophysiological characteristics of the four β-blockers on the endogenous currents in nonexcitable cancer cells, are not known (see Supplemental Table 2 for details on the electropharmacology of β-blockers in other cell types). We sought to establish a basic characterization of the effects of β-blockers on the endogenous whole-cell currents in PC3 cells using medium-throughput recording. Six concentrations of each β-blocker were tested in the cells, based on the pharmacologic doses (Supplemental Fig. 1; Joint Formulary Committee, 2017) and the IC50 values described previously (Supplemental Table 2), following a voltage-clamp protocol with a command ramp from −120 to +120 mV. Figure 8 shows the concentration dependence of whole-cell current regulation by β-blockers in PC3 cells at positive (+100 mV) and a negative (−100 mV) potential. β-Blockers regulate whole-cell currents with distinct features (Fig. 8, A–D); atenolol inhibited whole-cell currents at positive, but not at negative potentials, and metoprolol activated currents at negative and positive potentials. Both these effects were concentration-dependent.

β-Blocker regulation of endogenous whole-cell currents in PC3 cells. Single cells were recorded under whole-cell configuration using the QPatch automated cell patch-clamp system, which implements microfluidic control of drug delivery. (A–D) Endogenous whole-cell currents were measured by adding an increasing concentration (0–500 µM, with solution changes within 500 milliseconds) of each β-blocker to the same cell clamped at a Vh of −10 mV and voltage ramped from −120 to +120 mV every 3 seconds. Currents at +100 and −100 mV were measured with or without β-blockers at six different concentrations and are shown as mean ± S.E.M. (n = 5–19 single cells per β-blocker). Changes in cell currents with respect to control (0 µM) were analyzed using the Wilcoxon test; only significant changes are indicated (*P < 0.05; **P < 0.01; ***P < 0.001). (A) Atenolol, (B) metoprolol, (C) propranolol, and (D) sotalol. (E) Representative current-voltage (I–V) curves of control (gray lines) and with β-blockers (colored lines): atenolol at 38 µM (magenta), metoprolol at 56 µM (yellow), propranolol at 100 µM (green), and sotalol at 50 µM (blue) (n = 5–30 cells; mean ± S.E.M.).
In contrast, propranolol enhanced whole-cell currents at both positive and negative potentials at concentrations <8.4 µM. At higher concentrations (i.e., 56–500 µM), currents were inhibited, which we ascribe to nonspecific effects at these very high (>56 µM) levels of the drug. Sotalol did not cause significant alterations to endogenous currents at any of the concentrations that we tested. The representative current-voltage (I-V) curves are shown in Fig. 8E. These results indicate that endogenous currents in PC3 cells can be modulated differently by the β-blockers investigated.
Discussion
In this study, we investigated the direct activation of calcium stores by atenolol, metoprolol, propranolol, and sotalol. We have shown that β-blockers have differential characteristics of Ca2+i mobilization in human cancer cell lines. Propranolol activates free Ca2+i release, whereas atenolol, metoprolol, and sotalol do not. We propose that CICR is a mechanism by which propranolol activates free Ca2+i release from intracellular stores.
Free Ca2+i is an important second messenger owing to its regulatory role of normal (Berridge et al., 2000) and malignant cell function (Prevarskaya et al., 2011). To the best of our knowledge, the regulation of Ca2+i mobilization by β-blockers in cancer cells remains largely unknown (Supplemental Table 1). We found that propranolol activates the release of Ca2+i from the cellular stores with distinct kinetics of rise, dwell, and fall times in both PC3 and MCF7 cancer cell lines. In comparison with previous research analyzing the kinetics of Ca2+i mobilization in response to Wnt ligands (Thrasivoulou et al., 2013) and other MPRCs in clinical use (Petrou et al., 2017), the propranolol-induced Ca2+i time constants (Fig. 5) suggest that this β-blocker activates a slow exhaustion and replenishment of Ca2+i stores, with a short dwell time (i.e., 20 ± 6 seconds, mean ± S.D. of n = 8 imaging experiments).
The propranolol-induced Ca2+i release does not follow a classic pattern. A measurable Ca2+i release (i.e., a waveform with a well time of >15 seconds), is observed in around 30% of cells (n = 518 cells, from n = 6 experiments) at 35 µM propranolol, a variability reflected in the box plot (Fig. 3); however, at 50 µM, >98% of cells show an increase in propranolol-induced Ca2+i release. The data suggest that activation of Ca2+i release in PC3 cells occurs between 35 and 50 µM (Fig. 3) with no significant, observable response to concentrations <35 µM. It should be noted that the Ca2+i release readout is not a direct assay of propranolol binding to effective receptors, which generally follows a classic Michaelis-Menten kinetics. The readout may reflect cooperativity within the numerous intermediate steps until a threshold is achieved to activate the intracellular calcium stores.
Extracellular calcium triggering CICR, an autocatalytic mechanism found in muscle (Endo, 2009) and nonmuscle cells (Verkhratsky and Shmigol, 1996; Petrou et al., 2017), is likely to be at least one of the signals that triggers the release of Ca2+i in response to propranolol. Our dantrolene results (Fig. 6A) suggest that type 1 and/or 3 RyRs are also likely to be involved in the calcium efflux from the ER (Zhao et al., 2001), although there are other RyRs inhibitors, such as ruthenium red (Xu et al., 1999), that we have not tested in our experiments. EGTA and dantrolene did not inhibit the propranolol-induced Ca2+i release completely (Fig. 6B); this may indicate that there are other mechanisms by which stores are activated by propranolol or may be due to a partial calcium chelation and RyR inhibition by these agents.
Depletion of ER Ca2+i with thapsigargin, which inhibits the SERCA pump (Thastrup et al., 1990), or 4-CmC, which discharges the ER via activation of RyR channels (Zorzato et al., 1993; Herrmann-Frank et al., 1996), completely abolished the Ca2+i release induced by propranolol, suggesting that the ER is the main store from which calcium is released in response to this β-blocker (Fig. 6; Supplemental Fig. 3). We do not, however, exclude the possibility that other intracellular calcium stores (e.g., mitochondria) may also contribute to the propranolol-induced Ca2+i release (Michelangeli et al., 2005).
These data also suggest that IP3 is involved in the propranolol-induced Ca2+i release. Here, 2-APB, an agent largely used as an inhibitor of IP3-induced Ca2+i release (Maruyama et al., 1997; Choi et al., 2010; Saleem et al., 2014), abolished the Ca2+i release activated by propranolol (Fig. 6; Supplemental Fig. 4). 2-APB is also thought to block store-operated calcium entry channels (Gregory et al., 2001), the SERCA pump (Missiaen et al., 2001), and some members of the TRP family (Xu et al., 2005; Togashi et al., 2008), although these interactions are complex (Prakriya and Lewis, 2001; Xu et al., 2005) and vary across cell types (Bootman et al., 2002). It is possible that the blockade of TRP channels may also contribute to the inhibition of the propranolol-induced Ca2+i release caused by 2-APB, presuming that TRP channels may be involved in the activation of CICR pathways in response to propranolol (see later). Based on our observations, we suggest that inhibition of the propranolol-induced Ca2+i release by 2-APB indicates the involvement of IP3 as an intracellular transducer that is produced upon propranolol-receptor binding and contributes to the activation of a calcium influx through IP3 receptors in the ER, in agreement with our observations using thapsigargin since IP3-responsive Ca2+i pools are thapsigargin-sensitive (Tanaka and Tashjian, 1993; Tribe et al., 1994).
Class II antiarrhythmics are β-AR antagonists primarily, but they are also known to act upon potassium (Sakuta et al., 1992; Xie et al., 1998; Dupuis et al., 2005; Kawakami et al., 2006; Tamura et al., 2009) and sodium ion channels (Desaphy et al., 2003; Bankston and Kass, 2010; Wang et al., 2010a) (Supplemental Table 2). I–V curves of whole-cell patch-clamp recordings (Fig. 8E) showed the characteristics of an outward rectifying K+ current, a current that has been previously described for PC3 cells (Laniado et al., 2001). Propranolol inhibits these endogenous currents similarly to those described for antiarrhythmic MPRCs, such as dofetilide (Petrou et al., 2017). At low concentrations (between 0.04 and 8.4 µM), there is an increase in the inward and outward currents, which are inhibited at higher concentrations (>56 µM). In view of the absence of a measurable Ca2+i release at <35 µM propranolol, we speculate that this observation may reflect differences in the experimental design in which drugs are applied to the bath as a bolus for Ca2+i imaging compared with rapid microfluidic application in the QPatch recordings. The amplitude of the propranolol-induced Ca2+i release plateaus between 50 and 250 µM propranolol (Fig. 3), indicating that once the threshold concentration of 50 µM propranolol is reached, the Ca2+i stores are activated. The 50 µM propranolol is an order of magnitude greater than the concentration at which cell membrane currents are activated by propranolol (Fig. 8C). We have previously suggested that there may be a small number of TRP channels (∼100 channels, calculated based on the conductance of control cells’ inward currents; Petrou et al., 2017) that may be responsible for the calcium influx required for the activation of Ca2+i stores, and this may also be the case for propranolol. It is also plausible that there may be independent inhibition of whole-cell currents (based on the multiple interactions of propranolol with sodium and potassium ion channels; Supplemental Table 2), as well as an interdependent CICR-mediated mechanism of Ca2+i release. Such cellular functions for a commonly used antiarrhythmic agent may have implications on the pharmacology of this drug.
The addition of neither sotalol, which did not alter the endogenous whole-cell currents, nor metoprolol, which conversely activated these currents, led to no increase in Ca2+i release. We speculate that atenolol, which does inhibit the endogenous currents but also does not activate Ca2+i release, may not be able to activate the downstream IP3 pathway. The electrophysiological properties described here represent only a beginning of what appears to be intricate and complicated mechanisms regulating intracellular signals transduced by these drugs. Our results give an interesting first insight into how β-blockers might alter the electrical properties of nonexcitable cells, although their effects are complex and require further investigation.
Excitable and nonexcitable cells are thought to have different cell electrical properties but similar intracellular calcium signaling mechanisms (Putney, 1993). The pharmacology of β-blockers is often linked to the regulation of intracellular calcium, most notably in cardiac cells (Weiss et al., 2013). Our observation that β-blockers regulate Ca2+i release differently may reveal new mechanistic aspects of the action of these compounds. Atenolol and metoprolol, for example, are used mostly as antianginal and antihypertensive drugs; propranolol has a wider spectrum of applications besides its cardiovascular use (Joint Formulary Committee, 2017), such as the treatment of essential tremor (Zesiewicz et al., 2002) or anxiety (Steenen et al., 2016). Furthermore, if activation of Ca2+i stores by propranolol is initiated via the β2-AR (see later), this may imply that other tissues expressing this receptor, such as the lungs or blood vessels (Daly and McGrath, 2011), may undergo Ca2+i release events similar to those described in the cell lines used in this study.
Based on the data presented in our study, we propose a model of action for propranolol acting on intracellular calcium stores via a CICR-facilitated mechanism. Extracellular calcium contributes to the opening of the stores and the release of calcium through RyRs, and IP3 acts as an intracellular transducer for the activation of Ca2+i release from the ER (Fig. 9). Our observation that the β2-AR is the main subtype in PC3 cells at the gene expression level (Supplemental Table 3), as has been reported in human prostate tissue (Goepel et al., 1997; Suzuki et al., 2016) and other prostate cancer cell lines (Nagmani et al., 2003; Kasbohm et al., 2005), suggests that propranolol may be exerting its function via this receptor. This suggestion is supported by the observation that blockade of β-ARs with the nonselective β-blockers sotalol and timolol (Baker, 2005) inhibited the propranolol-induced Ca2+i release (Fig. 7), although this could also be attributed to nonselective cellular targets common to sotalol, timolol, and propranolol (Supplemental Table 2). MCF7 cells also express β-ARs (Supplemental Table 3; Shi et al., 2011; Işeri et al., 2014); even if the expression of the β-ARs is not as high in MCF7 cells as in other breast cancer cell lines (Vandewalle et al., 1990), radioligand binding assays have estimated that there are ∼80,000 binding sites per cell in MCF7 cells (Gargiulo et al., 2014).
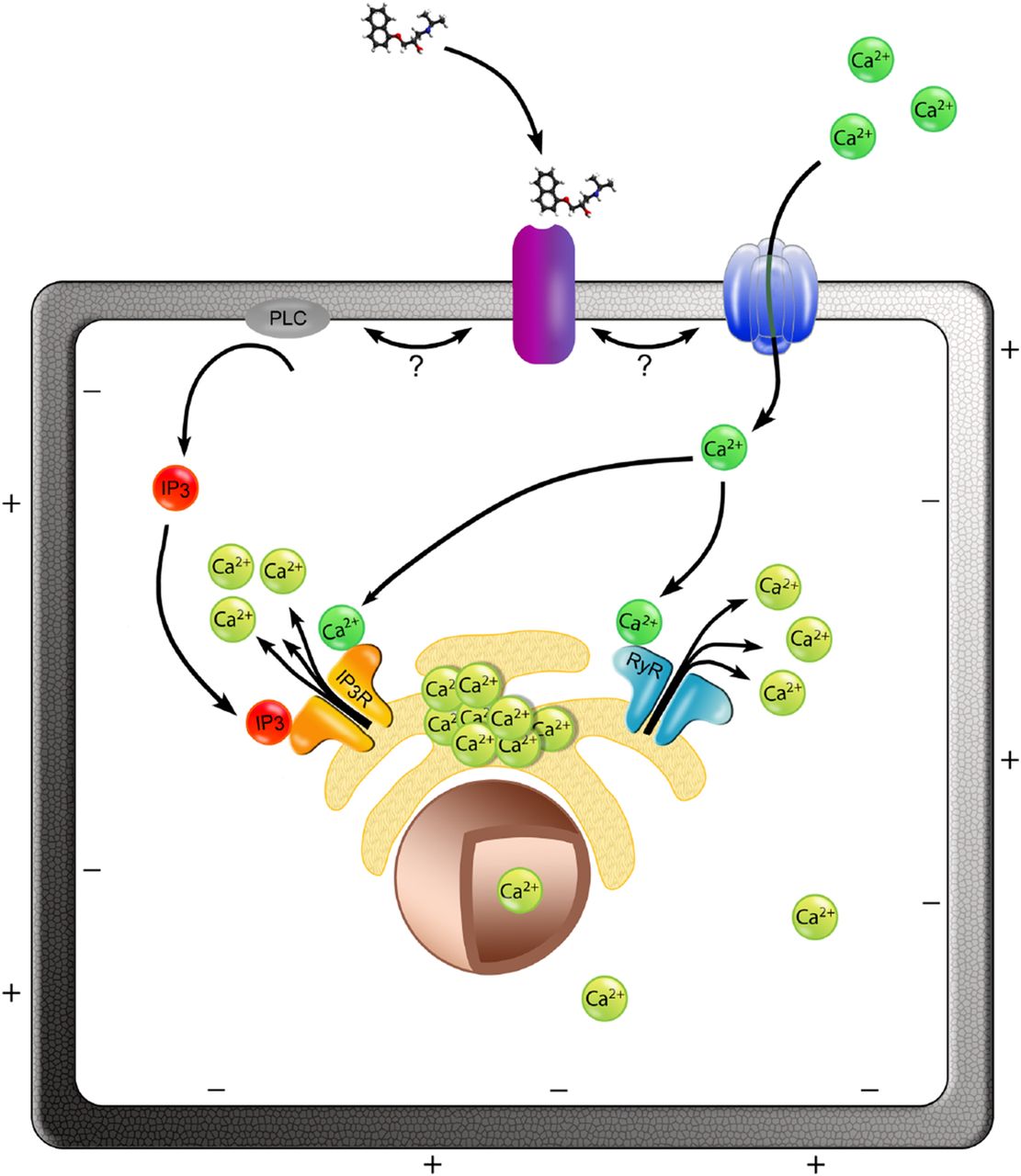
Proposed model for the activation of Ca2+i release by propranolol via a CICR-facilitated mechanism. We propose that the binding of propranolol to its receptor in the cell membrane (purple) triggers the downstream activation of Ca2+i release from cellular stores (e.g., the ER depicted in the model). Extracellular calcium (dark green) contributes to the propranolol-induced Ca2+i release; small extracellular calcium influx may enter the cell via a calcium channel or a permeable cation channel (dark blue), which is activated by propranolol-receptor binding. Extracellular calcium promotes the release of Ca2+i from ER via RyRs (light blue). Additionally, propranolol-receptor binding results in the production of IP3 (red) through the phospholipase C (PLC; gray), and this initiates a Ca2+i efflux via the IP3 receptors (IP3R; orange) present in the ER surface. Once released, the free cytosolic calcium (light green) can enter the nucleus depolarizing the nuclear envelope.
In the model we propose (Fig. 9), binding of propranolol to its receptor may activate extracellular calcium entry, which can trigger store-operated calcium release via CICR. Routes for calcium entry can include specific calcium channels as well as nonspecific cation channels that have significant calcium permeability, such as members of the TRP family (Clapham et al., 2001), some of which are known to be expressed in cancer cells (Bödding, 2007), including prostate cancer (Wissenbach et al., 2004; Bidaux et al., 2007; Prevarskaya et al., 2007). The focus of our future studies will be to investigate the receptors, ion channels, and downstream pathways responsible for the propranolol-induced Ca2+i release described here and examine the effects in the transcriptional profile of cancer cells.
Propranolol is known to inhibit cell migration in several in vitro and in vivo models of cancer, including breast (Campbell et al., 2012; Işeri et al., 2014; Pon et al., 2016), colon (Masur et al., 2001; Işeri et al., 2014), angiosarcoma (Stiles et al., 2013), and prostate cancer (Palm et al., 2006). Free Ca2+i is an important regulator of tumor metastasis, and several Ca2+-dependent mechanisms contribute to malignant cell migration (Prevarskaya et al., 2011). Propranolol has shown greater antimigratory effects than other β-blockers, such as atenolol (Masur et al., 2001; Işeri et al., 2014). In cancer cell lines, including PC3 cells, propranolol (between 100 and 200 µM) was shown to inhibit proliferation and induce apoptosis (Zhang et al., 2010; Brohée et al., 2015; Coelho et al., 2015; Wrobel and Le Gal, 2015; Chin et al., 2016; Wei et al., 2016; reviewed in Pantziarka et al., 2016). The concentration at which propranolol exerted these antiproliferative and proapoptotic effects is within the range at which we observe propranolol-induced Ca2+i release in PC3 and MCF7 cells. It is also notable that in the same studies (Zhang et al., 2010; Coelho et al., 2015; Wrobel and Le Gal, 2015; Chin et al., 2016; Wei et al., 2016) neither atenolol nor metoprolol inhibited cell proliferation or induced apoptosis.
Our results point to a novel action of propranolol and its potential as a regulator of the magnitude and duration of Ca2+i release in vitro. Our finding that propranolol mobilizes free Ca2+i, which distinguishes this drug from other commonly used β-blockers, opens new possibilities into how propranolol may contribute to the inhibition of malignant cell migration and proliferation, whereas other β-blockers that do not activate Ca2+i release may not exert the same effect. This mechanism may thus be relevant patients who are treated with these drugs (Baker et al., 2011; Pantziarka et al., 2016).
Acknowledgments
We thank Jane Pendjiky, University College London, for help with the preparation of Fig. 9.
Authorship Contributions
Participated in research design: Ahmed, Reyes-Corral, Sørensen.
Conducted experiments: Reyes-Corral, Sørensen.
Contributed to new reagents or analytical tools: Thrasivoulou, Dasgupta, Ashmore.
Performed data analysis: Reyes-Corral, Sørensen, Ashmore, Ahmed.
Wrote or contributed to the writing of the manuscript: Reyes-Corral, Ashmore, Ahmed.
Footnotes
- Received October 12, 2018.
- Accepted January 2, 2019.
This work was supported by the Prostate Cancer Research Centre [UK Charity no. 1156027] (A.A.) and a Ph.D. studentship sponsored by the National Institute for Health Research Biomedical Research Centre at Guy’s and St. Thomas’ National Health Service Foundation Trust and King’s College London (M.R.-C.).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- 2-APB
- 2-aminoethoxydiphenyl borate
- β-AR
- β-adrenoceptor
- Ca2+i
- free intracellular calcium
- CICR
- calcium-induced calcium release
- 4-CmC
- 4-chloro-m-cresol
- DMSO
- dimethyl sulfoxide
- ER
- endoplasmic reticulum
- IP3
- inositol-1,4,5-trisphosphate
- MPRC
- membrane potential regulating compound
- PBS
- phosphate-buffered saline
- RyR
- ryanodine receptor
- SERCA
- sarco/endoplasmic reticulum Ca2+ ATPase
- TRP
- transient receptor potential channels
- Copyright © 2019 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}