


Abstract
Diabetic nephropathy remains an area of high unmet medical need, with current therapies that slow down, but do not prevent, the progression of disease. A reduced phosphorylation state of adenosine monophosphate-activated protein kinase (AMPK) has been correlated with diminished kidney function in both humans and animal models of renal disease. Here, we describe the identification of novel, potent, small molecule activators of AMPK that selectively activate AMPK heterotrimers containing the β1 subunit. After confirming that human and rodent kidney predominately express AMPK β1, we explore the effects of pharmacological activation of AMPK in the ZSF1 rat model of diabetic nephropathy. Chronic administration of these direct activators elevates the phosphorylation of AMPK in the kidney, without impacting blood glucose levels, and reduces the progression of proteinuria to a greater degree than the current standard of care, angiotensin-converting enzyme inhibitor ramipril. Further analyses of urine biomarkers and kidney tissue gene expression reveal AMPK activation leads to the modulation of multiple pathways implicated in kidney injury, including cellular hypertrophy, fibrosis, and oxidative stress. These results support the need for further investigation into the potential beneficial effects of AMPK activation in kidney disease.
Introduction
The incidence of metabolic syndrome is rising across the world, presenting patients and society with an increasing need for transformative therapies to treat this cluster of comorbidities. Patients who suffer from diabetic nephropathy (DN), a form of chronic kidney disease that is linked mechanistically to diabetes and metabolic syndrome, are in acute need of disease-modifying treatment options that will delay or prevent the progression to end stage renal disease (Gallagher and Suckling, 2016). The etiology of this disease progression is complex because of the interplay between systemic metabolic and vascular dysfunctions that involves both tubular and glomerular renal systems; this has made the rational identification of novel disease-modifying therapeutic targets difficult (Vallon and Komers, 2011). We have chosen to focus on targets in which there is a concordance between genetic loci associated with disease and evidence of efficacy in preclinical models; one potential therapeutic target that satisfies these two criteria is the protein kinase, adenosine monophosphate-activated protein kinase (AMPK) (Hallows et al., 2010; Köttgen et al., 2010).
AMPK is a heterotrimeric kinase that is activated by changes in cellular energy levels, leading to phosphorylation of its target proteins, which include acetyl-CoA carboxylase (ACC), TSC2, raptor, TBC1D1, and PGC1α, resulting in increased energy production and reduced energy consumption (Mihaylova and Shaw, 2011). AMPK has been studied in many organ systems and has been actively pursued as a therapeutic target for metabolic syndrome, in part because of the observation that elevated nutrient status results in low AMPK activity (Steinberg and Kemp, 2009). Reduced phosphorylation of the AMPK α subunit at the activating threonine 172 (Thr172) site has been observed in renal glomeruli of diabetic patients, suggesting that low AMPK activity in the diabetic state could be part of the etiology of DN progression (Dugan et al., 2013). Multiple interventional studies in rodent models of diabetic kidney disease have suggested the potential benefits of activating AMPK for DN. Many of these studies used activators of AMPK that also have AMPK-independent activities, which could impact kidney function autonomously or nonautonomously and are limited in their capacity to activate AMPK at clinically relevant doses (Lee et al., 2007; Sharma et al., 2008; Dugan et al., 2013; Kim et al., 2013; Yuan et al., 2014). Therefore, we set out to identify novel, specific, small molecule activators of AMPK that hold the potential for clinical development as a treatment option for DN.
Materials and Methods
Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) Protection-Activation Assay.
AMPK allosteric activation and/or protection from dephosphorylation was determined using a novel two-step TR-FRET kinase assay designed to measure both changes in intrinsic activity (activation) and phosphorylation at the Thr172 site of the α-subunit (protection). The first step of the assay involves treatment with a phosphatase to dephosphorylate pAMPK by approximately 50%. This condition was chosen since it reduced the population of pAMPK sufficiently to generate an acceptable protection assay window, while preserving enough pAMPK to measure activity and allosteric activation. Following the dephosphorylation step, the remaining phosphatase activity was terminated with okadaic acid, and kinase activity was monitored using a TR-FRET assay format as previously described utilizing a Cy5-labeled SAMS peptide and a europium-labeled phospho-ACC [serine 79 (Ser79)] mouse monoclonal antibody (Calabrese et al., 2014).
All assays were performed in 384-well low-volume white opaque plates (Corning, New York) with a 20 μl total reaction volume. Reactions were carried out in assay buffer containing 50 mM HEPES, pH 7.5, 1 mM EGTA, 10 mM MgCl2, 0.25 mM DTT, 0.01% Tween-20, and 0.01% bovine serum albumin. Compound was tested at 11 different doses against human AMPK α1β1γ1, rat AMPK α1β1γ1, human AMPK α1β2γ1, human AMPK α2β1γ1, human AMPK α2β2γ1, and human AMPK α2β2γ3. All assays were performed with 50 nM Cy5-labeled SAMS peptide and at an ATP concentration equal to the Km value for each isoform. (20, 20, 40, 20, 40, and 100 μM final for AMPK α1β1γ1, rat AMPK α1β1γ1, human AMPK α1β2γ1, human AMPK α2β1γ1, human AMPK α2β2γ1, and human AMPK α2β2γ3, respectively).
Compound was serially diluted in 100% dimethylsulfoxide (DMSO) and 1 μl was spotted in duplicate into a compound plate. For each compound plate, corresponding positive control wells (40 μM AMP) and negative control wells (DMSO) were also added. The compounds were then diluted to 8% DMSO in buffer and 5 μl was transferred to a white assay plate. To this mixture, 5 μl of AMPK enzyme diluted in assay buffer was added to the assay plate (0.25, 0.25, 0.5, 2, 1, and 1 nM final for human AMPK α1β1γ1, rat AMPK α1β1γ1, human AMPK α1β2γ1, human AMPK α2β1γ1, human AMPK α2β2γ1, and human AMPK α2β2γ3, respectively) and incubated at room temperature for 15 minutes. Next, 5 μl of PP2a (2.5, 2.5, 5.0, 0.25, 0.25, and 0.25 nM final for AMPK α1β1γ1, rat AMPK α1β1γ1, human AMPK α1β2γ1, human AMPK α2β1γ1, human AMPK α2β2γ1, and human AMPK α2β2γ3, respectively) diluted in assay buffer was added and the plate was incubated for an additional 60 minutes. PP2a was selected not only for its activity against pAMPK, but also for its ability to be inhibited by okadaic acid, a potent inhibitor that does not interfere with the assay technology. The phosphatase treatment was terminated and the kinase reaction initiated with the addition of 5 μl of okadaic acid (50 nM final; Calbiochem, San Diego, CA), Cy5-SAMS peptide, and ATP diluted in assay buffer. The kinase reactions were incubated for 60 minutes at room temperature and terminated by the addition of 10 mM EDTA and 2 nM Eu-pACC antibody in LANCE Detection Buffer (Perkin Elmer, Waltham, MA). Kinase activity was monitored by excitation at 320 nm and measuring emission at 665 and 615 nm, respectively, using the EnVision Multilabel Reader (Perkin Elmer) in TR-FRET mode. The final ratio of fluorescence emissions at wavelengths 665/615 nm (emission from phosphorylated SAMS peptide/emission from the europium antibody) was used to calculate the percentage of the effect relative to the AMP and DMSO controls. The percentage of the effect values were then plotted against the concentrations of the compound and the EC50 values were calculated using GraphPad prism software (GraphPad Software Inc., La Jolla, CA).
Protection Assay.
The ability of AMPK activators to protect pAMPK from dephosphorylation was determined using dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) technology. Briefly, test compounds were incubated with fully phosphorylated biotin acceptor peptide–tagged AMPK in assay buffer consisting of 50 mM HEPES, pH 7.5, 1 mM EGTA, 10 mM MgCl2, 0.25 mM DTT, 0.01% Tween-20, and 0.01% bovine serum albumin. PP2a was then added to the mixture and allowed to proceed for 60 minutes at room temperature to dephosphorylate the AMPK protein by about 70%. Reactions were terminated by the addition of okadaic acid and the mixture was transferred to a DELFIA streptavidin-coated 384-well white plate and allowed to incubate with gentle shaking for 1 hour at room temperature. Wells were washed 6× and the captured AMPK was probed with primary anti-α subunit-pThr172 antibody for 45 minutes with gentle shaking at room temperature, and then washed 5× as previously described. Wells were probed with DELFIA Eu-N1-labeled anti-rabbit antibody for 45 minutes with gentle shaking and washed as previously described. The signal was developed by the addition of DELFIA-enhancement solution and time-resolved fluorescence was measured using an EnVISION plate reader (Perkin Elmer) with the factory-set DELFIA europium protocol (excitation at 340 nm and emission at 615 nm).
Determination of the Mechanism of Activation.
Steady-state kinetic experiments to determine the mechanism of allosteric activation of the compounds were performed by monitoring incorporation of 33P-phosphate from 33P-ATP into the SAMS peptide substrate as previously described (Calabrese et al., 2014; Rajamohan et al., 2015).
Crystallization and Refinement.
The α1β1γ1 crystallization construct of rat AMPK was expressed and purified as previously described with the exception of a single mutation of Gln109 in the β1 subunit to His as is found in the human protein (Calabrese et al., 2014). His109 lies in the vicinity of the allosteric binding site; however, the limited resolution of X-ray diffraction data do not permit detailed analysis of the subtle differences in the structure caused by this mutation. Lysine 29 of the α subunit is modeled in a conformation similar to previous observations although electron density is not well-defined for the side chain.
Crystallization was carried out by sitting drop vapor diffusion by mixing equal volumes of protein (∼11 mg/ml) and precipitant. Crystallization conditions consisted of a grid around the base condition of 100 mM trisodium citrate, 500 mM ammonium sulfate, 900 mM lithium sulfate, and 4% glycerol. Crystals grew over the course of 3–10 days at approximately room temperature.
To determine the structure with PF-249, crystals were soaked overnight in mother liquor supplemented with 400 μM AMP, 400 μM staurosporine, and 500 μM PF-249 with 10% glycerol. For cryoprotection, crystals were transferred to an identical solution containing 28% glycerol. Data were collected at the IMCA-CAT beamline at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL) and were processed using HKL2000 (Otwinowski and Minor, 1997). Structure was solved by molecular replacement using our previous structure (4QFR) as a search model. Refinement and rebuilding were carried out using autoBUSTER and Coot and figures were generated using PyMOL (Schrödinger, New York, NY) (Blanc et al., 2004; Emsley and Cowtan, 2004). Nucleotides on the γ subunit were modeled as previously described and subjected to the same caveats (Calabrese et al., 2014).
Quantification of β1 AMPK.
Kidney samples were collected from C57Bl6J mice (The Jackson Laboratory, Bar Harbor, ME), Wistar Han rats (Charles River Laboratories, Wilmington, MA), cynomologus nonhuman primates, and post mortem human (Analytical Biological Services, Inc., Wilmington, DE). The rodent tissue samples were immediately snap-frozen in liquid nitrogen. Primate samples were collected during necropsy and frozen on dry ice. Human kidney samples were acquired in collaboration with the Cleveland Clinic (Cleveland, OH) and Cornell University (Ithaca, NY). The amount of β1 and β2 AMPK was quantified using a modified version of the ThermoFisher Scientific (Waltham, MA) AMPK phospho-enzyme-linked immunosorbent assay (ELISA) kit. Kidney lysate from three species was added to the microwells of two plates precoated with an AMPKα mouse antibody. Recombinant human α1β1γ1 AMPK was used as a standard curve on one plate. To detect β1 AMPK, rabbit anti-AMPK β1 (4 μg/ml) was added to each well. On the second plate, recombinant human α2β2γ1 was used as a standard curve. To detect β2 AMPK, rabbit anti-AMPK β2 (4 μg/ml) was added to each well. Anti-rabbit IgG, horseradish peroxidase (HRP)–linked antibody, was then used to recognize the bound detection antibody. The HRP substrate, tetramethylbenzidine, was added to develop color. The magnitude of the absorbance was measured at 450 nm. The standard curve was plotted for each plate and data were interpolated to the β1 or β2 standard curves, respectively, using SoftMax Pro (Molecular Devices, LLC, Sunnyvale, CA). Picomolar per gram total protein values were calculated for the β1 isoform in Excel, as well as the relative percentage of β1 versus β2.
In Vitro Knockdown of pACC/tACC.
The 293FT cells were grown in Dulbecco’s modified Eagle’s medium prepared with 10% fetal bovine serum, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 6 mM L-glutamine, and 1% penicillin-streptomycin. The siRNA duplexes purchased from Life Technologies (Carlsbad, CA) (s11060 for PRKAB1 knockdown and negative control 1) were complexed with RNA MAX following the manufacturer’s protocol. The control and PRKAB1 siRNA were added to cells and incubated overnight. After 48 hours, cells were serum starved in Opti-MEM (ThermoFisher Scientific, Waltham, MA) reduced serum medium for 2 hours. PF-249 and PF-06409577 were prepared in DMSO and cells were treated with a final concentration of 1 and 3 μM with 0.1% final DMSO concentration. Cells pretreated with both the control-scrambled siRNA and the PRKAB1 siRNA were dosed with either DMSO or compound. After 1 hour, cells were rinsed with phosphate-buffered saline and lysed in ice-cold lysis buffer. Protein content was quantified by a bicinchoninic acid based protein assay.
Lysate was prepared for western blot analysis by adding reducing agent and lithium dodecyl sulfate and boiling for 10 minutes. Samples were loaded onto two identical Bis-Tris gels and run at 200 V for 60 minutes. The Bis-Tris gels were blotted onto a nitrocellulose membrane for 60 minutes at 30 V, following a standard wet transfer protocol. Membranes were blocked in 5% nonfat milk prepared in Tris-buffered saline. After 1 hour, membranes were washed and antibody prepared in 5% bovine serum albumin prepared in Tris-buffered saline with 0.1% Tween-20. Rabbit anti-Pan α AMPK antibody and Rabbit anti-β1 and β2 AMPK antibodies were incubated overnight at 4°C. Membranes were washed and incubated with anti-rabbit IgG, HRP-linked antibody diluted in 5% nonfat milk in Tris-buffered saline. After 1 hour, membranes were washed, enhanced chemiluminescent substrate was added, and membranes were developed in bright-field white light.
The cell culture and siRNA treatment was repeated. Protein content was quantified. Phosphorylated (Ser79) ACC and total ACC protein levels were quantified using two electrochemiluminescent immunoassays based on the technologies provided by Meso Scale Discovery (Rockville, MD). Cell lysates were added to streptavidin-coated plates and incubated for 2 hours at 37°C. To detect phosphorylated (Ser79) ACC and total ACC levels, anti-phospho-ACC (Ser79) rabbit polyclonal antibody (Millipore, Billerica, MA) at 2 μg/ml and anti-ACC rabbit monoclonal antibody at 1 mg/ml (Cell Signaling Technology, Danvers, MA) were used, respectively. SULFO-TAG-labeled goat anti-rabbit antibody was then used to recognize the bound detection antibody. MSD Read Buffer (Meso Scale Diagnostics, Rockville, Maryland) was added to the initial electrochemiluminescent reaction. The magnitude of the luminescence was measured using the SECTOR S600 (Meso Scale Diagnostics, Rockville, Maryland) plate reader.
ZSF1 Rat Studies.
All animal experiments were conducted following study protocols and procedures reviewed and approved by the Pfizer Institutional Animal Care and Use Committee. The facilities that supported this work are fully accredited by AAALAC International (Frederick, MD). For each study, 6-week-old, male ZSF1 lean and ZSF1 obese rats (Charles River Laboratories) were acclimated to the vivarium for 14 days prior to measurement of body weight and baseline evaluation of urine. Six days later, obese rats were assigned to one of five groups based on the urine albumin creatinine ratio (Advia 2400, Siemens, Tarrytown, NY), urine protein creatinine ratio (Advia 2400, Siemens), body weight, 24-hour urine albumin (Advia 2400, Siemens), and urine volume (Supplemental Table 4). Daily administration of 0.5% methylcellulose (by mouth), PF-06409577 at 10, 30, or 100 mg/kg (by mouth), PF-249 at 3, 10, or 30 mg/kg (by mouth), or ramipril in drinking water (∼1 mg/kg/d) (Zoja et al., 2011) was initiated and continued for 68 days. Urine was collected for 24 hours and volume recorded from all lean and obese rats after 14, 28, 42, and 60 days of dosing. On day 63 all rats were administered a final dose after 16-hour overnight fasting. One hour following the final dose, blood glucose was measured by glucometer (AlphaTrak 2, Zoetis, Parsippany, New Jersey) and a 100 μl tail vein blood sample collected and processed for determination of insulin levels (ALPCO, Salem, NH) and total protein (Advia 2400, Siemens). Each rat was then anesthetized with isoflurane. The right kidney was collected and immediately freeze-clamped and transferred to liquid nitrogen storage; the left kidney was fixed in 10% formalin. Rats were then euthanized by exsanguination from the vena cava.
In a separate study, blood pressure was monitored continually in 14-week-old, telemeterized male, obese ZSF1 rats that were administered vehicle or PF-06409577 at 10, 30, or 100 mg/kg (by mouth) (n = 10/group) for 14 days. Fifteen-minute intervals of data from each animal were grouped into blocks of time: 0–2, 2–4, 4–8, 8–12, 12–16, 16–20, and 20–24 hours postdose. Statistical analysis compared the variances of data for drug- and vehicle-treated animals at each time block (adjusted for baseline) using analysis of variance at days 1, 6, and 14.
Kidney Gene Expression.
Tissue was pulverized under frozen conditions and 30 mg lysed in 1 ml of Qiazol reagent (Qiagen, Hilden, Germany) using MP Biomedical Lysing Matrix D tubes. Next, 0.2 ml of chloroform (Sigma Aldrich, St. Louis, MO) was added and tubes were disrupted by vortexing. Samples were then spun at 10,300 rpm at 4°C for 15 minutes to ensure phase separation, and 0.2 ml of the upper aqueous phase was then placed in a gDNA eliminator column (Qiagen) and eluted. The sample was mixed with 0.2 ml of fresh 70% ethanol, and the sample was added to a 96-well RNeasy plate (Qiagen) vacuum system. The wash procedure included in the RNeasy kit was then followed, and the final sample was eluted in 60 ml of RNase-free water. RNA concentration and purity were evaluated using a NanoDrop ND-8000 spectrophotometer (ThermoFischer, Waltham, MA). RNA was then diluted for a concentration of 100 ng per 10 μl, matched with 10 μl of High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, Foster City, CA). cDNA was then processed on a polymerase chain reaction (PCR) GeneAmp System 9700 (Applied Biosystems, Foster City, CA) according to the kit directions. Next, 400 ng of cDNA sample, Taqman primers (Applied Biosystems) and TaqMan Fast Universal PCR Master Mix (Applied Biosystems) was loaded onto 384-well PCR reaction plates (Applied Biosystems) and run on a Viia 7 PCR system (ThermoFisher Scientific, Waltham, MA). Analysis was performed to determine reference gene outliers, and the Ct values were transformed into relative quantification data. The ΔCt value was generated by removing the background of the housekeeping gene from the sample of interest. Results were then generated using the comparative ΔΔCt method and were normalized to the obese vehicle-treated animals. Genes were normalized to PPIa mRNA.
Urine Analysis.
All urine samples were centrifuged to pellet sediment and stored at −80°C prior to analysis. Kim1/Tim1 was measured using the Quantikine RAT TIM-1/KIM-1 Immunoassay (R&D Systems, Minneapolis, MN). Samples were diluted 4-fold and the manufacturer’s protocol was followed with sample values calculated based on the provided kidney injury marker-1 standards. Thiobarbituric acid reactive substance (TBAR) assay was purchased from R&D Systems and used at a 1:2 dilution of urine according to the manufacturer’s protocol. Tissue inhibitor of metalloproteinases 1 (TIMP1) was measured with Sigma Aldrich ELISA using a 20-fold dilution of urine and following the manufacturer’s protocol. Creatinine, total protein, and albumin were measured by a clinical analyzer. Urine markers measured by ELISA were corrected for daily urinary excretion by expression as the ratio with urinary creatinine.
Kidney Histology and Phospho-S6 Quantification.
Rat kidneys were trimmed in a coronal fashion before being immersed in 10% neutral buffered formalin. After fixation, kidneys were processed to paraffin blocks following standard tissue processing protocols. Five micron sections of kidney were sectioned using a rotary microtome and mounted on glass slides for subsequent staining. All immunohistochemistry steps were performed on the Leica Bond III (Leica Microsystems, Buffalo Grove, IL). Briefly, slides were deparaffinized utilizing Leica’s Bond dewax solution and pretreated by using Epitope Retrieval Solution 1(Leica Microsystems) for 30 minutes with heat. Following pretreatment, nonspecific binding was blocked by using Dako’s serum-free protein block (Dako, Carpinteria, CA). A rabbit anti-human phosphor-S6 ribosomal protein ser235–236 monoclonal antibody (Cell Signaling Technology) was incubated at a dilution of 1/200 for 15 minutes at room temperature. Positive staining was detected by using Leica’s Refine Polymer kit, which includes an anti-rabbit polymer and diaminobenzidine for visualization. All slides were counterstained with hematoxylin, dehydrated through a series of graded alcohols and xylenes, and mounted with cover slips. Additionally, a rabbit isotype control (Vector Laboratories, Burlingame, CA) was run in parallel using all of the same pretreatment and detection steps.
Microscope slides with kidney sections stained for pS6 were scanned on the Leica/Aperio AT2 whole slide digital scanner at the 20× objective setting. Images were saved in .svs format and stored in the Aperio eSlide Manager Image Database. An image analysis rule set was created in Definiens Tissue Studio (Definiens, Cambridge, MA) that allowed for the manual outline of individual glomeruli. Manual outlines of glomeruli were sampled from three uniform 10× fields from each section. At least 20 glomeruli from each section were sampled. A threshold for pS6 stain was applied to each glomerulus. A measurement of the percentage of pS6 stain area per glomerulus was transferred to Excel. A mean pS6 stain percentage area per glomerulus was calculated for each group. Statistics and graphing were completed in the GraphPad Prism software.
Determination of pAMPK/tAMPK in Tissues.
Frozen kidney samples were pulverized over liquid nitrogen and transferred to homogenization tubes containing ceramic beads. Ice-cold lysis buffer with protease and phosphatase inhibitors was added to each tube. Samples were secured on the FastPrep-24 benchtop tissue homogenizer and lysed at 6.5 m/s for 60 seconds at 4°C. The homogenate was spun in a table-top microcentrifuge at 20,800 relative centrifugal force for 10 minutes at 4°C. The supernatant was collected and the protein content of the lysate was measured. Lysate was stored at −80°C for future use.
pAMPK/tAMPK Assay Methods.
Phosphorylated (Thr172) AMPK and total AMPK protein levels were quantified using a modified version of the solid-phase sandwich ELISA from Cell Signaling Technology. Kidney tissue lysates were added to microwells precoated with an AMPKα rabbit antibody. Fully phosphorylated recombinant human α2β2γ1AMPK was used as a standard curve. To detect Thr172 phosphorylation or total AMPK levels, anti-phospho AMPK (Thr172) mouse antibody or anti-AMPK β mouse antibody were used, respectively. Anti-mouse IgG, HRP-linked antibody was then used to recognize the bound detection antibody. The HRP substrate, 3,3′,5,5′-tetramethylbenzidine, was added to develop color. The magnitude of the absorbance was measured at 450 nm. The standard curve was plotted and data were interpolated to the standard curve using SoftMax Pro. The ratios of phosphorylated to total AMPK protein were calculated.
Statistical Analysis.
Statistics were calculated in GraphPad Prism. One-way or two-way analysis of variance was performed and multiple comparison analysis was done to evaluate significance between indicated groups; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
PF-06409577 and PF-249 are Selective Activators of AMPK β1-Containing Heterotrimers.
Using a novel AMPK assay format to measure the activation and protection of the AMPK α1β1γ1 heterotrimer, we determined the EC50 values for PF-06409577 to be 6 nM (human) and 5 nM (rat), and for PF-249 to be 9 nM (human) and 5 nM (rat). Both compounds were also tested against the human α1β2γ1, α2β2γ1, and α2β2γ3 heterotrimers, showing weak activity for each (Fig. 1, A and B). PF-06409577 and PF-249 exhibited minimal off-target pharmacology based on a broad panel of other receptors, channels, phosphodiesterases, and kinases (see Supplemental Tables 1–3 and Cameron et al., 2016). Both compounds activated AMPK through direct allosteric activation by reducing the Km value for the peptide substrate from 28 to 6.3 μM for PF-249 and by decreasing dephosphorylation of the activation loop Thr172, catalyzed by the phosphatase PP2A (Supplemental Fig. 1). To further characterize protein-ligand interactions, we determined the crystal structure of PF-249 bound to the α1β1γ1 isoform of AMPK at ∼3.5 Å resolution, observing strong electron density corresponding to the ligand that maps to the interface between the N-lobe of the kinase domain of the α1 subunit and the carbohydrate binding motif of the β-subunit (Fig. 1, C and D).
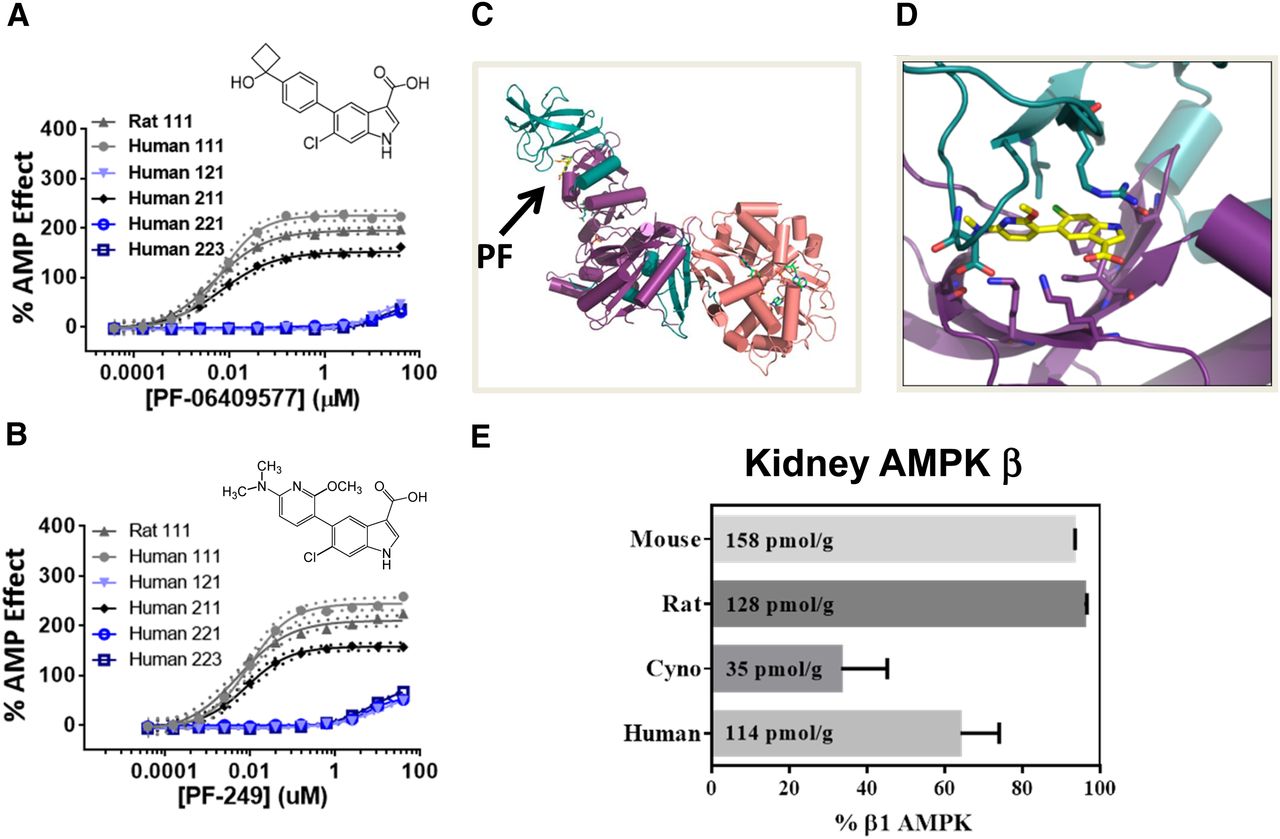
(A) In vitro AMPK activity for selected AMPK heterotrimers (αβγ) activated with PF-06409577. (B) In vitro AMPK activity for selected AMPK heterotrimers (αβγ) activated with PF-249. (C) Ribbon representation of crystal structure of AMPK α1β1γ1 bound to PF-249. (D) Close-up view of the ligand-protein interface. (E) AMPK β subunit levels in kidney tissue were measured by quantitative ELISA and plotted as percentage of AMPK heterotrimer containing the β1 subunit. The β1 subunit protein content (based on the summation of both β1 and β2 subunits) is also shown for each species in tissue (pmol/g). The coordinates of the X-ray structure are available from the Protein Data Bank (accession code: 5T5T).
AMPK β1 Is Highly Expressed in Rodent and Human Kidneys.
The selectivity of the AMPK activators for β1-containing isoforms prompted us to quantify both the total and relative expression of AMPK isoforms across several species of interest. We observed that the β1 subunit is present in 93.1%, 95.5%, 33.3%, and 64.0% of all AMPK heterotrimers in the normal C57Bl6J mouse, Wistar Han rat, nonhuman primate, and human kidney, respectively (Fig. 1E). Across the four species we tested, mouse (158 pmol/g) contained the highest concentration of β1-containing isoforms, followed by rat (128 pmol/g), human (114 pmol/g), and nonhuman primate (35 pmol/g).
PF-06409577 and PF-249 are Selective for AMPK β1 in Cells.
To confirm PF-06409577 and PF-249 could activate AMPK in intact cells and that the β1-selectivity was preserved, experiments were performed in 293FT cells with an RNAi knockdown of the corresponding gene, PRKAB1. The AMPK β1 subunit protein was significantly reduced in cells transfected with RNAi duplexes targeting PRKAB1 mRNA (Fig. 2A). Additionally, RNAi knockdown of PRKAB1 resulted in a lowering of the total AMPK α protein in addition to AMPK β1, which is likely attributable to the high contribution of AMPK β1 to AMPK heterotrimers in the untransfected cells. Both compounds significantly increased phosphorylation of ACC in control cells transfected with the scrambled RNA duplexes, indicative of activation of intracellular AMPK (Fig. 2, B and C). PF-06409577 and PF-249 exhibited reduced ACC phosphorylation in cells with reduced AMPK β1 expression (Fig. 2, B and C).
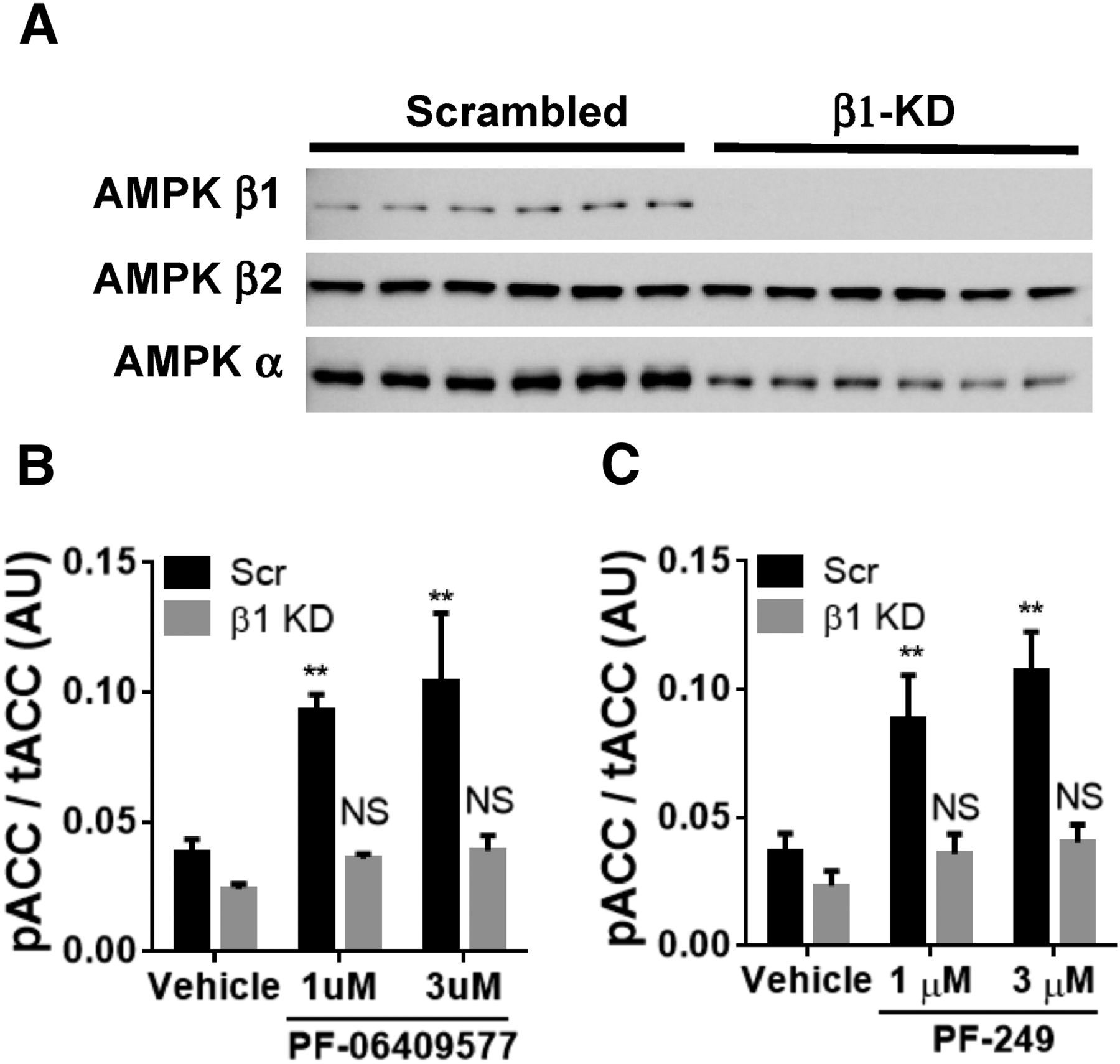
(A) Western blot for AMPK β1, β2, and pan-α in 293FT cells transfected with scrambled siRNA or siRNA targeting AMPK β1. (B) ELISA quantification of ACC phosphorylation status in 293FT treated with siRNA and PF-06409577. (C) ELISA quantification of ACC phosphorylation status in 293FT treated with siRNA and PF-06409577. **P < 0.01.
Both PF-06409577 and PF-249 Are Orally Bioavailable and Dose Dependently Increase the Phosphorylation of AMPK in the Kidney.
The pharmacokinetic and pharmacodynamic properties of PF-06409577 and PF-249 were initially evaluated in male Wistar Han rats. The plasma half-lives of PF-06409577 and PF-249 were 1.06 and 27.2 hours, respectively, with oral bioavailabilities ranging from 47% to 52% (Supplemental Table 4). Maximal doses for subsequent in vivo studies were selected by measuring kidney phosphor-AMPK/total AMPK (pAMPK/tAMPK) ratios following a single, oral dose of either PF-06409577 or PF-249. Both compounds dose dependently increased the pAMPK/tAMPK, with PF-06409577 achieving a maximal response at 100 mg/kg (Cameron et al., 2016); the PF-249 response plateaued at the 30 mg/kg dose (Supplemental Fig. 2).
Activation of AMPK β1-Containing Heterotrimers in a Rat Model of DN Altered the Progression of Proteinuria and Improved Urine Biomarkers of Kidney Disease.
The ZSF1 rat is commonly used as a preclinical model of DN since it exhibits many characteristics of the human disease (Bilan et al., 2011; Boustany-Kari et al., 2016). We employed this model to evaluate whether direct activation of AMPK has an effect on proteinuria and overall kidney function. Obese ZSF1 rats exhibited mild proteinuria at 8 weeks of age and progressed rapidly to more severe kidney dysfunction (Fig. 3A). Oral administration of PF-06409577 (10, 30, and 100 mg/kg every day) resulted in dose-dependent reductions in proteinuria in the obese ZSF1 animals, with greater than 2-fold reduction in 24-hour urinary albumin loss compared with vehicle control after 60 days of treatment (Fig. 3A). Histologic assessment of the kidneys from these rats revealed a reduction in the amount of protein casts in the tubules of both PF-06409577- and Ramipril-treated animals; however, we failed to rigorously identify improvements in glomerular structures of treated animals (Supplemental Fig. 3). Additionally, administration of PF-06409577 had no significant effect on the hyperglycemia and insulin levels displayed by all groups of obese ZSF1 rats (Fig. 3, C and D). Furthermore, when the improvement in proteinuria was compared with the observed plasma drug exposure and corrected for plasma protein binding, the IC50 value for the effect was calculated to be 5.5 nM (Supplemental Figs. 3–5). Moreover, the inhibitory effect on proteinuria correlated with a dose-dependent increase in kidney pAMPK levels (Fig. 3B).

(A) Cumulative urinary albumin excretion over a 24-hour period at multiple time points during a dosing study in obese ZSF1 rats treated with vehicle or PF-06409577; 10 or 11 animals per group. (B) Kidney AMPK phosphorylation status in terminal samples after 8 weeks of dosing, 1 hour after the last dose. Values are normalized such that the obese vehicle group is equal to 100. Significance is indicated compared with the vehicle; 10 to 11 animals per group. (C and D) Fasting blood glucose and insulin after 8 weeks of dosing. (E and F) Mean blood pressure in telemeterized ZSF1 rats treated with vehicle or 10, 30, or 100 mg/kg of PF-06409577. Data are presented as the fold change from the vehicle group during defined periods throughout days 6 and 14. *P < 0.05, ***P < 0.001.
We also assessed the impact of PF-06409577 on systemic blood pressure as a potential explanation for the improvement in kidney function in the ZSF1 rat in telemeterized animals. PF-06409577 delivered at 100 mg/kg had some modest reductions in systemic blood pressure at times during the day, but treatment did not cause a sustained and consistent effect (Fig. 3, E and F). To replicate our initial observation, the related AMPK activator PF-249 was compared with PF-06409577 and the angiotensin-converting enzyme inhibitor, ramipril, in the same model. As observed previously progressive elevation in proteinuria in obese ZSF1 rats was attenuated by both AMPK activators PF-06409577 and PF-249, the latter in a dose-dependent manner following 68 days of dosing (Fig. 4A). Modeling of the dose-dependent improvements in proteinuria observed in this study, using the observed plasma drug exposure corrected for plasma protein binding, revealed IC50 values for the effect of 32 nM for PF-249 and 14.5 nM for PF-06409577 (Supplemental Figs. 3, 6, and 7). The top dose tested for both AMPK activators resulted in a reduction of the urinary albumin creatinine ratio, urinary protein creatinine ratio, and 24-hour urinary albumin excretion equivalent to or greater than that achieved with ramipril (Fig. 4A; Table 1). These improvements in urinary albumin excretion were accompanied by AMPK activation in the kidney for animals treated with AMPK activators (Fig. 4B).

(A) Cumulative albumin excretion over a 24-hour period at multiple time points during a dosing study in ZSF1 rats treated with vehicle, PF-249, PF-06409577, or ramipril (1 mg/kg/d in drinking water); 12 animals per group. (B) Kidney AMPK phosphorylation status in terminal samples after 8 weeks of dosing, 1 hour after the last dose. Values are normalized such that the obese vehicle group equals 100. Significance is indicated compared with the vehicle; 10 to 11 animals per group. **P < 0.01.
Measurements of urinary biomarkers of kidney function after 8 weeks of dosing with vehicle, PF-249, PF-06409577, or Ramipril
Samples are measured from urine samples collected over a 24-hour period; 11–12 animals per group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
PF-06409577 and PF-249 Improve Mechanistic Markers of Kidney Function.
We explored additional endpoints to increase the understanding of the biologic consequences of AMPK activation in the kidney that might account for the improvements in renal function. Urinary levels of kidney injury marker-1, a marker of general tissue damage, were reduced to a similar extent, >40% (P < 0.01), by PF-249, PF-06409577, and ramipril (Table 1). Urinary levels of TIMP1, a tissue metalloprotease inhibitor that is elevated in states of fibrosis, were elevated in obese animals with impaired kidney function and reduced (P < 0.01) by PF-249 and PF-06409577 treatment, with the highest dose groups approaching the levels detected in lean animals with normal kidney function (Table 1). Ramipril also lowered (P < 0.05) TIMP1, although to a lesser degree than the AMPK activators. Urinary TBARs are an indicator of reactive oxygen species–mediated production of reactive lipid species in the setting of kidney dysfunction. As expected, the obese animals in these studies exhibited elevated urinary TBARs. Treatment with PF-249, PF-06409577, and ramipril reduced (P < 0.01) TBAR levels toward those observed in lean animals. In aggregate, these results demonstrated improvements to multiple facets of the diseased ZSF1 kidney, while revealing subtle differences in biologic responses to AMPK activators compared with ramipril.
PF-06409577 Lowers Glomerular mTOR Signaling.
Glomerular mechanistic target of rapamycin (mTOR) activity is an established driver of podocyte hypertrophy and compromised filtration barrier (Gödel et al., 2011; Inoki et al., 2011; Inoki and Huber, 2012; Eid et al., 2013). AMPK is well-known to be negative regulator of mTORC1, raising the possibility that this mechanism could be responsible for the improvements in renal function observed in obese ZSF1 rats treated with AMPK activators. The phosphorylation state of ribosomal S6 kinase (p-S6) is a marker of mTOR activation that has been used histologically to evaluate glomerular mTOR signaling (Gödel et al., 2011). Histologic assessment of obese ZSF1 kidneys revealed kidneys with increased p-S6 levels within the glomeruli (Fig. 5, A–D), consistent with the previous hypothesis. Treatment of obese ZSF1 animals with 100 mg/kg PF-06409577 reduced p-S6 within glomeruli to levels comparable to lean animals and significantly less than vehicle-treated obese ZSF1 rats (Fig. 5, A–D).
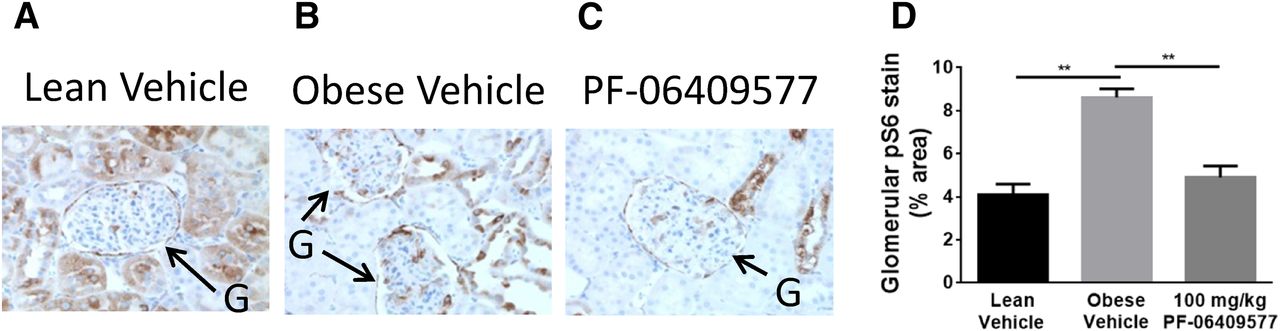
(A–C) Representative histology images of phospo-S6 (p-S6) reactivity in kidneys collected after 8 weeks of dosing vehicle or PF-06409577 to ZSF1 rats; 12 animals per group. Glomeruli are indicated by the arrows and stain area was quantified. (D) Quantitative measure of p-S6 stain area in glomeruli of five lean ZSF1 treated with vehicle, seven obese ZSF1 treated with vehicle, and seven obese ZSF1 animals treated with PF-06409577. **P < 0.01.
PF-06409577 and PF-249 Impact Kidney mRNA of Mechanistic Markers.
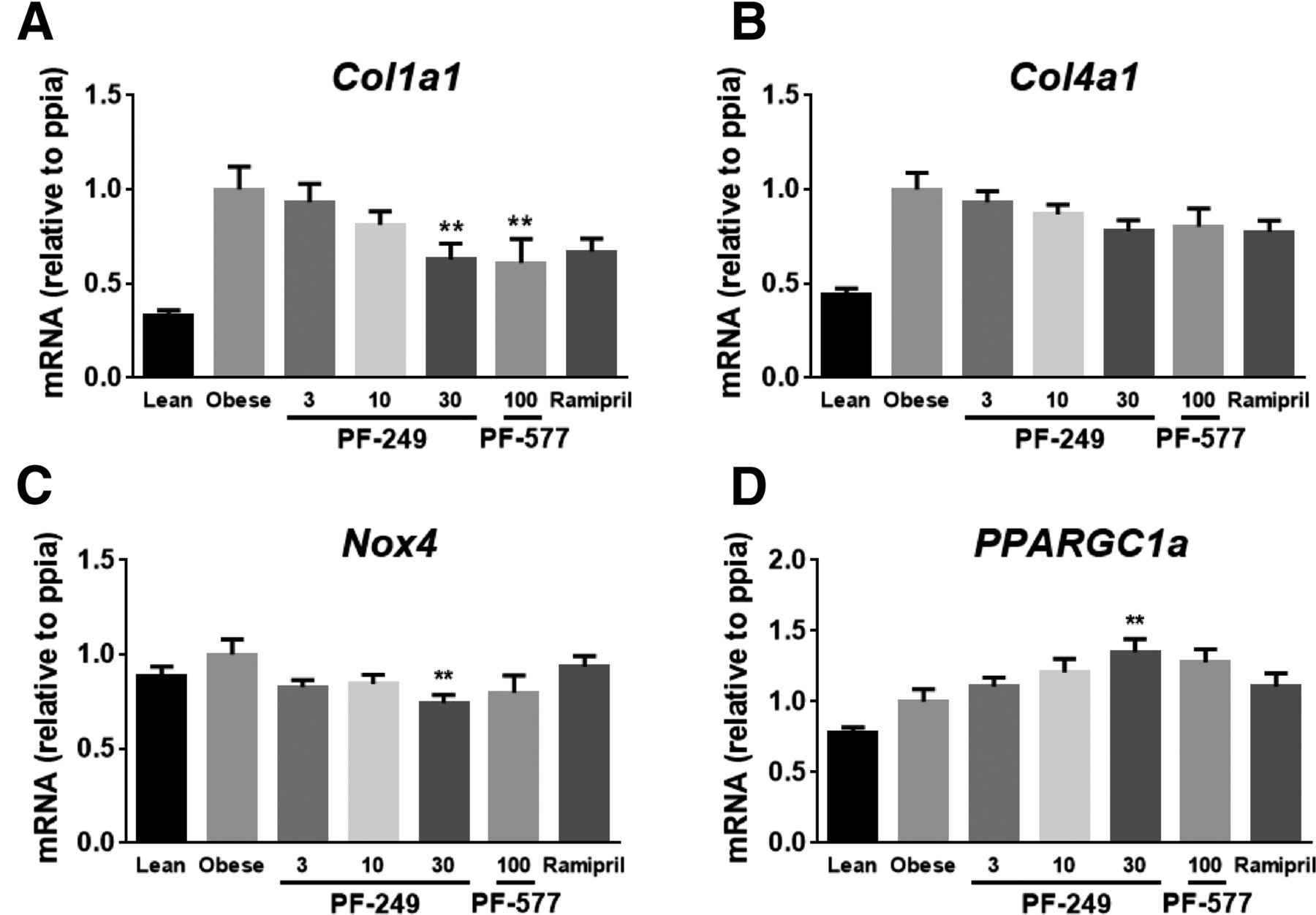
To gain further insight into the mechanism(s) underlying the improvement in kidney function upon AMPK activation, mRNA expression was also evaluated in kidney tissue samples from the chronically treated ZSF1 rats. The potential of AMPK activation to modify fibrosis was assessed by exploring the expression of the Col1a1 and Col4a1 mRNAs that encode the major collagens. Both transcripts were elevated in diseased animals, with PF-249 and PF-06409577 treatment reducing Col1a1 (P < 0.01) but not Col4a1 expression (Fig. 6, A and B). It has been reported that AMPK activation reduces levels of the NADPH-oxidase Nox4 and this may be responsible for improvement of kidney function (Sharma et al., 2008; Eid et al., 2013). In our studies Nox4 mRNA was not significantly elevated in the obese ZSF1 animals, implying Nox4 is not a major contributor to renal dysfunction in this model (Eid et al., 2013). Nevertheless, Nox4 mRNA was modestly decreased in the 30 mg/kg PF-249 dose group (P < 0.05) (Fig. 6C).

Quantitative PCR measurements of mRNA for (A) Col1a1, (B) Col4a1, (C) Nox4, and (D) Ppargc1a in kidney samples from animals treated with 3, 10, and 30 mg/kg PF-249, 100 mg/kg PF-06409577 (PF-577), or ramipril for 8 weeks; 8–10 animals per group. Significance compared with the obese vehicle group. **P < 0.01.
In diabetic kidney models the transcriptional regulator PGC1α, a well-known target of AMPK, has been linked to the modulation of mitochondrial function and the accompanying changes in the oxidative potential and reactive oxygen species generation (Dugan et al., 2013). While PGC1α mRNA was similar in the obese and lean ZSF1 groups, an increase in the PGC1α transcript was observed in the 30 mg/kg PF-249-treated animals (Fig. 6D). The modest changes in whole tissue mRNA levels of the markers of kidney disease in this study may be ascribed to the multiple cell types comprising the nephron. Consequently, these mRNA-based transcriptomics studies were inconclusive in defining a specific mechanism downstream from AMPK activation responsible for the observed improvements in kidney function.
Discussion
Disease-modifying therapies are needed to stem the tide of renal disease in the burgeoning population of individuals with metabolic syndrome. To this end, AMPK activation has shown some promise in this therapeutic area based on the use of imperfect indirect activators that are unlikely to be effective clinically. Here, we describe the identification of PF-06409577 and PF-249, and demonstrate the ability of each to selectively activate AMPK heterotrimers that contain the β1 subunit and modify the rate of disease progression in the ZSF1 rat model of DN.
Both PF-06409577 and PF-249 improve kidney function, with robust reductions in proteinuria that are stable over treatment duration of more than 2 months. Interestingly, the profile of proteinuria improvement appears distinct compared with the angiotensin-converting enzyme inhibitor ramipril, which lowers the urinary albumin creatinine ratio more rapidly but fails to alter the disease progression slope. In contrast, AMPK activation by either compound resulted in an effect that was not evident at 2 weeks of treatment but appeared to alter the rate of increased proteinuria. However, the relationship of the observed effects in the animal model to true delay in disease progression in man is unclear. Nevertheless, the potential for beneficial effects of AMPK activation in multiple relevant cell types in the kidney suggests advantages of this novel approach for the treatment of DN compared with the current standard of care. The reduced mTOR signaling in the glomeruli following PF-06409577 treatment suggests that suppression of this hypertrophic pathway by AMPK activation could be one source of the AMPK-mediated benefit in this DN model. We observed improvement in urinary markers of specific disease processes, including the oxidative stress marker TBARs, fibrotic marker TIMP1, and the general kidney injury marker kidney injury marker-1, in addition to reduction of proteinuria. While treatment did not cause a large drop in systemic blood pressure, there were modest decreases observed at some time periods that could contribute to the observed improvements in disease. Nevertheless, because of the complexity of renal pathology, which includes inflammation, fibrosis, tubular damage, systemic blood pressure dynamic, and glomerular sclerosis, it is unlikely that a single protective mechanism underlies the observed pharmacological effects.
The selective small molecule activators of AMPK β1-containing heterotrimers described here dramatically improve renal function in a rodent model of DN. These results suggest that activators of AMPK warrant further testing as a treatment of human DN, and potentially other kidney diseases that remain inadequately treated by existing medicines.
Authorship Contributions
Participated in research design: Salatto, Miller, Cameron, Ward, Birnbaum, DaSilva-Jardine, Rolph.
Conducted experiments: Salatto, Miller, Cokorinos, Reyes, Ward, Kalgutkar, Tess, Jatkar, Maciejewski, Amaro, Gandhok, Monetti, Cialdea, Bollinger, Kreeger, Coskran, Opsahl, Boucher.
Contributed new reagents or analytic tools: Cameron, Calabrese, Rajamohan, Shavnya, Genung, Edmonds.
Wrote or contributed to the writing of the manuscript: Salatto, Miller, Cameron, Cokorinos, Reyes, Ward, Calabrese, Kurumbail, Kalgutkar, Edmonds, Monetti, Birnbaum, Rolph.
Footnotes
- Received December 10, 2016.
- Accepted March 6, 2017.
All authors were employees of Pfizer Inc. at the time these experiments were conducted.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ACC
- acetyl-CoA carboxylase
- AMP
- adenosine monophosphate-activated protein
- AMPK
- AMP kinase
- DELFIA
- dissociation-enhanced lanthanide fluorescent immunoassay
- DMSO
- dimethylsulfoxide
- DN
- diabetic nephropathy
- ELISA
- enzyme-linked immunosorbent assay
- HRP
- horseradish peroxidase
- mTOR
- mechanistic target of rapamycin
- PCR
- polymerase chain reaction
- PP2A
- Protein phosphatase 2A
- Ser79
- serine 79
- TBAR
- thiobarbituric acid reactive substance
- Thr172
- threonine 172
- TIMP1
- tissue inhibitor of metalloproteinases 1
- TR-FRET
- time-resolved fluorescence resonance energy transfer
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}