


Abstract
Ibrutinib (IBT), the first-in-class inhibitor of Bruton’s tyrosine kinase (BTK), has demonstrated clinical activity against various B-cell malignancies. Aside from its therapeutic mechanism through BTK inhibition, IBT has other target sites reported for cancer therapy, leading us to investigate whether IBT has unreported targets. Our study revealed that IBT can inhibit SMMC-7721 cells through irreversible inhibition of mammalian thioredoxin reductase enzymes. Further study demonstrated that IBT can cause cellular reactive oxygen species elevation and induce cancer cell apoptosis. The discovery of a new target of IBT sheds light on better understanding its anticancer mechanisms and provides a theoretical foundation for its further use in clinical therapy.
Introduction
Liver cancer has become a serious threat to the health of humans. It was called “the king of cancer” because of properties such as diagnostic difficulties, rapid development, high malignancy, easy metastasis, and high mortality. Liver cancer was ranked by the World Health Organization as the third highest mortality cancer in 2018 (Bray et al., 2018). The treatment of liver cancer has a poor prognosis, with only 6- to 12-month median survival, and the recurrence rate is as high as 19%–25% in the first year (Torzilli et al., 2013). Due to the aforementioned properties, clinical treatments for liver cancer have not made remarkable progress. However, chemotherapy is still the main clinical method for liver cancer therapy; systemic chemotherapy can obviously extend patients’ lives, especially patients in advanced stage whose cancer has metastasized. Hence, finding a low-toxicity and high-efficiency drug has become the top priority.
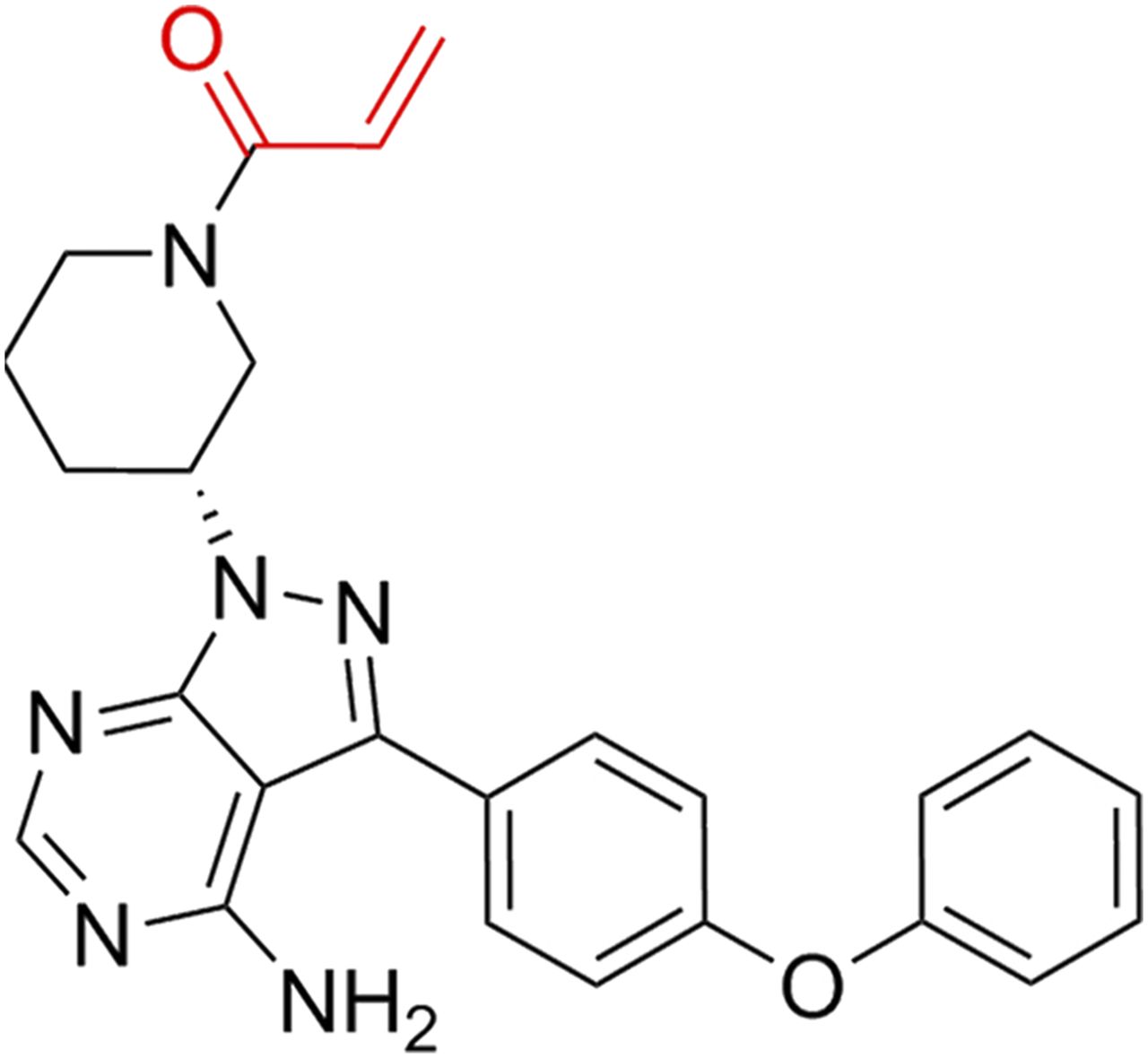
Ibrutinib (IBT) (Fig. 1), also named PCI-32765, is a first-in-class inhibitor of Bruton’s tyrosine kinase (BTK). It was initially modified from the Src family kinase selected inhibitor PP1 by Arnold and Pan to treat rheumatoid arthritis (Arnold et al., 2000; Pan et al., 2007). IBT was the second approval through the US Food and Drug Administration Breakthrough Therapy Designation Pathway; it was also approved by the European Medicines Agency to treat mantle cell lymphomas and chronic lymphocytic leukemia. Currently, numerous phase I and phase II clinical studies have demonstrated that IBT can significantly reduce the tumor burden and slow the progression of various lymphomas (Harrison, 2012; Byrd et al., 2013). It has an overall response rate of 70% in both relapsed/refractory and newly diagnosed chronic lymphocytic leukemia (CLL) patients (Herman et al., 2011). It has been reported that the therapeutic effect for B-cell receptor malignancies was through its irreversible inhibition for BTK (Byrd et al., 2013), further leading to blocking the expression of downstream of BCR, including NF-κB, v-akt murine thymoma viral oncogene homolog (Akt), and extracellular signal-regulated kinase (Herman et al., 2011; Chong et al., 2018). It has been reported that IBT could also inhibit ErbB receptor phosphorylation and stimulate the ATPase activity of ATP- binding cassette subfamily B member 1 (ABCB1) (Grabinski and Ewald, 2014; Zhang et al., 2017a) and has therapeutic effects on glioblastoma, breast cancer, and gastric cancer. These reports make us consider whether IBT possesses other molecular targets for cancer therapy.

The structure of IBT.
Thioredoxin reductase (TrxR) is a component of the thioredoxin (Trx) system, which also contains Trx and NADPH and is one of the most important systems to balance the redox state. Not only can Trx scavenge reactive oxygen species (ROS), it also has an abroad substrate such as ASK1 caspase3 P53 and NF-κB in vivo (Zhang et al., 2017b). As the main electron receptor of TrxR, Trx activity is directly dependent on the activity of TrxR. It has been reported that TrxR was highly expressed in most cancer cells (Arner and Holmgren, 2006). High expression of TrxR can help scavenge ROS and resist cell death. And various recent studies in the literature have reported that irreversible inhibition of TrxR could induce cancer cell apoptosis (Stafford et al., 2018; Zhuge et al., 2018; Zhang et al., 2019). Our studies have provided preliminary evidence that IBT could target mammalian TrxR for cancer therapy, which will give us more viewpoints to understand the cancer therapy mechanism.
Materials and Methods
Materials.
IBT (molecular mass 440.50 Da, 99% purity) was purchased from Easybio (Shanghai, China), and 100 mM IBT in dimethylsulfoxide (DMSO) was stored at −20°C as a stock solution. Dulbecco’s modified Eagle’s medium (DMEM), dithiothreitol (DTT), bovine insulin, yeast glutathione reductase (GR), reduced glutathione (GSH), oxidized glutathione (GSSG), hydrogen peroxide (H2O2), N-acetyl-l-cysteine (NAC), 2′,7′-dichlorofluorescein diacetate (DCFH-DA), l-buthionine-(S,R)-sulfoximine (BSO), 2-vinylpyridine, 3-(3-cholamidopropyl)dimethylammonio-1-propanesulfonate, Hoechst 33342, and N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide were purchased from Sigma-Aldrich (St. Louis, MO). Antiactin antibody (AA128-1), trypan blue, phenylmethylsulfonyl fluoride, sodium orthovanadate (V) (Na3VO4) and bovine serum albumin (BSA) were purchased from Beyotime (Nantong, China). 5′,5′-dithiobis-2-nitrobenzoic acid (DTNB) was obtained from J&K Scientific (Beijing, China). Polyvinylidene difluoride (PVDF) membrane and NADPH were obtained from Millipore (Billerica, MA) and Roche (Mannheim, Germany). The Annexin V–fluorescein isothiocyanate (Annexin V-FITC)/propidium iodide (PI) double stain apoptosis kit were obtained from Zoman Biotech (Beijing, China). The Western blotting chemiluminescence kit was purchased from GE Healthcare Life Science. The cell cycle kit (BB-4104) was purchased from Shanghai BestBio Science. Streptomycin, penicillin, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Amresco (Solon, OH). Anti-rabbit IgG-horseradish peroxidase (sc-2004), anti-mouse IgG-horseradish peroxidase (sc-2031), and TrxR1 primary antibody (sc-28321) were products of Santa Cruz Biotechnology (Santa Cruz, CA). Fetal bovine serum (FBS) was a product of Sijiqing (Hangzhou, China). All other reagents were analytical reagents.
Cell Cultures.
SMMC-7721, HepG2, HeLa, A549, and HEK 293T cells were purchased from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. DMEM was supplemented with 100 U/ml penicillin/streptomycin, 2 mM glutamine, and 10% FBS with 5% CO2 to culture the cells at 37°C. HeLa-shNT and HeLa-shTrxR1 cells stably transfected with shNT plasmid and shTrxR1 plasmid, which were generated in our laboratory (Duan et al., 2014), were incubated with the same conditions as the aforementioned cells with an additional supplement of 1 μg/ml puromycin in DMEM.
Trypan Blue Exclusion Assay.
The indicated concentrations of IBT were used to culture 4 × 104 SMMC-7721 cells at 37°C in 24-well plates for 48 hours, and cells in the control group were treated with 0.1% DMSO alone for 48 hours. After incubation for 48 hours, 0.4% trypan blue was used to trypsinize and incubate the cells. Dead cells were stained while live cells were nonstained, and cell viability was determined as follows: (cell viability) % = living cells/(control living cells) × 100.
MTT Assay.
Cells (8 × 103) were cultured with the aforementioned concentrations of IBT in a 96-well plate in triplicate at 37°C for the indicated times until the final volume was 100 μl. The same cells incubated with 0.1% DMSO alone were used as control, and the blank group was incubated with 0.1% DMSO alone without cells. Then, 10 μl of 5 mg/ml MTT was added to DMEM for a 4-hour incubation. After adding 100 μl of extraction buffer (0.1% HCl, 5% isobutanol, and 10% SDS), the cells were further incubated at 37°C for 8 hours. A microplate reader (Thermo Scientific Multiskan GO, Finland) was used to assay the absorbance at 570 nm. The viability of cells was calculated as follows: (cell viability) % = (AIBT − ABlank)/(AControl − ABlank) × 100.
Cell Cycle Assay.
After being seeded in a six-well plate, 5 × 105 SMMC-7721 cells were incubated overnight at 37°C and then cultured with the indicated concentrations of IBT for 24 hours. The cells washed with phosphate-buffered saline (PBS) were trypsinized. They were suspended with iced PBS and fixed with iced ethanol with a final concentration of 70%. Then, cells were harvested in −20°C for more than 4 hours. PBS was used to wash the cells twice, which were incubated with 400 μl of PI solution. The samples were finally analyzed using a FACSCanto flow cytometer (BD Biosciences).
Wound Healing Assay.
SMMC-7721 cells (1 × 106) seeded in a six-well plate were incubated at 37°C for 24 hours. Even scratches were made by 10-μl pipette tips. After aseptic PBS was used to wash the plate twice, the cells were cultured with the indicated concentrations of IBT in 1% FBS medium. Before and after 24-hour incubation, bright field images were obtained by a Floid Cell Imaging Station (Life Technologies).
Molecular Docking Simulation.
Covalent docking by Schrödinger Software Suite (Schrödinger, LLC, New York, NY) was carried out to reveal the probable interaction between compound IBT and TrxR1. A crystal structure of rat TrxR1 with Sec498 nonmutant (Protein Data Bank accession number: 3EAN, Chains A and B) (Cheng et al., 2009) was downloaded from the Protein Data Bank. To investigate the interaction, the structure was prepared by the Protein Preparation Wizard module. The active site reacting with IBT was the residue Sec498, which was set to be centroid of the binding site. The docking simulation was implemented by using the default parameters.
Cell Lysis.
Cells (2 × 106) were seeded in a 100-mm-diameter plastic Petri dish to grow overnight at 37°C. Afterward, they were cultured with indicated concentrations of IBT for 24 hours and washed with fresh PBS. If necessary, they can be stored at −20°C for further study. Radioimmunoprecipitation assay buffer [50 mM Tris-HCl (pH 7.5), 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1% Triton X-100, 150 mM NaCl, 0.1% SDS, 2 mM EDTA, and 0.5% deoxycholate] was used to lyse the samples for 30 minutes on ice, and the homogenate was centrifuged (Thermo Scientific Sorvall Legend Micro21R Centrifuge) at 13,000 rpm for 10 minutes to remove cellular debris. The cell suspension was collected, and the Bradford procedure was used to measure the protein quantification. Samples were stored at −20°C for further study if necessary.
Endpoint Insulin Reduction Assay.
The TrxR activity of cells was determined using the endpoint insulin reduction assay, which has been reported in our published methods (Duan et al., 2014). The cells were extracted and measured as mentioned earlier. Tris-HCl buffer (100 mM, pH 7.6) was added with the samples which contained 20 μg of total proteins to a final volume of 50 μl (containing 3 mM EDTA, 660 μM NADPH, 15 μM Escherichia coli Trx, and 0.3 mM insulin) for 30 minutes at 37°C. Then, 200 μl of 6 M guanidine hydrochloride (pH 8.0 including 1 mM DTNB) was added to the systems to terminate the reaction. The blank group, containing everything but Trx, was incubated in the same way as experimental groups. The TrxR activity was calculated by the absorbance at 412 nm as follows: (TrxR activity) % = (AIBT − ABlank)/(AControl − ABlank) × 100.
Fluorescence Microscopy Imaging by TrxR fluorogenic substrate-Green.
The fluorogenic probe TrxR fluorogenic substrate-Green was reported by our laboratory to selectively determined intracellular TrxR activity (Zhang et al., 2014). SMMC-7721 cells (5 × 105) were cultured in a 12-well plate overnight at 37°C, and then they were cultured with indicated concentrations of IBT for 20 hours. The cell culture medium was added with 1 μl of 10 mM TRFS-green for 4 hours. Floid Cell Imaging Station obtained the images.
Gpx Activity Assay.
The samples were extracted and measured as mentioned earlier. Samples containing 20 μg of protein were added to 65 μl of Tris-HCl EDTA (TE) buffer (0.2 mg/ml BSA). Then, 2 μl of 50 mM NADPH (or TE buffer for the control group), 20 μl of 10 mM GSH, and 10 μl of 40 U/ml GR were added to the samples. After 100 μl of 1 mM H2O2 was added to the mixture, the absorbance at 340 nm was immediately read every 10 seconds for 5 minutes. The Gpx activity was reflected by the rate of increase in absorbance of 340 nm. The Gpx activity was calculated as follows: (Gpx activity) % = (RIBT − RBlank)/(RControl − RBlank) × 100 (Zheng et al., 2017).
GR Activity Assay.
The samples were extracted and measured as mentioned earlier. Samples containing 30 μg of protein were added to 95 μl of TE buffer (0.2 mg/ml BSA) for 30 minutes at 37°C. Then, 2 μl of 50 mM NADPH or TE buffer was added to the mixture. As soon as 100 μl of 10 mM GSSG was added to the groups, the absorbance at 340 nm was immediately read every 30 seconds for 10 minutes. The GR activity was reflected by the rate of increase in absorbance of 340 nm (Zhang et al., 2018).
Western Blot Analysis.
The expressions of TrxR1 in the control and experimental group were detected through western blot analysis. The cells were extracted and measured as mentioned earlier. Each sample contained an equal quantity of protein and was heated at 100°C for 10 minutes to denature. Twelve percent SDS-PAGE was used to separate 50 mg of protein in the presence of DTT (100 mM). Next, the protein was transferred to PVDF membranes. Five percent nonfat milk was resolved in Tris-buffered saline/Tween 20 [TBST; 10 mM Tris-HCl (pH 7.5) containing 0.1% Tween-20 and 150 mM NaCl] to block the PVDF membranes at 25°C for 2 hours. Five percent nonfat milk containing 0.1% (v/v) specific primary antibodies was used to incubate the PVDF membranes at 4°C for 8 hours. The PVDF membranes were washed with TBST three times and incubated with the 0.025% secondary antibodies for 1 hour at 25°C. Finally, TBST was used to wash the PVDF membranes for 10 minutes three times. The immunoreactive bands were obtained by an enhanced chemiluminescence kit.
Measurement of Intracellular ROS Levels.
The cellular ROS levels caused by IBT were determined by fluorescence microscopy imaging. SMMC-7721 cells (5 × 105) were cultured in a 12-well plate overnight at 37°C and cultured with indicated concentrations of IBT for 1 hour. After the medium was removed, the plate was washed with PBS three times and replaced with FBS-free medium. One microliter of 10 mM ROS probe DCFH-DA was added to each well, which was incubated for another 30 minutes in the dark. The images were obtained by the Leica inverted fluorescence microscope.
Assessment of Intracellular Thiols.
Total cellular thiol content was measured via a DTNB titration assay. The cells were extracted and measured as mentioned earlier. Cell lysate (10 μl) was mixed with 90 μl of 6 M guanidine hydrochloride (pH 8.0, containing 1 mM DTNB) in a 96-well plate. After incubating at 25°C for 5 minutes, the absorbance at 412 nm was assayed by a microplate reader. The total thiol levels were obtained by the calibration curve generated from several standards of GSH.
Measurement of Cellular Glutathione.
An enzymatic recycling assay (Rahman et al., 2006; Zhang et al., 2018) was used to measure the cellular GSH and GSSG. The cells were cultured and harvested as mentioned earlier and were extracted by precooling extraction buffer (5 mM EDTA, 0.6% sulfosalicylic acid Triton X-100 in potassium phosphate buffer, pH 7.5 [KPE buffer]). After the iced buffer was sonicated for 30 seconds twice, the homogenate was centrifuged at 3000g for 5 minutes at 4°C, so that the cellular debris could be removed. The supernatant was collected as soon as possible, and the Bradford procedure was carried out to measure the protein quantification. To determine the total GSH level, 20 μl of sample was added to a 120-μl KPE solution which contained 1.66 U/ml GR and 0.83 mM DTNB. The buffer was supplemented with 60 μl of 0.66 mg/ml NADPH, and the absorbance at 412 nm was read at intervals of 15 seconds for 3 minutes. The total levels of GSH were linearly dependent on the rates of increase of the absorbance. The total levels of GSH were calculated by the calibration curve, which uses several concentrations of GSH as standard. To determine the GSSG level, 2 μl of 2-vinylpyridine was added to 100 μl of cell lysis and vortexed as soon as the supernatant was collected, and then the mixture was incubated at 25°C for 1 hour. The reaction system was supplemented with 6 μl of triethanolamine, which was incubated with the samples for 10 minutes at 25°C. Levels of GSSG were calculated as mentioned earlier for GSH.
Determination of Caspase 3 Activity.
Cells were extracted and measured as mentioned earlier. Lysis samples containing 50 μg of total proteins were added to 50 mM HEPES [containing 0.2 mM N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide, 10 mM DTT, 5% glycerol, 2 mM EDTA, and 0.1% 3-(3-cholamidopropyl)dimethylammonio-1-propanesulfonate, pH 7.5] to make the final volume 100 μl. The samples were incubated for 2 hours at 37°C. The blank group contained everything except the sample. The absorbance at 400 nm was read by microplate reader. The activity was calculated as follows: (caspase 3 activity) % = (AIBT − ABlank)/(AControl − ABlank) × 100.
Hoechst 33342 Staining.
After seeding in a 12-well plate overnight at 37°C, SMMC-7721 cells (5 × 105) were cultured with indicated concentrations of IBT for 48 hours. After adding 1 μl of 5 mg/ml Hoechst 33342 to DMEM, the mixture was incubated for 15 minutes at 37°C. The images were obtained by the Leica inverted fluorescence microscope.
Flow Cytometry Analysis.
Cell apoptosis was assayed using the Annexin V-FITC/PI double staining kit. After seeding in a 12-well plate overnight at 37°C, SMMC-7721 cells (5 × 105) were cultured with indicated concentrations of IBT. Then, the cells were trypsinized and washed by fresh PBS, and the original DMEM was added to the 12-well plate to inhibit the trypsinization. PBS was used to wash the cells again, which was resuspended by 500 μl of binding buffer. Finally, the samples were incubated with 5 μl of Annexin V-FITC and 10 μl of PI at 25°C for 30 minutes, avoiding light, and were analyzed by a FACSCanto flow cytometer.
Results
Inhibiting the Growth of SMMC-7721 Cells by IBT.
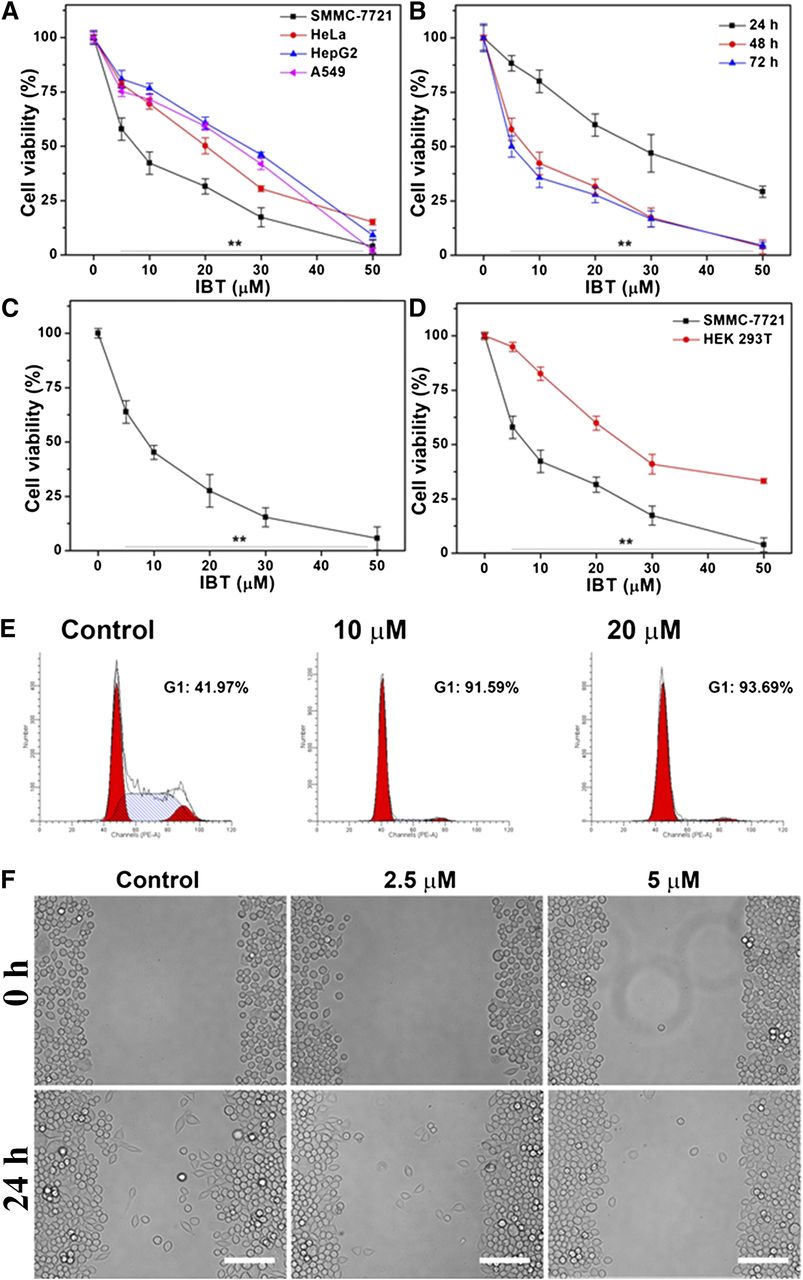
To evaluate the therapeutic efficacy, we determined the cytotoxicity of IBT against several cancer cell lines (SMMC-7721, HepG2, HeLa, and A549) by MTT assay. Among those cell lines, IBT exhibited the highest cytotoxicity toward the human hepatoma SMMC-7721 cells, with the IC50 value lower than 10 μM after 48-hour treatment, while the IC50 values from other cancer cell lines were all above 20 μM (Fig. 2A). Our study also showed the time and dose dependency of IBT cytotoxicity on SMMC-7721 cells (Fig. 2B). In our work, trypan blue exclusion assay results (Fig. 2C) were confirmed with fairly consistent results using MTT assay. IBT displayed a low cytotoxicity toward the normal HEK 293T cell line (Fig. 2D), which indicated its feasibility for cancer therapy.

Inhibition of cell growth by IBT. (A) Cytotoxicity of IBT toward HeLa, A549, HepG2, and SMMC-7721 cells. IBT with the indicated concentrations was used to treat the cells for 48 hours, and MTT assay was used to determine the cell viability. (B) Time-dependent cytotoxicity of IBT toward SMMC-7721 cells. IBT with the indicated concentrations was used to incubate the SMMC-7721 cells for 24, 48, or 72 hours, and MTT assay was carried out to determine the cell viability. (C) Cytotoxicity of SMMC-7721 cells treated with IBT for 48 h was determined by the trypan blue exclusion assay. (D) Cytotoxicity of IBT toward HEK 293T cells compared with SMMC-7721 cells. The viability of the cells treated by IBT with the indicated concentrations for 48 hours was determined using the MTT assay. (E) IBT inhibits proliferation of SMMC-7721 cells. Ten or 20 μM IBT was used to incubate the SMMC-7721 cells for 24 hours, and the cell percentage in G0/G1, S, and G2/M phase was analyzed by flow cytometry. (F) IBT suppresses migration of SMMC-7721 cells. Before (upper) and after (lower) SMMC-7721 cells were incubated with 2.5 or 5 μM IBT for 24 hours, the images were acquired by fluorescence microscope. Scale bar, 100 μm. Data are expressed as the average ± S.E. in the three experiments. **P < 0.01 vs. the control groups in (A)–(C); **P < 0.01 vs. the normal cell groups in (D).
Further studies demonstrated that IBT can arrest the cell proliferation and migration. The cell cycle result was analyzed by flow cytometry. It can be seen from Fig. 2E that SMMC-7721 cells were arrested at the G0/G1 phase by IBT. The effects of IBT on cell migration were examined via the wound healing assay. As illustrated in Fig. 2F, the scratch area was obviously wider in the IBT-treated group after 24-hour incubation, which revealed inhibition of SMMC-7721 cell migration by IBT. Our results conclusively demonstrated that IBT caused significant inhibition against SMMC-7721 cells.
Molecular Docking Simulation.
To investigate the potential mechanism of IBT cytotoxicity, we analyzed its chemical structure first to glean some preliminary insights. We found that IBT has an α,β-unsaturated ketone reactive group which has been demonstrated to be an acceptable recognition site of the mammalian TrxR inhibitor. The approved structure, also including naphthoquinone, α-methylene-γ-lactone moiety, and α,β-γ,δ-unsaturated lactone moiety, can inhibit TrxR activity due to its electrophilic propensity toward the nucleophilic groups of TrxR (Cai et al., 2012; Zhang et al., 2019).
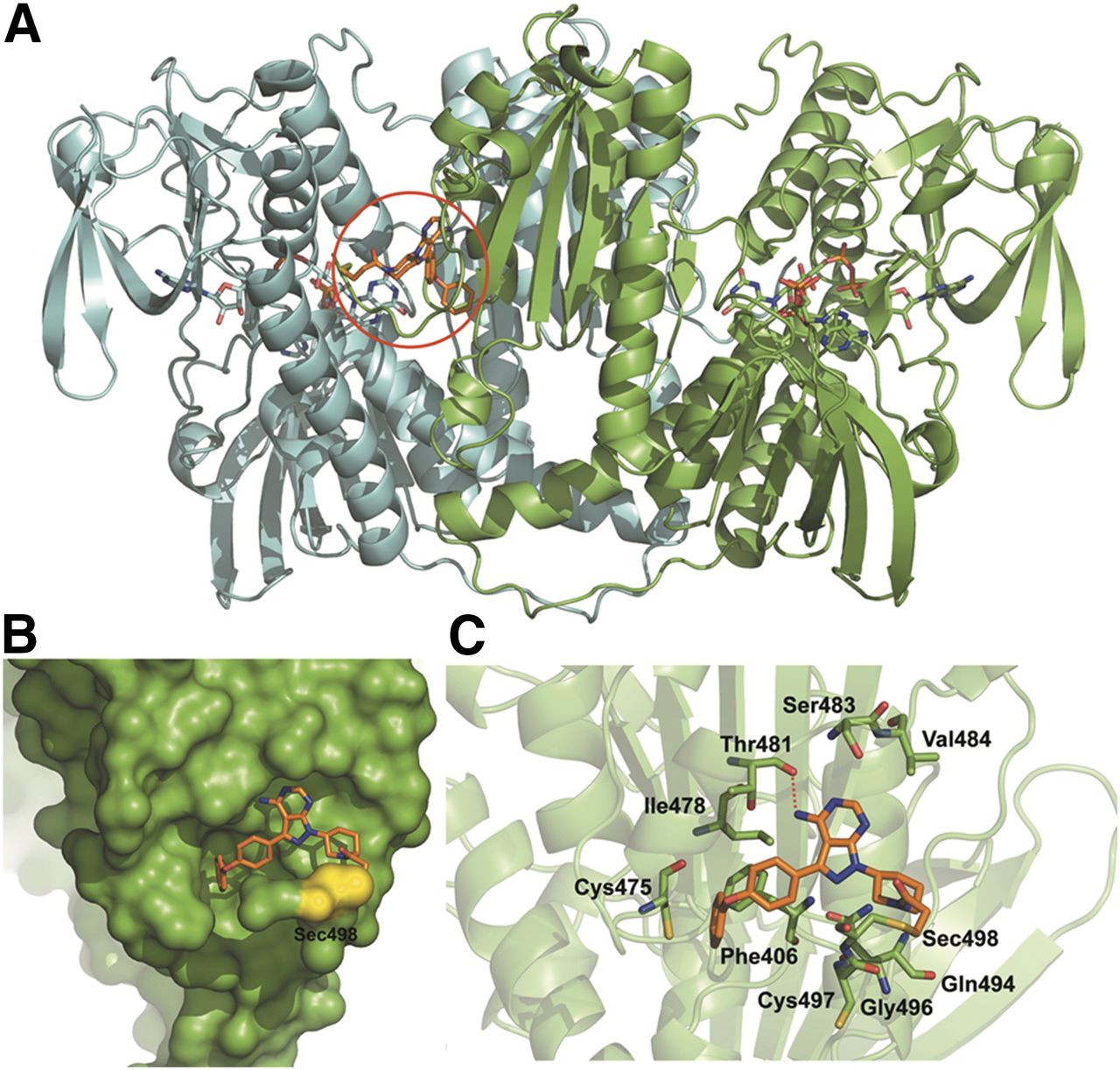
To verify whether IBT could be an irreversible inhibitor of TrxR, we adopted a molecular docking approach to simulate the possible binding mode of IBT interactions with mammalian TrxR1. As shown in Fig. 3A, IBT mainly bound to the pocket of TrxR1 with good complementarity, and the α,β-unsaturated carbonyl group of IBT formed a covalent bond with the selenol of Sec498 by Michael addition. The surface presentation indicated good complementarity between the pocket and the structural scaffold of IBT (Fig. 3B). As illustrated in Fig. 3C, the binding pocket of IBT was formed by Phe406, Cys475, Ile478, Thr481, Ser483, Val484, Gln494, Gly496, Cys497, and Sec498, and a hydrogen bond was formed between the amino group of IBT and the carboxyl group of Thr481. The docking score of the nonbonding interaction was −5.38 kcal/mol. All of these results present a clear view that IBT can irreversibly interact with mammalian TrxR1 and connect a covalent bond with Sec498 of TrxR1. Moreover, the nonbonding interactions between the residues around Sec498 tend to form a suitable binding site for IBT.

The binding mode of IBT with mammal TrxR1. (A) The binding site of the TrxR1 dimer with IBT is labeled by a red circle. Chain A and chain B of the TrxR1 dimer are shown in green and cyan illustration, respectively. (B) The surface representation of the binding site of IBT on TrxR1. Sec498 is shown in yellow. (C) The surrounding residues of IBT in the pocket. The structural scaffold of IBT is shown in orange sticks.
Inhibition of Cellular TrxR Activity.
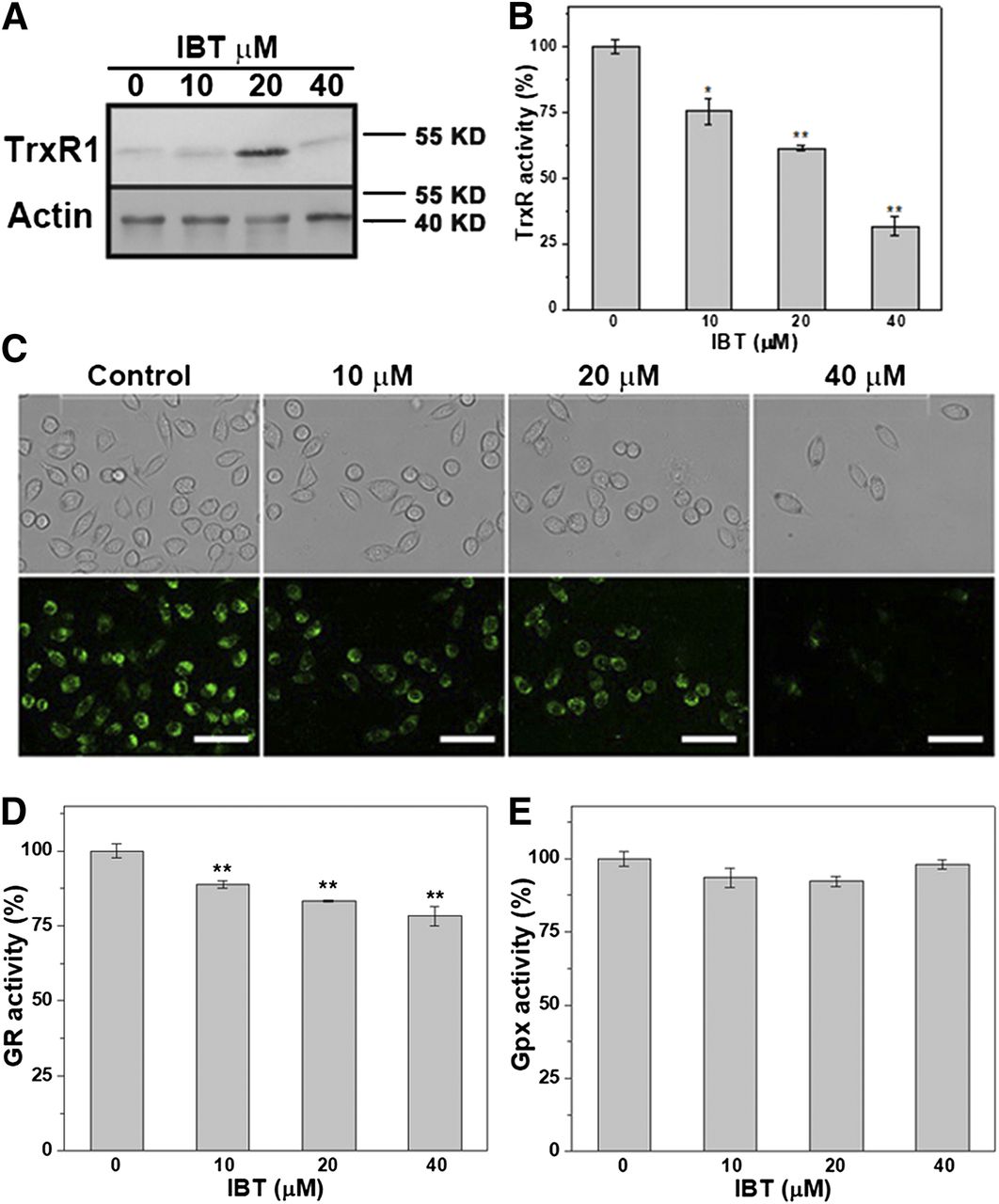
The aforementioned results demonstrated that IBT could be an irreversible inhibitor of mammalian TrxR. To extend this discovery, we adopted the classic Trx-mediated insulin reduction assay to investigate it. SMMC-7721 cells were selected to research the physiologic function between IBT and TrxR. The control group was treated with the maximum DMSO content of the experimental groups without IBT. It can be seen from Fig. 4A that IBT upregulated the expression of cellular TrxR1, which was suspected to be caused by activation of the Keap1-Nrf2-antioxidant response element pathway (Kumar et al., 2011; Wilson et al., 2013), while the decrease of TrxR1 expression of the 40 μM group was suspected to be caused by overcytotoxicity-induced inhibition of gene expression (Lopez-Alonso et al., 2013). It can be seen from Fig. 4B that incubation of SMMC-7721 cells with IBT caused a significant decrease of cellular TrxR activity in a dose-dependent manner. Only approximately 25% of TrxR activity remained with 40 μM IBT treatment. To further confirm the insult of cellular TrxR by IBT, we used a specific TrxR indicator, TRFS-green (Zhang et al., 2014), to measure the intracellular TrxR activity in SMMC-7721 cells. The results showed that the fluorescence of SMMC-7721 cells, which was correlated with cellular TrxR activity, decreased dose-dependently (Fig. 4C). Despite increased expression of cellular TrxR1, total cellular TrxR activity significantly decreased, which indicated that IBT inhibited enzymatic activity through its direct inhibition of TrxR. As shown in Fig. 4, D and E, IBT had a faint inhibition of GR and Gpx compared with the significant inhibition of TrxR, which indicated that IBT might have selectively inhibited the activity of TrxR in our experimental conditions.

Change of intracellular TrxR1 expression and TrxR, GR, and Gpx activity by IBT. (A) TrxR1 protein levels in SMMC-7721 cells changed by IBT. IBT with different concentrations was used to incubate SMMC-7721 cells for 24 hours, and cell lysis was conducted with western blot assay. Endpoint insulin reduction assay (B) and cell imaging (C) were used to determine the intracellular TrxR activity. After SMMC-7721 cells were incubated with IBT with different concentrations for 24 hours, the intracellular TrxR activity was assayed through the aforementioned method. The fluorescence images (bottom panel) and the bright filed images (top panel) were obtained by fluorescence microscope. Scale bar, 50 μm. After SMMC-7721 cells were treated with IBT for 24 hours, the GR activity (D) and Gpx activity (E) were determined by the enzyme dynamic method. Data are expressed as average ± S.E. in three experiments. *P < 0.05; **P < 0.01 vs. the control groups in (B), (D), and (E).
Inhibition of TrxR Contributes to the Cytotoxicity of IBT.
To confirm whether cytotoxicity to SMMC-7721 cells by IBT was associated with the inhibition of cellular TrxR activity, we investigated the cytotoxic effects in the TrxR inhibition situation. ShTrxR1 vectors, which can specifically target the TrxR1 gene and knock down TrxR1 expression, were transfected into HeLa cells to generate HeLa-shTrxR1 cells, while HeLa-shNT cells were transfected with shNT vectors. ShTrxR1 and shNT vectors were obtained from Constantinos Koumenis from the University of Pennsylvania School of Medicine (Javvadi et al., 2010). As shown in Fig. 5, A and B, shTrxR1 vectors significantly decreased the expression of TrxR1 and the activity of TrxR. Treatment of HeLa-shNT cells and HeLa-shTrxR1 cells with the same concentration of IBT caused different cytotoxicity; IBT exhibited higher potency toward HeLa-shTrxR1 cells than HeLa-shNT cells (Fig. 5C). Pharmacological inhibition of TrxR also gave constant results. Pretreatment with auranofin (AF) for 4 hours significantly reduced the TrxR activity (Fig. 5D), and the pretreatment group was more sensitive to IBT-induced cell death (Fig. 5E). All of the aforementioned results indicated that the cytotoxicity of IBT toward SMMC-7721 cells was related to TrxR inhibition.

Pharmacological intervention of TrxR and TrxR1 knockdown increases the cytotoxicity by IBT. TrxR1 expression (A) and TrxR activity (B) in HeLa-shNT/HeLa-shTrxR1 cells. The endpoint insulin reduction assay was implemented to determine the TrxR activity, and the expression of TrxR1 in both cells was analyzed by western blot. (C) IBT had cytotoxic effects on HeLa-shTrxR1 and HeLa-shNT cells, both of which were incubated through IBT with indicated concentrations for 48 hours. The MTT assay was implemented to measure the cell viability. (D) SMMC-7721 cells were incubated with indicated concentrations of AF for 4 hours. The TrxR activity was determined by endpoint insulin reduction assay. (E) After culture with indicated AF for 4 hours, the SMMC-7721 cells were incubated with the aforementioned concentrations of IBT for 44 hours. The cell viability was determined by MTT assay. The data are expressed as average ± S.E. in three experiments. *P < 0.05; **P < 0.01 vs. the HeLa-shNT cells in (B), **P < 0.01 vs. the 0 μM AF group in (D) and (E).
Induction of ROS by IBT.
The Trx system is one of the most important in vivo systems to scavenge ROS generated during cell metabolism and balance intracellular redox homeostasis. TrxR inhibitors, such as securinine (Zhang et al., 2017c) and alantolactone (Zhang et al., 2016), have been reported to interfere with TrxR to induce the accumulation of oxidized Trx, which subsequently reduced the burst of ROS. These compounds can also react with TrxR to generate modified TrxR, which can transfer electrons from NADPH to molecular oxygen and produce superoxide anions (Fang et al., 2005). Thus, we examined whether IBT has an ROS-generating effect in SMMC-7721 cells.
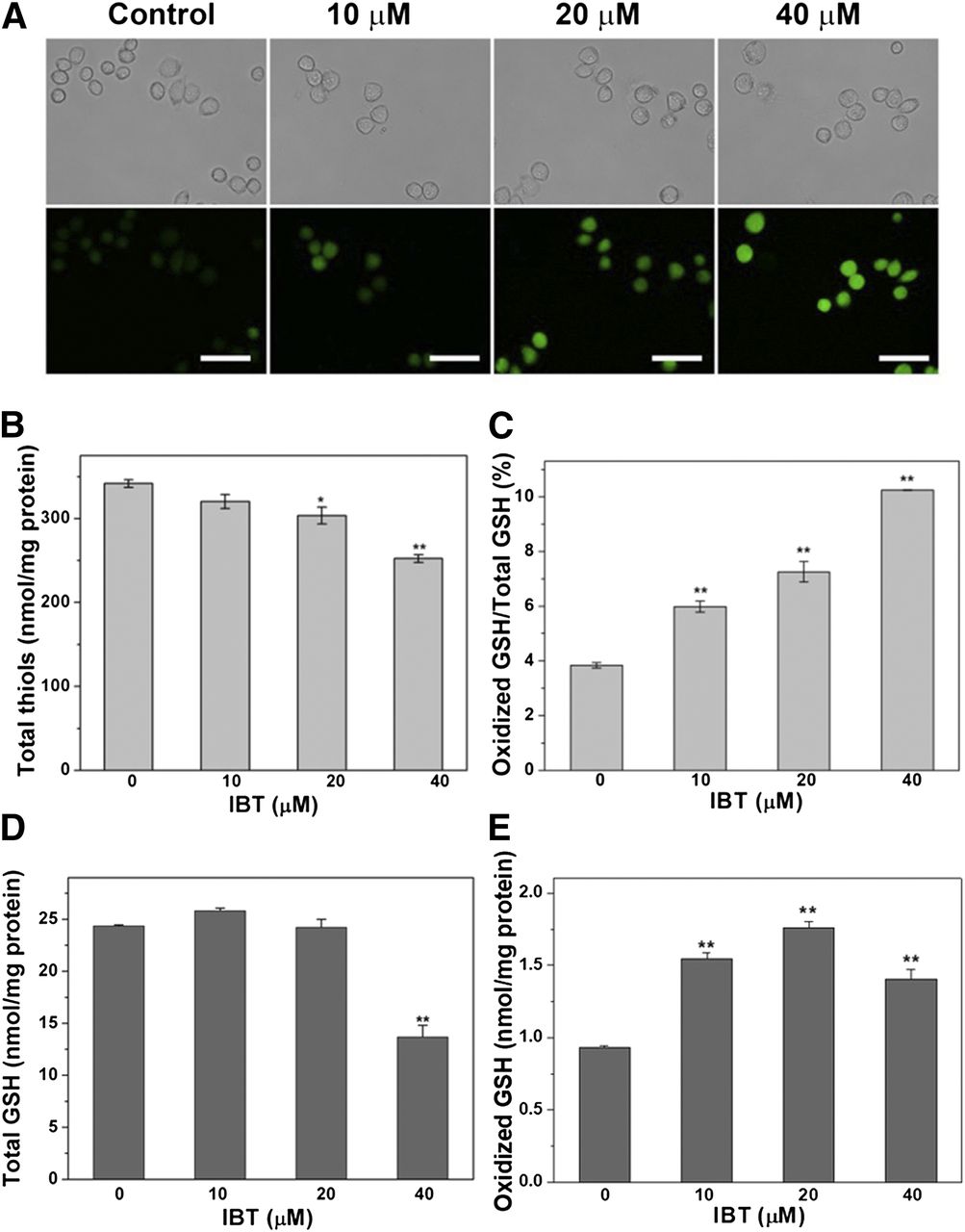
We first used the commercial probe DCFH-DA to detect the overall ROS levels. The membrane-permeable dye DCFH-DA can be deacetylated to DCFH, which is membrane-impermeable and nonfluorescent, by intracellular esterase. DCFH can be subsequently converted to fluorescent 2′,7′-dichlorofluorescein by intracellular ROS, and the cellular ROS content can therefore be reflected by the intensity of fluorescence. As shown in Fig. 6A, the fluorescence intensity increased with the rising of IBT concentration. The fluorescence of cells under basal conditions was undetectable, while the intense fluorescence in the experimental groups indicated a burst of ROS, especially the 40 μM group. Taken together, our findings indicated that IBT could promote ROS generation in SMMC-7721 cells.

Oxidative stress induced by IBT in SMMC-7721 cells. (A) Imaging of intracellular ROS level in SMMC-7721 cells by DCFH-DA. IBT with the aforementioned concentrations was used to treat SMMC-7721 cells for 1 hour. The fluorescence images (bottom panel) and the bright filed images (top panel) were obtained by fluorescence microscope. Scale bar, 50 mm. (B) The alteration of total thiols level by IBT in SMMC-7721 cells. IBT with the aforementioned concentrations was used to treat SMMC-7721 cells for 24 hours, and DTNB titration quantified the intracellular total thiols. (C) Increased GSSG/GSH ratio in SMMC- 7721 cells. The SMMC-7721 cells were treated by IBT with the aforementioned concentrations for 24 hours, and an enzymatic recycling method was adopted to assay the intracellular GSSG and GSH level; the GSSG/GSH ratio is shown. Scale bar, 50 μm. GSH (D) and GSSG content (E) in SMMC-7721 cells treated with IBT for 24 hours. Data are expressed as average ± S.E. of three experiments. **P < 0.01 vs. the control groups in (A), (B), (D), and (E).
Change of Cellular Thiols and Glutathione.
Because the results of DCFH-DA staining assays are prone to artifacts, as adequately discussed (Kalyanaraman et al., 2012), we assayed overall thiol contents to confirm the redox state in SMMC-7721 cells. The balance of thiols and disulfides is a key indicator of cellular redox state, and cellular thiols are also important antioxidants to resist ROS accumulation (Bindoli et al., 2008). Overall intracellular thiol levels were determined via the DTNB titration assay. It can be seen from Fig. 6B that total thiol levels in treated cells significantly decreased. To further confirm the results, we measured the change of cellular GSH homeostasis, another sensitive indicator of the cellular redox state. As illustrated in Fig. 6C, the ratio of GSSG/GSH in SMMC-7721 cells also showed a dose-dependent increase, and the ratio of the 40 μM group increased to more than 2.5 times that of the control group. Taken together, all of the aforementioned results confirmed that IBT could increase the intracellular oxidation level in SMMC-7721 cells.
Protection of Cell Death by NAC and Promotion of Cell Death by BSO.
The aforementioned results indicated that IBT could induce ROS generation and disrupt the balance of cellular redox state. We further determined the effects of NAC supplementation and GSH depletion on IBT-induced cell death to explore whether the cytotoxicity of IBT was related to the oxidative stress. After SMMC-7721 cells were treated with indicated concentrations of IBT and NAC for 48 hours, the results were detected via MTT assay. As shown in Fig. 7A, the cytotoxicity of IBT was gradually lost as the concentration of NAC increased; 5 mM NAC enhanced the survival rate to twice that of the control, while 10 mM NAC tripled it. Then, we measured the effect of GSH depletion on the cytotoxicity of IBT. The GSH system is an important backup for the Trx system and plays a significant role when the Trx system is dysfunctional (Du et al., 2012). SMMC-7721 cells were pretreated with 50 μM BSO for 24 hours, and the cells were then washed and replaced with new medium before incubation with IBT. In our conditions, cellular GSH content decreased to less than 20% of the control group (Fig. 7B). As shown in Fig. 7C, pretreatment with BSO obviously enhanced the cytotoxicity of IBT, while incubation with BSO alone resulted in faint cytotoxicity. Both the NAC-induced decreased cytotoxicity and the GSH depletion–induced promoted to cytotoxicity further support the notion that oxidative stress is involved in the process of IBT-induced cell death.

(A) Alleviation of IBT cytotoxicity against SMMC-7721 cells by NAC. IBT and NAC with the indicated concentrations were used to incubate the SMMC-7721 cells for 48 hours, and the MTT assay was carried out to measure the cell viability. (B) Total GSH content in SMMC-7721 cells treated with 50 mM BSO for 24 hours. (C) Enhancement of IBT cytotoxicity against SMMC-7721 cells by GSH depletion. The SMMC-7721 cells were incubated with 50 μM BSO for 24 hours and incubated by IBT with the indicated concentrations for 48 hours. The viability of cells was measured through the MTT assay. Data are expressed as average ± S.E. in three experiments. *P < 0.05; **P < 0.01 vs. the groups without NAC in (A); **P < 0.01 vs. the groups without BSO in (C).
Induction of Apoptosis by IBT.
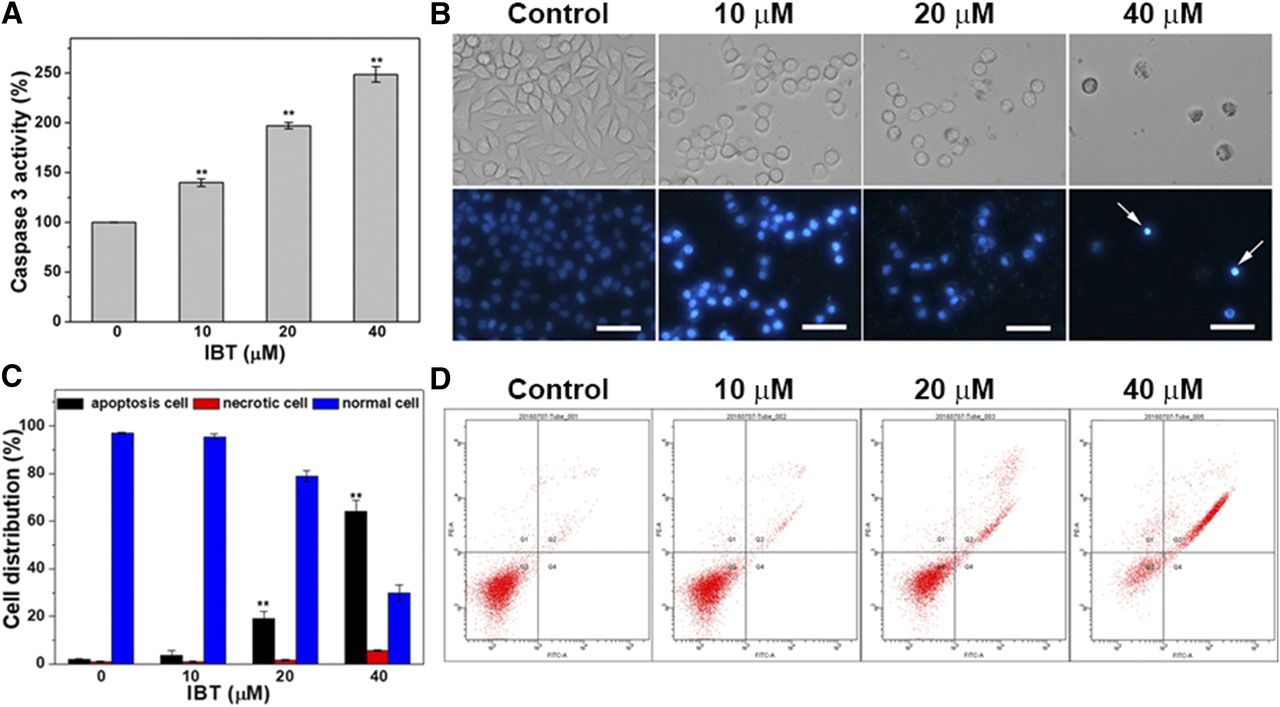
Apoptosis is a component of normal physiologic processes and is controlled by a series of complicated events. Inhibition of apoptosis has been thought to be the fundamental pathway through which cancer cells prolong cell viability (Hanahan and Weinberg, 2011). ROS are a double-edged sword to cells; at a low level, ROS can function as signaling molecules (D’Autréaux and Toledano, 2007), but excess ROS are cytotoxic and can induce cell apoptosis (Davis et al., 2001). As described earlier, we demonstrated that IBT can induce oxidative stress, contributing to cytotoxicity. We further wondered whether IBT killed SMMC-7721 cells through apoptosis. Chemotherapeutic drugs and oxidative stress can induce an intrinsic apoptosis pathway. Mitochondrion-dependent apoptosis is initiated by the release of cytochrome C and recruitment of caspase 9, which subsequently activates the effector caspases (caspases 3, 6, and 7) (Boatright and Salvesen, 2003; Fink and Cookson, 2005). Therefore, we first assessed the activation of caspase 3, the most common hallmark of early apoptosis. It can be seen from Fig. 8A that IBT obviously activated cellular caspase 3 in a dose-dependent manner. Then, we used Hoechst 33342, a membrane-penetrating double-stranded DNA–binding stain, to characterize the nuclear morphologic changes to confirm the results. The control group showed homogeneous round nuclei with diffuse weak fluorescence, while the experimental group showed condensed nuclei with more intense fluorescence (Fig. 8B), which was considered to be a characteristic morphology of apoptosis. Finally, an Annexin V-FITC/PI apoptosis detection assay was performed to determine the specific amounts of apoptosis. Living and apoptotic cells were quantified via flow cytometry. It can be seen from Fig. 8D that the control group consisted almost entirely of all living cells, while the experimental groups exhibited an increasing rate of apoptotic cells as the IBT concentration increased. All of the aforementioned results suggest that IBT predominantly triggered SMMC-7721 cell apoptosis.

Apoptotic cell death induced by IBT. (A) Activation of caspase 3 by IBT in SMMC-7721 cells. The SMMC-7721 cells were incubated with IBT with the indicated concentrations for 24 hours. Based on the fluorescence intensity of free pNA, the activity of caspase 3 activity was assayed. (B) The nuclear morphologic changes in SMMC-7721 cells by IBT. SMMC-7721 cells were incubated with IBT with the indicated concentrations for 48 hours, and the culture was added with the Hoechst 33342 staining. The fluorescence images (bottom panel) and the bright filed images (top panel) were obtained by a fluorescence microscope. Scale bar, 50 μm. (C) Quantification of the population of normal cells, apoptotic cells, and necrotic cells from (D). (D) Apoptotic cell death analyzed by Annexin V/PI double staining assay. The SMMC-7721 cells were incubated by IBT with indicated concentrations for 24 hours. Flow cytometry was used to analyze the percentage of apoptotic cells, necrotic cells, and live cells. Cells were divided into four different cell populations in the scatter grams: PI-positively and FITC-negatively stained cells were necrotic cells (upper left, Q1). FITC and PI double-positive (stained) cells were late apoptosis (upper right, Q2). FITC and PI double-negative (unstained) cells were live cells (lower left, Q3). FITC-positive and PI-negative (stained) cells were early apoptosis (lower right, Q4). Data are expressed as average ± S.E. in three experiments. **P < 0.01 vs. the control groups in (A and C).
Discussion
IBT has been approved for the treatment of several B-cell malignancies (Dreyling et al., 2016), and numerous phase I and phase II trials have demonstrated its clinical effects (Harrison, 2012; Byrd et al., 2013). It has been reported that IBT could block downstream B-cell receptor activation (Herman et al., 2011), abolish cell proliferation, and induce cell apoptosis both in vitro and in vivo (Herman et al., 2013). As a clinical BTK inhibitor for various B-cell malignancy therapies, IBT has been proved by extensive in vivo and in vitro studies. Although the IC50 for BTK was at the nanomole level, the IC50 of IBT in CLL cells was in the micromole level (Herman et al., 2011; Ponader et al., 2012), which made us consider whether IBT has another target for its clinical therapy. For example, ErbB receptor and ABCB1 were also reported to be targeted by IBT (Grabinski and Ewald, 2014; Zhang et al., 2017a). Our results demonstrated that IBT can target mammalian TrxR to inhibit SMMC-7721 cell growth. TrxR is an antioxidant enzyme widely existing in all organisms, and it has recently received more and more attention in cancer therapy (Urig and Becker, 2006; Zhang et al., 2017b). Because BTK was mainly expressed in bone marrow, spleen, and hematopoietic cells (Smith et al., 2001; Mohamed et al., 2009), the anticancer effect we researched may not be related to BTK inhibition. Only in this condition can we investigate other targets of IBT. Novel targets of IBT could contribute positively to understanding its anticancer mechanism, and it is easier to apply IBT in clinical therapy without a large number of clinical trials.
Among several cancer cell lines, the human hepatoma SMMC-7721 cell line was found to be the most sensitive cell line to IBT with an IC50 value of 7.3 μM (Fig. 2A). The results also showed that IBT could arrest the cell cycle and migration (Fig. 2, E and F), which further indicated that IBT can significantly inhibit SMMC-7721 cell growth. As shown in Fig. 2D, the low cytotoxicity of IBT toward the HEK 293T cell line demonstrated its potential for cancer therapy.
The mechanism by which IBT inhibited BTK was due to its similar structure with ATP, and it can bind the ATP-binding site of BTK. Since there was a Cys residue in the binding site, the α,β-unsaturated ketone group was added to enhance the binding effect (Arnold et al., 2000; Pan et al., 2007). Our molecular docking results also suggested that IBT could be an irreversible inhibitor of mammalian TrxR (Fig. 3). Intracellular TrxR activity in SMMC-7721 cells was obviously inhibited by IBT (Fig. 4, B and C). The IC50 of CLL cells was also at a micromole level (Herman et al., 2011; Ponader et al., 2012), which indicated that TrxR could also be a potential target of IBT for cancer therapy. Under our experiential conditions, IBT caused obvious inhibition of cellular TrxR activity (Fig. 4B) but mild inhibition of cellular GR and Gpx activity (Fig. 4, D and E), which indicated that IBT showed a high preference for mammalian TrxR compared with other sulfhydryl proteins. There was a pocket around the active site GCUG of TrxR (Cheng et al., 2009), and IBT binding with the pocket can inhibit the activity of TrxR. Although Gpx is also a selenoprotein which can directly scavenge the H2O2 produced from superoxide anion (Dey et al., 2016), Gpx can exert the function only in the presence of extensively reduced GSH. Because of the amount of ROS in living cells, especially cancer cells, IBT could not react with the oxidized Gpx. Low inhibition of GR was due to the deficiency of Sec residues (Berkholz et al., 2008), which reduced the reactivity to IBT. The aforementioned results supposed that IBT may target mammal TrxR for cancer therapy. Due to the high homology between mammal TrxR1 and mammal TrxR2 (Mustacich and Powis, 2000), IBT should also equally inhibit both TrxRs, which was proven in our previous investigation (Duan et al., 2016). Inhibition of both TrxRs contributed to the cancer therapy (Urig and Becker, 2006; Topkas et al., 2016). It has also been reported that AF and IBT have a synergistic effect on epidermal growth factor receptor-mutant non–small-cell lung cancer (Hu et al., 2018). AF was also a reported irreversible inhibitor of TrxR (Becker et al., 2000). According to our results, IBT could inhibit the TrxR activity. Therefore, the synergistic effect could also relate to oxidative stress. Further cellular-level evidence was obtained to support our hypothesis. Knockdown of TrxR1 and pharmacological inhibition of TrxR both sensitized cancer cells to IBT (Fig. 5, C and E). It has been reported that inhibition of the Trx system can lead to arrest of cell cycle and migration (Boronat et al., 2017; Xie et al., 2017), which was also obtained in our experimental results (Fig. 2, E and F). In conclusion, our results demonstrate that TrxR is a crucial target in the cellular effects of IBT.
Recently, the oxidative stress strategy has received increasing interest in relation to cancer therapy. Tumor cells are more sensitive to ROS promotion because they are more subject to oxidative stress than normal cells. Thereby, oxidative stress can kill cancer cells at lower levels of cytotoxicity than are required to kill normal cells (Gorrini et al., 2013; Glasauer and Chandel, 2014). It has been reported that IBT can induce oxidative stress in several B-cell malignancies (Rotin et al., 2014). In our study, we demonstrated that treatment with IBT can obviously insult the TrxR activity. Since the Trx system is a vital antioxidant for scavenging ROS in vivo, the significant ROS elevation by IBT was also determined through various methods (Fig. 6, A–C). NAC can act as a source of Cys for GSH biosynthesis in a situation of severe GSH depletion, whereas it acts as an antioxidant in the situation of no severe GSH depletion (Gleixner et al., 2017). Some literature has also reported that additional NAC usually acts as an antioxidant but not a Cys provider for GSH biosynthesis (Patriarca et al., 2005; Gleixner et al., 2017). Treatment of SMMC-7721 cells with IBT did not lead to a significant GSH decrease (Fig. 6D) or GR activity inhibition (Fig. 4D), so sensitization of cells to IBT with BSO pretreatment could be caused by synergistic effects of oxidative stress (Fig. 7C).
Upregulation of TrxR is one of the characteristics of cancer cells (Cha et al., 2009; Park et al., 2014). Not only can Trx, the substrate of TrxR, scavenge excess ROS produced by a vigorous metabolism, but TrxR can also help cancer cells resist apoptosis (Sasada et al., 1996; Kim et al., 2005). Resistance to apoptosis is the main way cancer cells evade cell death (Hanahan and Weinberg, 2011); thus, inducing the apoptosis of cancer cells has been a feasible scheme for cancer therapy. Our study has determined that IBT can induce the apoptosis of SMMC-7721 cells (Fig. 8), which has been previously reported (Schwamb et al., 2012; Amin et al., 2017). Inhibition of TrxR obviously weakened resistance to apoptosis and induced an increase in ROS (Fig. 6A), and excess ROS can further induce cellular apoptosis (Simon et al., 2000; Valko et al., 2007). It has been reported that compounds with electrophilic groups can connect with the Sec residue of mammalian TrxR to form selenium-compromised thioredoxin reductase–derived apoptotic proteins, which can also induce cell apoptosis (Anestal et al., 2008).
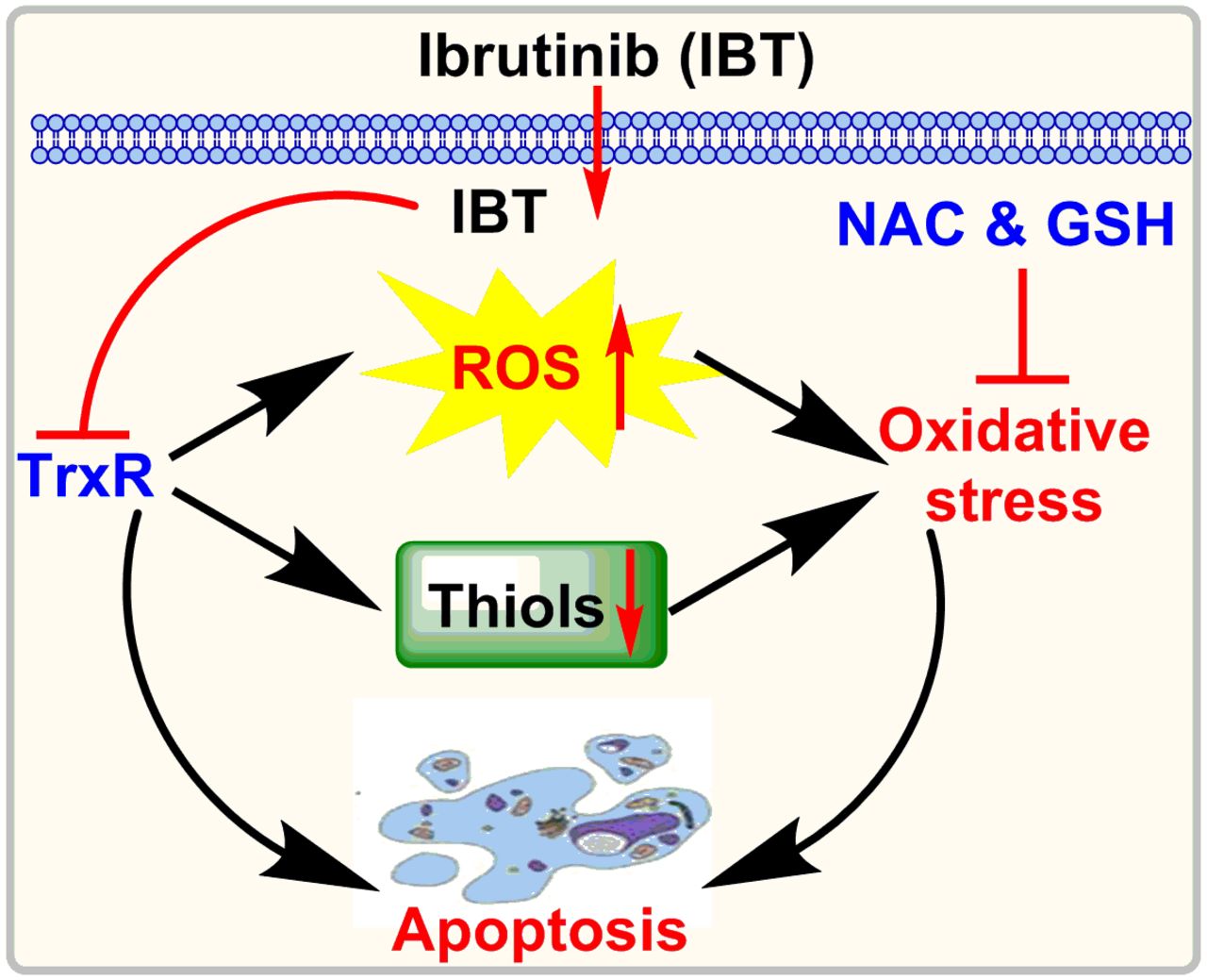
In conclusion, our findings proved that IBT could interfere with cellular TrxR and induce oxidative stress to promote the apoptosis of SMMC-7721 cells (Fig. 9). The discovery of a new target of IBT sheds light on better understanding its anticancer mechanisms and provides a theoretical foundation for its further use in clinical therapy.

Proposed mechanism for the apoptosis-inducing ability of IBT.
Acknowledgments
We thank Constantinos Koumenis for providing the shTrxR and shNT vectors.
Authorship Contributions
Participated in research design: Han, Zhang, Xu, Yao, Fang.
Conducted experiments: Han, Shi.
Performed data analysis: Han, Zhang, R. Liu, T. Liu, Shi, Wu, Fang.
Wrote or contributed to the writing of the manuscript: Han, Zhang, Wu, Fang.
Footnotes
- Received November 5, 2018.
- Accepted February 11, 2019.
This work was supported by grants from the National Natural Science Foundation of China [21572093 and 21778028], Natural Science Foundation of Gansu Province [18JR3RA296], Lanzhou University [Fundamental Research Funds for the Central Universities, lzujbky-2018-39], and Dalian University of Technology [Fundamental Research Funds for the Central Universities and DUT17RC (4) 27], and the 111 projects are greatly acknowledged.
Abbreviations
- AF
- auranofin
- BSA
- bovine serum albumin
- BSO
- l-buthionine-(S,R)-sulfoximine
- BTK
- Bruton’s tyrosine kinase
- CLL
- chronic lymphocytic leukemia
- DCFH-DA
- 2′,7′-dichlorofluorescein diacetate
- DMEM
- Dulbecco’s modified Eagle’s medium
- DMSO
- dimethylsulfoxide
- DTNB
- 5′,5′-dithiobis-2-nitrobenzoic acid
- DTT
- dithiothreitol
- FBS
- fetal bovine serum
- FITC
- fluorescein isothiocyanate
- GR
- glutathione reductase
- GSH
- glutathione
- GSSG
- oxidized glutathione
- IBT
- ibrutinib
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC
- N-acetyl-l-cysteine
- PBS
- phosphate-buffered saline
- PI
- propidium iodide
- ROS
- reactive oxygen species
- TBST
- Tris-buffered saline/Tween 20
- TE
- Tris-HCl EDTA
- TRFS
- XXX
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}