


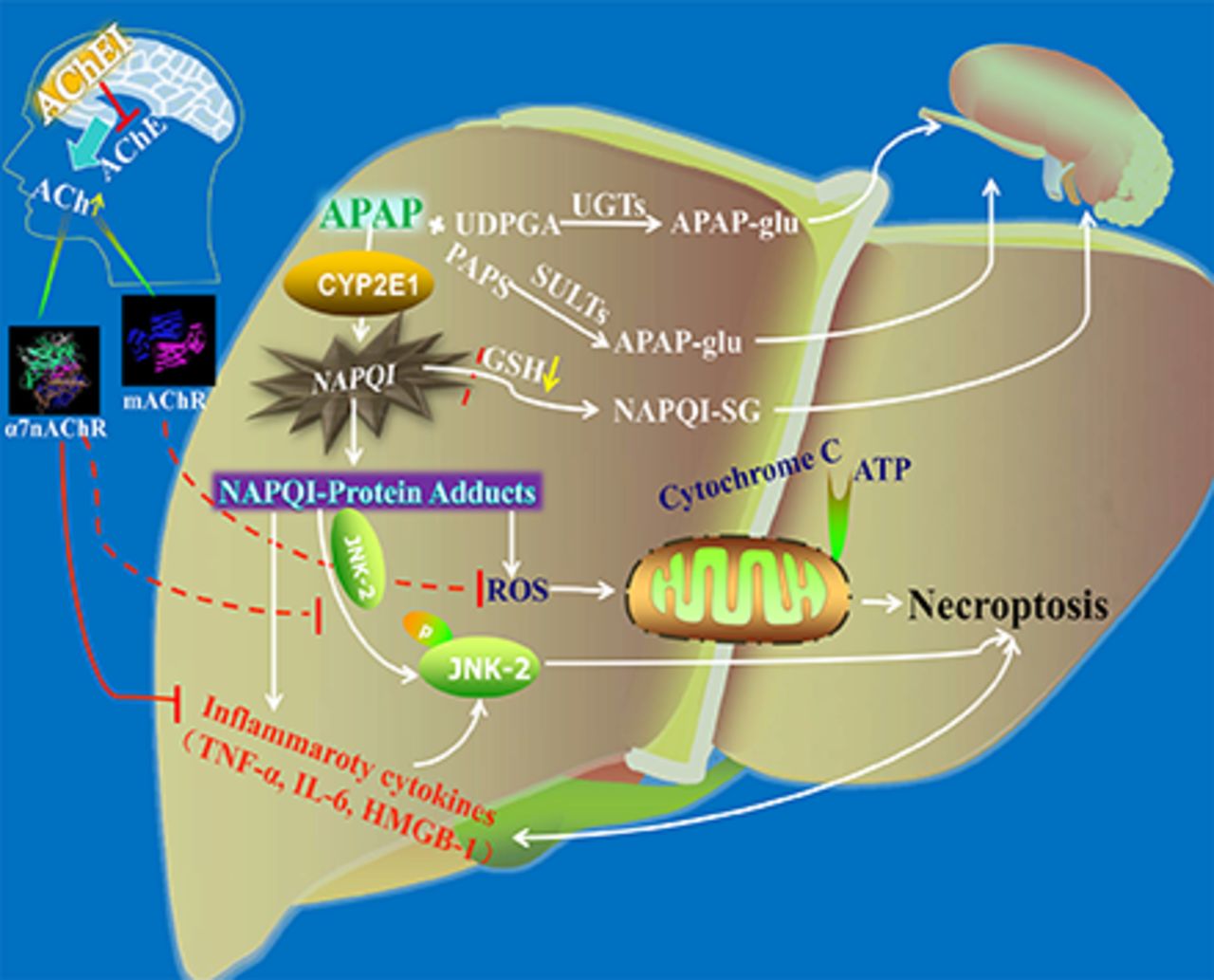
Visual Overview

Abstract
Acetaminophen (APAP) is widely used as an analgesic and antipyretic agent, but it may induce acute liver injury at high doses. Alzheimer’s disease patients, while treated with acetylcholinesterase inhibitor (AChEI), may take APAP when they suffer from cold or pain. It is generally recognized that inhibiting acetylcholinesterase activity may also result in liver injury. To clarify whether AChEI could deteriorate or attenuate APAP hepatotoxicity, the effects of AChEI on APAP hepatotoxicity were investigated. Male C57BL/6J mice were administrated with the muscarinic acetylcholine receptor (mAChR) blocker atropine (Atr), or classic α7 nicotine acetylcholine receptor (α7nAChR) antagonist methyllycaconitine (MLA) 1 hour before administration of AChEIs—donepezil (4 mg/kg), rivastigmine (2 mg/kg), huperzine A (0.2 mg/kg), or neostigmine (0.15 mg/kg)—followed by APAP (300 mg/kg). Eight hours later, the mice were euthanized for histopathologic examination and biochemical assay. The results demonstrated that the tested AChEIs, excluding neostigmine, could attenuate APAP-induced liver injury, accompanied by reduced reactive oxygen species formation, adenosine triphosphate and cytochrome C loss, c-Jun N-terminal kinase 2 (JNK2) phosphorylation, and cytokines. However, Atr or MLA significantly weakened the protective effect of AChEI by affecting mitochondrial function or JNK2 phosphorylation and inflammation response. These results suggest that central mAChR and α7nAChR, which are activated by accumulated acetylcholine resulting from AChEI, were responsible for the protective effect of AChEIs on APAP-induced liver injury. This indicates that Alzheimer’s patients treated with AChEI could take APAP, as AChEI is unlikely to deteriorate the hepatotoxicity of APAP.
Introduction
Acetaminophen (APAP) is used globally as an analgesic and antipyretic agent (Graham et al., 2005). The liver injury induced by APAP overdose is a critical issue, as it is the most frequent cause of acute liver injury in different regions of the world (Gow et al., 2004; Larson et al., 2005; Wei et al., 2007; Craig et al., 2011; Li and Martin, 2011; Lee, 2012). Patients with the most severe symptoms may develop hepatic encephalopathy and require liver transplantation (Ostapowicz et al., 2002; Russo et al., 2004; Lee, 2012).
Alzheimer’s disease (AD) is a neurodegenerative disorder that affects predominantly the elderly (Stephan and Phillips, 2005; Harold et al., 2009). Currently, over 35 million people worldwide live with AD, and this number is expected to double by 2030 (Prince et al., 2013). One of the most effective treatments for AD is to enhance cholinergic neurotransmission by administrating an acetylcholinesterase inhibitor (AChEI) to suppress the breakdown of released acetylcholine (ACh) (Lopez et al., 2002). Liver function is modulated by both sympathetic and parasympathetic nerves, which are cholinergic nerve systems (Lautt, 1980; Shimazu, 1981; Berthoud, 2004). Consequently, liver function can be affected by stimulating cholinergic nerves via ACh accumulated by AChEI administration.
AChEIs, such as donepezil (Don) and rivastigmine (Riv), are drugs approved by the U.S. Food and Drug Administration and are widely used for AD treatment. Huperzine A (Hup A) is another AChEI suitable for AD patients (Zangara, 2003; Zhang and Tang, 2006) and is approved in the People’s Republic of China. However, neostigmine (Neo), as a peripheral AChEI, is administered to treat myasthenia gravis. Previous studies have demonstrated that inhibiting acetylcholinesterase (AChE) might result in liver injury (Uzunhisarcikli and Kalender, 2011; Uzun and Kalender, 2013). Don-induced hepatotoxicity also has been reported in an elderly adult taking fluoxetine (Chew et al., 2014). Furthermore, it has been proven that the cholinergic system plays an important role in Hup A–induced abnormal transaminase activity enhancement (Ma et al., 2003b).
Some drug interactions with APAP have been reported; for instance, acarbose and ethanol potentiate the hepatotoxicity of APAP (Zimmerman and Maddrey, 1995; Wang et al., 1999). AChEI and APAP, or a compound containing APAP, are most often used simultaneously to treat AD patients with fever or pain. However, the potential effects of AChEI on APAP-induced liver injury—whether AChEI diminishes the hepatotoxicity of APAP—remain unclear and need to be elucidated. This study was conducted to investigate whether AChEI, administered before APAP, could affect APAP-induced liver injury in mice, which might guide future clinical applications of APAP with AChEI for optimal safety.
Materials and Methods
Drugs and Reagents.
Eisai Pharmaceutical (Tokyo, Japan) produced Don, and Riv was manufactured by Novartis (Basel, Switzerland). Hup A was purchased from the Shanghai Tauto Biotech Company (Shanghai, People’s Republic of China). Neo was acquired from Shanghai Pharmaceuticals (Shanghai, People’s Republic of China). Methyllycaconitine (MLA) was obtained from Tocris Bioscience/R&D Systems (Minneapolis, MN). Interleukin-6 (IL-6) and tumor necrosis factor α (TNF-α) enzyme-linked immunosorbent assay (ELISA) kits were bought from R&D Systems (Minneapolis, MN). The high mobility group box-1 protein (HMGB1) ELISA kit was purchased from the Shanghai Westang Bio-tech Company (Shanghai, People’s Republic of China). Total c-Jun N-terminal kinase (JNK) antibodies (sc-7345) and phosphorylation JNK antibodies (sc-6254) were obtained from Santa Cruz Biotechnology (Dallas, TX). Alanine transaminase (ALT) and aspartate transaminase (AST) kits, reduced glutathione (GSH) kit, adenosine triphosphate (ATP), reaction oxygen species (ROS), and cytochrome c kits were obtained from Nanjing Jiancheng Bioengineering Institute (Jiangsu, People’s Republic of China). Other chemicals—including APAP, atropine (Atr) and mecamylamine (Mec)—were purchased from Sigma-Aldrich (St. Louis, MO).
Animal Treatment and Experimental Design.
Male C57BL/6J mice (8 weeks) weighing 20 ± 2 g were purchased from Beijing HFK Bio-Technology and maintained in accordance with the guidelines for the care and use of laboratory animals at Yantai University. Mice were kept in the animal facility under standard conditions (22 ± 2°C, 50% ± 5% relative humidity, 12-hour light/dark cycle) with free access to food and water until completion of the study. Mice were randomly divided into 10 groups (n = 10): group 1 (normal control group, Con), groups 2–5 (AChEI control groups), group 6 (model group, APAP), and groups 7–10 (AChEI-treated groups). Groups 7, 8, 9, and 10 were, respectively, administrated with Hup A, Don, Riv, and Neo at doses of 0.2, 4.0, 2.0, and 0.15 mg/kg once daily for 4 consecutive days.
One hour after the final administration, the mice in groups 6–10 were injected intraperitoneally (i.p.) with APAP (300 mg/kg), and the mice in groups 1–5 received only appropriate vehicle. Afterward, in another experiment, mice in AChEI-treated groups were pretreated subcutaneously with muscarinic acetylcholine receptor (mAChR) antagonist Atr (1 mg/kg), nicotine acetylcholine receptor (nAChR) blocker Mec (4 mg/kg, i.p.), and α7 nicotine acetylcholine receptor (α7nAChR) blocker MLA (4 mg/kg, i.p.) 1 hour before AChEI treatment. One hour after the last administration, the mice were injected with a APAP solution that was prepared at 15 mg/ml in warmed saline (42°C).
Eight hours after the APAP injection, blood was harvested and centrifuged at 5000 rpm for 10 minutes. Aliquots of serum were transferred to Eppendorf tubes and stored at −80°C until analysis. The brain and liver were separated on ice and homogenized with 0.1 M ice-cold phosphate-buffered saline buffer (pH 7.4) to yield a 10% (w/v) homogenate. The homogenate was centrifuged at 3500 rpm for 10 minutes at 4°C, after which the supernatant was stored at −80°C until subsequent assay. The remaining liver was partially collected by snap-freezing or fixed in 10% formalin for H&E staining.
Determination of AChE Activity and Aminotransferase Activity.
AChE activities in the blood and brain were determined according to the methods of Ellman et al. (Ellman et al., 1961). ALT and AST activities were measured using commercially available kits using the microplate-modified Reitman Frankel assay method. The absorption of the final solution was measured by an automated ELISA reader (Molecular Devices, Sunnyvale, CA).
Histopathologic Examination.
Immediately after the mice were euthanized, their livers were removed for histologic analysis. Liver specimens were fixed in 10% phosphate-buffered formalin, dehydrated, and embedded in paraffin. Each paraffin-embedded block was cut into 5-μm sections and then stained with H&E for histologic observation under a light microscope. A pathologist blinded to the treatment groups or the corresponding liver biochemistries assessed the liver histology.
Preparation of Mice Liver Subcellular Fractions.
Ten mice from each group were decapitated. The livers were then removed. The liver homogenate (25% w/v in 1.15% KCl–0.01 M phosphate buffer, pH 7.4) was centrifuged at 10,000g at 4°C for 20 minutes. A portion of the supernatant was used to measure the GSH content. The remaining supernatant was mixed with 0.1 ml of 88 mM CaCl2 buffer at 4°C and incubated for 5 minutes, then further centrifuged at 27,000g at 4°C for 15 minutes to harvest microsomal pellets. The pellets were suspended in phosphate buffer and centrifuged again at 27,000g for 15 minutes. The microsomal pellets were resuspended with the buffer, and the protein content was measured by the Lowry method (Lowry et al., 1951). The protein content was then adjusted to a concentration of 10 mg/ml with the buffer solution, and the suspension then was stored at −80°C until analysis.
Liver mitochondria and cytoplasm were isolated according to standard methods, and were supplemented with protease and phosphatase inhibitors according to methods described elsewhere (Schneider and Hogeboom, 1950). Protein concentration was determined using the Bradford assay, with albumin serving as the reference protein. Mitochondria and cytoplasm were immediately snap-frozen in liquid nitrogen and kept at −80°C until analysis, except for the measurement of mitochondrial ATP content, for which freshly isolated mitochondria were used.
Measurement of GSH Content and CYP2E1 Activity.
The GSH content was measured by the dithiobisnitrobenzoic acid method as described by the manufacturer’s directions. Cytochrome P450 isoform 2E1 (CYP2E1) activity was assessed using high-performance liquid chromatography (C18 column, 250 × 4.6 mm; 5 μm; Shimadzu LC-20A; Shimadzu, Kyoto, Japan) (Supplemental Methods, Supplemental Fig. 1, and Supplemental Table 1).
Proinflammatory Cytokines Assay.
Liver IL-6, TNF-α, and HMGB1 levels were measured by ELISA kits according to the standard protocols.
ATP and Cytochrome C Assessment.
The ATP content was measured in isolated mitochondria according to the manufacturer’s instructions with a hexokinase reaction kit. The absorption was measured at 636 nm by ELISA reader. The concentration of cytochrome c was determined by ELISA kit per the manufacturer’s instructions.
ROS Assay.
ROS generation was determined in liver mitochondria using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) levels as an index of the peroxide production by cellular components (1 mM). Briefly, the liver mitochondria were added to the standard medium, and the fluorescence was determined at 488 nm for excitation and 525 nm for emission.
Protein Immunoblotting.
Equal amounts of denatured protein were loaded per lane and separated on a 12% SDS-PAGE gel, followed by subsequent transferred to nitrocellulose membranes. The membranes were blocked in 5% nonfat dry milk prepared with Tris-buffered saline/Tween 20 for 1 hour at 25°C. Anti-phosphorylated JNK2 (P-JNK2; 1:1000), anti-JNK (1:1000), and anti-β-actin (1:1000) were used as the primary antibodies. The protein bands were visualized by enhanced chemiluminescence after incubation with horseradish peroxidase–conjugated secondary antibody (1:2000). Anti-β-actin was used as a reference for protein loading.
Statistical Analysis.
The results were presented as mean ± S.D. Comparisons between more than two groups were performed by analysis of variance (one-way ANOVA), followed by the Student’s t test. P ≤ 0.05 was considered statistically significant, unless indicated otherwise.
Results
AChEI Treatment of AD Attenuated APAP-Induced Liver Injury in Mice.
A significant decrease in brain AChE activities was observed in mice treated with several AChEIs, including Don, Riv, and Hup A in comparison with the control. This reduction in AChE activity was not observed in Neo (Fig. 1A).

Effect of AChEI on APAP-induced liver injury in C57BL/6J mice. Mice were treated with various AChEIs, including Hup A, Don, Riv, and Neo, 1 hour before APAP treatment. The brain and blood AChE levels (A), serum ALT and AST (B) activities were detected 8 hours after i.p. administration of APAP. (C) The histopathology was assessed at 200× magnification. Data were expressed as mean ± S.D. (n = 10). *P < 0.05 and **P < 0.01 compared with control (Con); #P < 0.05 and ##P < 0.01, compared with APAP.
ALT and AST activities were significantly increased in APAP-treated mice compared with the control group. The addition of AChEIs markedly decreased the ALT and AST activities induced by APAP treatment. However, Neo did not show any protective effects (Fig. 1B). These results were confirmed by histopathologic examination, whereas APAP alone caused centrilobular parenchymal necrosis in all mice. Treatment with AChEIs afforded nearly complete protection from hepatic necrosis in some mice and attenuated the extent of injury. However, Neo had no effect on liver injury (Fig. 1C). Several AChEIs that can penetrate the blood–brain barrier (BBB) (but not peripheral AChEIs) have demonstrated ameliorative effects on APAP-induced liver injury.
AChEI Affected Neither GSH Content nor Metabolic Enzymes by APAP Treatment.
Decline in hepatic GSH level is a widely accepted marker for the generation of toxic reactive intermediates in vitro or in vivo (Boess et al., 1998). Previous studies have reported that GSH concentration was significantly reduced 1 hour after toxic exposure, remained low until 3 hours after exposure, and then started to recover (McGill et al., 2012; Williams et al., 2014). Hepatic GSH levels initially decreased but increased back to standard levels after the first 2 hours after treatment (Fig. 2A). This indicated that GSH was not the key point in the injury process. Mice that received AChEI before APAP treatment did not show a significant change in GSH levels compared with those treated with APAP alone.

The effects of AChEI on GSH content and CYP2E1 activity. Mice were treated with AChEI 1 hour before APAP treatment. The liver GSH concentrations (A) were determined at 0, 2, 4, and 8 hours, while CYP2E1 activities (B) were detected 8 hours after i.p. administration of APAP. Data were expressed as mean ± S.D. (n = 10). *P < 0.05 compared with control (Con).
To ascertain that the hepatic protection afforded by AChEI was not due to metabolic inhibition of CYP2E1, which converts APAP to N-acetyl-p-benzoquinone imine (NAPQI)—a hepatotoxic metabolite—CYP2E1 activity was measured. No obvious changes in CYP2E1 activity were observed with any AChEI treatments (Fig. 2B). According to earlier reports regarding the degradation of Hup A (metabolized via CYP1A2), Don (metabolized via CYP2D6), and Riv (Matsui et al., 1999; Wesnes et al., 2002; Ma et al., 2003a), these AChEIs should not interfere with CYP2E1 activity in mouse liver microsomes. Taken together, these data indicated that AChEI-induced hepatic protection was not accomplished by affecting GSH content and CYP2E1 activity, suggesting that this effect was not related to the production of NAPQI.
AChEI Protected against APAP-Induced Liver Injury via Central AChR.
In the present study, different AChR antagonists—including Atr (mAChR antagonist), Mec (nAChR antagonist), and MLA (selective α7nAChR antagonist)—were used to investigate their effects on AChEI protection against APAP-induced liver injury. Atr, Mec, or MLA were not observed to have a protective effect on APAP-induced liver injury. In contrast, Atr or MLA administration significantly altered the ALT and AST activities that were down-regulated by AChEIs in APAP-induced liver injury (Fig. 3A). Subsequently, the protective effect of AChEIs was slightly weakened by Mec, but only at the high dose of 4 mg/kg. However, mice treated with the high dose of Mec had adverse health effects, as this drug can cause severe hypotension (Supplemental Fig. 2).

The potential mechanisms of different cholinergic receptors antagonists on APAP-induced liver injury which is protected by AChEI. Muscarinic receptor antagonist (Atr), nicotine receptor antagonist (Mec), and α7 nicotine receptor blocker (MLA) were supplemented 1 hour before AChEI treatment to investigate the potential mechanism of AChEI protection against APAP-induced liver injury. ALT and AST activities were determined 8 hours after APAP treatment (A). The effect on histopathology was analyzed at 200× magnification (B). Data were expressed as mean ± S.D. (n = 10). *P< 0.05 compared with control (Con); #P < 0.05, ##P < 0.01 compared with Hup A or Riv.
The histopathology assessment also demonstrated similar alterations in accordance to changes in biochemical factors (Fig. 3B). The protective effect of AChEIs was markedly blocked by Atr and MLA treatment, but only slightly altered by Mec. This suggests that AChEI could prevent mice from APAP-induced liver injury via stimulation of mAChR and α7nAChR. Hence, Atr and MLA were chosen to explore the further mechanisms related to AChR.
AChEI Protected against APAP-Induced Mitochondrial Damage via mAChR Activation.
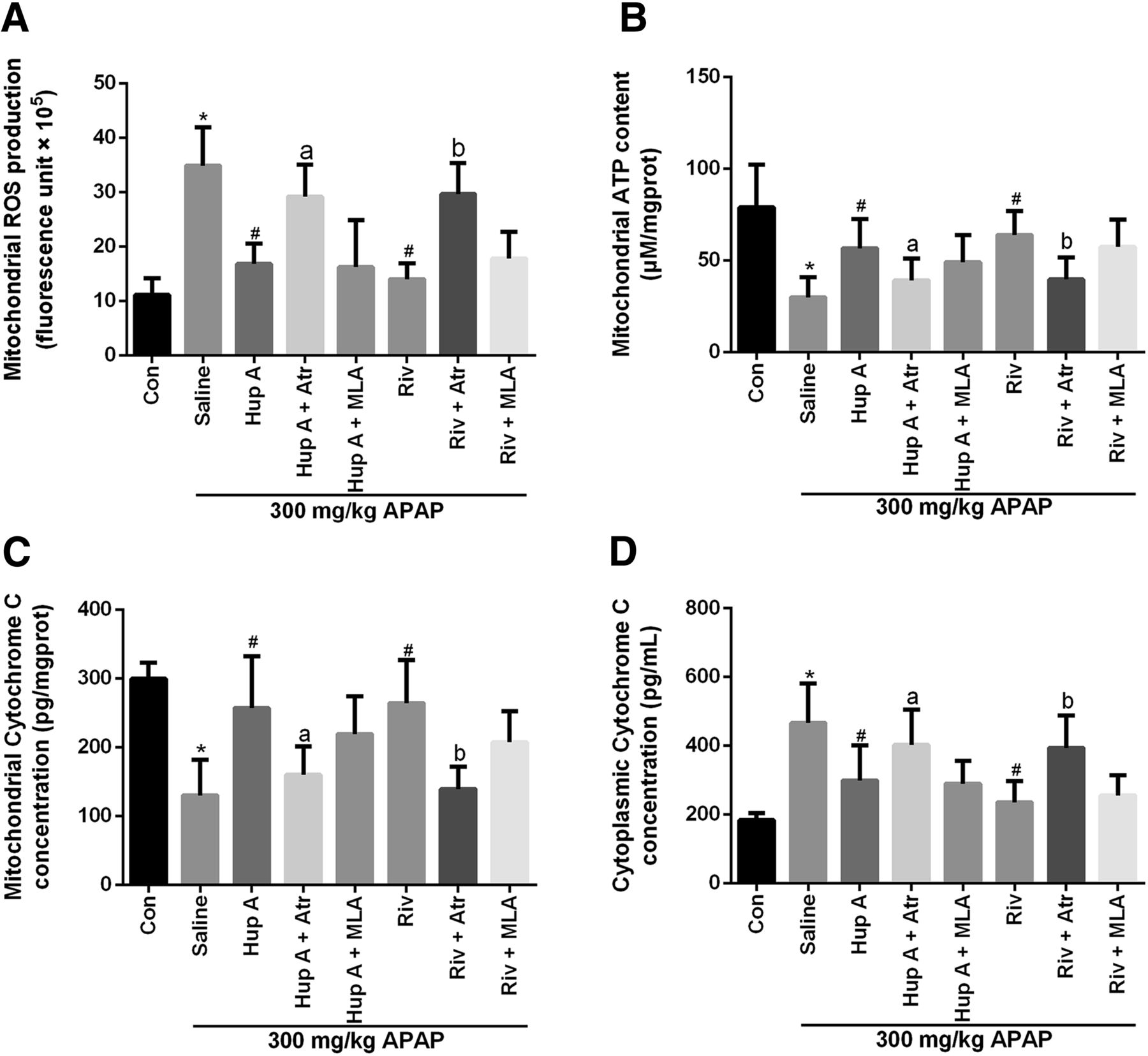
To determine whether AChEIs provided protection from liver necrosis originating from mitochondria, mitochondrial functions were measured. The high concentration of ROS induced by APAP in mitochondria was reduced by AChEI administration (Fig. 4A). APAP treatment significantly decreased ATP levels but addition of AChEIs generally returned ATP content to normal levels and protected the mitochondria from functional injury induced by APAP (Fig. 4B). In addition, the mAChR antagonist Atr greatly reduced the ameliorative effect of AChEIs on ATP decrease.

Protective effects of AChEI on APAP-induced liver injury were partly dependent on ameliorating mitochondrial damage via mAChR. Livers were harvested 8 hours after APAP treatment. The mitochondria and cytoplasm were isolated from the livers of mice in different groups for analysis. Mitochondrial ROS level (A), ATP content (B), cytochrome c concentration (C), and cytoplasmic cytochrome c concentration (D) were determined. Data were expressed as mean ± S.D. (n = 10). *P < 0.05 compared with control (Con); #P < 0.05 compared with saline; aP < 0.05 or bP < 0.05 compared with Hup A or Riv.
Cytochrome c was then chosen as a biomarker for mitochondrial membrane permeabilization, as measured by cytochrome c content in mouse mitochondrial and cytoplasmic fractions. APAP treatment resulted in a significant loss of cytochrome c from mitochondria to the cytoplasm, which was prevented in mice pretreated with AChEI (Fig. 4, C and D). However, the mitochondrial cytochrome c loss was again observed upon treatment with Atr in the AChEI group. Collectively, these results verified that the mitochondrial damage was involved in APAP-induced liver injury in mice. More significantly, AChEI afforded protection through mAChR activation.
AChEI Decreased Cytokine Levels and JNK2 Phosphorylation by α7nAChR.
MLA was chosen to ascertain the role of α7nAChR in the protective effect of AChEI on APAP-induced liver injury. Liver TNF-α, IL-6, and HMGB1 levels were initially increased by APAP treatment but were subsequently reduced by AChEI treatment. However, reversion by MLA suggested that AChEI treatment prevented APAP-induced liver injury via α7nAChR, in turn decreasing proinflammatory cytokine formation in the liver (Fig. 5, A–C).

AChEI prevented C57BL/6J mice from APAP-induced liver injury by reducing cytokine formation and JNK2 phosphorylation via α7nAChR. Livers were obtained 8 hours after APAP treatment. TNF-α (A), IL-6 (B), and HMGB1 (C) protein secretion were measured by ELISA. The level of JNK2 phosphorylation was determined by protein immunoblotting (D) and then analyzed with densitometry (E). Data were expressed as mean ± S.D. (n = 10). *P < 0.05 compared with control (Con); #P < 0.05 compared with saline; aP < 0.05 or bP < 0.05 compared with Hup A or Riv.
To further explore the possible target of interaction with AChEI, an immunoblot was performed to determine JNK2 activation. Previously, APAP was found to activate JNK in immortalized human hepatocytes (Latchoumycandane et al., 2007; Jaeschke et al., 2012a). Recently, studies in mice have suggested that JNK2 plays a crucial role in APAP-induced liver injury (Latchoumycandane et al., 2006; Gunawan and Kaplowitz, 2007; Bourdi et al., 2008). In the present study, JNK2 phosphorylation activated by APAP was greatly inhibited by AChEI treatment (Fig. 5D), indicating that JNK2 might be a protective target of AChEI against APAP-induced liver injury. To confirm that the apparently lower levels of P-JNK2 were not due to lower expression of the nonphosphorylated JNK protein, immunoblotting was used to assess expression of JNK (Fig. 5E). JNK expression was not affected significantly by treatment with either APAP or AChEI with APAP. Finally, P-JNK2 was observed to significantly increase when AChEI-treated mice were administrated with MLA. This result also implied that AChEI inhibited the JNK2 phosphorylation induced by APAP treatment via α7nAChR activation.
Discussion
APAP has become the leading cause of drug-induced acute liver failure worldwide (Bunchorntavakul and Reddy, 2013). The present study was performed to clarify whether AChEIs used in AD could deteriorate or attenuate APAP hepatotoxicity. Hup A, Don, and Riv, which all can penetrate the BBB and peripheral AChEI, and Neo, which cannot cross the BBB, were selected to investigate their effects on liver injury in APAP-treated mice. The AChE activity in the brain was significantly inhibited when mice were administrated Hup A, Don, and Riv; no inhibition was noted with Neo. Additionally, the results indicated that AChEI used in AD can reduce APAP-induced alterations in ALT and AST activities and ameliorated liver cell necrosis. These results demonstrated that AChEI treatment of AD patients did not exacerbate liver injury and could also attenuate the deleterious effects induced by APAP. This protective effect was not observed in Neo-treated mice, although this is in contrast to results from previous study, which observed that APAP-induced acute liver failure (ALF) could be alleviated by Neo in balb/c mice (Steinebrunner et al., 2014).
However, these results may be due to destruction of the BBB in APAP-induced ALF, which would allow Neo, a peripheral AChEI, access to the brain. In that study, mice were repeatedly administrated with Neo after APAP administration (600 mg/kg), which is a marked difference from the general APAP-induced liver injury model, which utilizes i.p. injection of 300 mg/kg APAP. APAP at 600 mg/kg can cause severe ALF, ultimately destroying the BBB (Lv et al., 2010; Wang et al., 2011; Bosoi and Rose, 2013; Jayakumar et al., 2013). In addition, Steinebrunner et al. (2014) only measured survival rates and analyzed liver biochemical markers. Some proinflammatory factors were also measured, but the mechanisms of Neo against APAP-induced ALF were only deduced, without any direct observations on the effect of cholinergic agonists or antagonists. AChE activity was not assayed, although the study’s objective was to observe the effect of pharmacologic cholinesterase inhibition on APAP-induced acute liver failure. The objective of our study was to establish a reasonable application of APAP for AD patients who were administrated AChEI. The findings suggested that AChEI might protect against APAP-induced liver injury, but only the AChEI can penetrate the BBB; regulation of the central cholinergic system played a critical role in the protective effect of AChEI on APAP-induced liver injury.
CYP2E1 activity and hepatic GSH levels are closely related to the production and elimination of NAPQI, the toxic metabolite of APAP. Previous studies have demonstrated that excessive NAPQI production depletes hepatic GSH, which is critical in initiating APAP-induced liver injury (Jaeschke et al., 2011; Bantel and Schulze-Osthoff, 2012). In this experiment, AChEI affected neither CYP2E1 activity nor hepatic GSH levels, thus indicating that the protective effects of AChEI on APAP-induced liver injury may be not involved in NAPQI production/elimination.
The subdiaphragmatic vagus is a pathway that signals inflammatory events from the liver to the brain (Goehler et al., 2000). Vagal efferent suppresses the release of TNF-α via releasing Ach to act on α7nAChR in resident macrophages in the abdominal organs, which includes the liver (Borovikova et al., 2000; Tracey, 2002; Wang et al., 2003). Down-regulation of α7nAChR contributes to the development of end-organ damage by inducing inflammation (Li et al., 2011; Chen et al., 2012). In addition to this intrinsic anti-inflammatory characteristic, α7nAChR agonist can also inhibit the severe sepsis related to the chemokine of HMGB1 (Wang et al., 2004). Moreover, sterile inflammation induced by proinflammatory cytokines—such as TNF-α, IL-6, and HMGB1—plays a significant role in APAP hepatotoxicity (Simpson et al., 2000; Martin-Murphy et al., 2010; Jaeschke et al., 2012b; Huebener et al., 2015).
Additionally, JNK activation is related to the pathophysiology of APAP-induced necrotic cell death (Jaeschke et al., 2012a). The JNK inhibitor leflunomide, as well as JNK2 knockout, have been shown to rescue mice from APAP-induced liver injury (Latchoumycandane et al., 2007; Saito et al., 2010). Previous studies have demonstrated that treatment of endothelial cells with carbachol decreases JNK phosphorylation through α7nAChR (Li et al., 2010). Suzuki et al. (2006) also demonstrated that the inhibition of TNF-α released by nicotine through α7nAChR is associated with suppression of the JNK signaling pathway.
In our study, the effect of activation of α7nAChR on APAP-induced chemokine formation and phosphorylation of JNK2 in liver injury was explored. Mice treated with AChEI had a significant decrease in IL-6, TNF-α, HMGB1 formation, and JNK2 phosphorylation, which was blocked by MLA. These results indicated that indirect activation of α7nAChR by AChEI could attenuate liver damage by reducing the inflammatory response and JNK2 phosphorylation.
Previous reports have also revealed that mAChR activation protects cells from oxidative stress and mitochondrial dysfunction (De Sarno et al., 2003). Mitochondrial dysfunction has emerged as the most critical event in APAP toxicity (Ramachandran et al., 2013). Mitochondrial dysfunction, which is induced by cytochrome c loss and ATP depletion, is a hallmark of APAP-induced cell death (Kon et al., 2004; Jaeschke et al., 2012a). The data in this study suggest that AChEI treatment in AD could attenuate mitochondrial cytochrome c loss, ATP depletion, and ROS production, indicating that central mAChR activation is involved in regulating mitochondrial dysfunction in APAP-induced liver injury.
In our experiments, both mAChR blocker Atr and classic α7nAChR antagonist MLA could weaken the protective effect of AChEI on APAP-induced liver injury. Interestingly, Mec had a less pronounced effect and only at a high dose of 4 mg/kg, which could also be involved in nonselective blockage of α7nAChR. Our present study suggests that central mAChR and α7nAChR are activated by accumulated ACh, which results from inhibition of AChE and is responsible for the protective effect of AChEIs on APAP-induced liver injury.
In conclusion, AChEIs used in treatment of AD, such as donepezil, rivastigmine, and huperzine A, could attenuate APAP-induced liver injury by the central mAChR and α7nAChR pathways. This finding suggests that AD patients, when being administrated AChEI, could take APAP without worrying about AChEI-induced deterioration and hepatotoxicity of APAP.
Acknowledgments
The authors thank Tongshen Liu for providing technical assistance in pathological analysis, Lon Clark for revising the English text, and LetPub (www.letpub.com) for its linguistic assistance.
Authorship Contributions
Participated in research design: Fu, J. Zhang, L. Zhang.
Conducted experiments: J. Zhang, Sun, Yang, Kong, Lu.
Performed data analysis: J. Zhang, L. Zhang, Lv.
Wrote or contributed to the writing of the manuscript: J. Zhang, Fu, T. Wang, H. Wang.
Footnotes
- Received March 24, 2016.
- Accepted August 12, 2016.
This work was supported by the Taishan Scholar Project, Technology Development Program Projects of Shandong Province [Grant 2011YD18075], and National Natural Science Foundation of China [Grant 81274125] (to F.H.-F.); Graduate Innovation Foundation of Yantai University [Grant YJSY201424] (to J.Q.-Z.).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- α7nAChR
- α7 nicotine acetylcholine receptor
- ACh
- acetylcholine
- AChE
- acetylcholinesterase
- AChEI
- acetylcholinesterase inhibitor
- AChR
- acetylcholine receptor
- AD
- Alzheimer’s disease
- ALF
- acute liver failure
- ALT
- alanine aminotransferase
- APAP
- acetaminophen
- AST
- aspartate aminotransferase
- Atr
- atropine
- CYP2E1
- cytochrome P450 isoform 2E1
- BBB
- blood–brain barrier
- Don
- donepezil
- ELISA
- enzyme-linked immunosorbent assay
- GSH
- glutathione
- HMGB1
- high mobility group box-1 protein
- Hup A
- huperzine A
- IL-6
- interleukin-6
- JNK
- c-Jun-N-terminal kinase
- mAChR
- muscarinic acetylcholine receptor
- Mec
- mecamylamine
- MLA
- methyllycaconitine
- nAChR
- nicotine acetylcholine receptor
- NAPQI
- N-acetyl-p-benzoquinone imine
- Neo
- neostigmine
- P-JNK
- phosphorylated JNK
- Riv
- rivastigmine
- ROS
- reactive oxygen species
- TNF-α
- tumor necrosis factor α
- Copyright © 2016 The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}