


Abstract
PGs derived from cyclooxygenase-2 (COX-2), in particular PGE2, play important roles in the initiation of inflammation and pain. In the present study, we evaluated the role of COX-2-derived PGE2 in an animal model of established hyperalgesia. Inflammation and hyperalgesia were first induced by injection of carrageenan into rat footpads. Then we investigated the effects of subsequent therapeutic treatment with a selective COX inhibitor, with a nonsteroidal anti-inflammatory drug and with anti-PGE2 antibody. Test compounds were administered 1 to 3 hr after carrageenan challenge, and inhibition of pain (hyperalgesia, measured by withdrawal from a thermal stimulus), and changes in paw edema and PG levels were evaluated. The i.v. administration of a nonselective COX inhibitor, ketorolac, caused a rapid reduction in hyperalgesia in the inflamed footpad, returning it to near-normal values within 1 hr. Normal (control) paw response times were not affected. Therapeutic administration of ketorolac prevented most further swelling caused by carrageenan but did not reverse edema already present at the time of dosing. Administered p.o., a selective COX-2 inhibitor (SC-58635) was as efficacious as ketorolac in reducing inflammatory hyperalgesia. Footpad PG levels returned to base line or below within 5 min of dosing with ketorolac, which suggests rapid turnover of PG in the inflamed tissue. Therapeutic treatment with a monoclonal anti-PGE2 antibody also fully reversed the hyperalgesia response. These studies suggest that continuous production of PGE2 by the COX-2 enzyme is a critical element in sustaining the hyperalgesic response at sites of tissue inflammation.
Currently available NSAIDs act by inhibiting the activity of the COX enzyme (also referred to as PGH synthase) that catalyzes the conversion of arachidonic acid to PGH2, the initial step in the formation of PGs and thromboxane (Vane, 1971; Smith and Willis, 1971; DeWitt, 1991). Systemic inhibition of COX leads to decreased production of PG at sites of inflammation (DeWitt, 1991; Masferrer et al., 1994; Seibert et al., 1994; Vane et al., 1994), and in the spinal cord (Ramwell et al., 1966; Yaksh, 1982;Coderre et al., 1990), as well as reduction of constitutive PGs in the digestive tract and kidney that are needed for normal function (Seibert et al., 1994). PGs play an important role in promoting the signs and symptoms of inflammation (Vane and Botting, 1994), and they enhance the response of C fibers to algesic stimuli (hyperalgesia) (Chahl and Iggo, 1977; Martin et al., 1987;Cohen and Perl, 1990). In animals, NSAIDs inhibit pain behavior induced by inflammation and prevent or reduce the swelling associated with an inflammatory stimulus (Seibert et al., 1994; Otterness and Bliven, 1985). In the human, NSAIDs are widely used to treat rheumatoid arthritis and other inflammatory conditions and are highly effective analgesics (Insel, 1996).
Two isoforms of COX have been identified (Xie et al., 1991;Kujubu et al., 1991; O’Banion et al., 1991;Smith and DeWitt, 1996): a widely distributed and constitutively expressed form, COX-1; and an inducible enzyme, COX-2, that is prominent at sites of inflammation. COX-2 is also constitutively expressed in the macula densa of the kidney (Harris et al., 1994) and in brain (Yamagata et al., 1993; Kaufmann et al., 1996; Marcheselli and Bazan, 1996; Chen et al., 1995). Pharmacological investigation of COX inhibition has shown that the commercially available NSAIDs inhibit both forms of COX enzymes (Copeland et al., 1994; Mitchell et al., 1993;Laneuville et al., 1994; Gierse et al., 1995). Highly selective inhibitors of COX-2 have recently been developed that demonstrate anti-inflammatory and analgesic activity equivalent to NSAIDs. (Seibert et al., 1994; Penning et al., 1997; Futaki et al., 1993; Chan et al., 1995)
The carrageenan paw inflammation model has long been used to evaluate the anti-inflammatory activity of NSAIDs (Winter et al., 1962), and there is a good correlation between efficacy in this model and activity in humans (Otterness and Bliven, 1985). This model has also been used to evaluate inflammatory hyperalgesia (Hargreaveset al., 1988). Because NSAIDs act via inhibition of PG formation by COX, we have used this model to investigate the role of COX-1 and COX-2, as well as that of PGs, in acute inflammation and pain (Seibert et al., 1994; Portanova et al., 1996). Previous studies showed that COX-2 expression was induced in the footpad by carrageenan and that inflammation and hyperalgesia could be blocked by a selective COX-2 inhibitor (Seibert et al., 1994) and by a monoclonal antibody to PGE2 (Portanovaet al., 1996). These results suggest a crucial role for COX-2-derived PGE2 in the initiation of acute inflammation. In these studies, drugs were administered before challenge with carrageenan, so no conclusions could be drawn concerning the role of PGs in the maintenance of inflammation. We have modified the carrageenan inflammation protocol by dosing therapeutically in order to evaluate the role PGs play in maintaining already established inflammation and hyperalgesia. This model provides an accurate measure of the onset of action of drugs and reveals any correlation between inflammatory symptoms and tissue levels of PGs. The results presented here indicate that inhibition of COX-2 caused rapid reversal of established hyperalgesia and marked reductions in PG levels at the site of inflammation. Furthermore, therapeutic administration of anti-PGE2 antibody completely reversed inflammatory hyperalgesia. These results indicate that maintenance of the hyperalgesic state requires the continuous production of PGE2 by COX-2.
Materials and Methods
Materials.
The following drugs and chemicals were used in this study: dexamethasone, indomethacin, MOPC-21 ascites fluid (mouse IgG1), methyl cellulose, Tween-20 (Sigma Chemical Co., St. Louis, MO), inflammatory-grade carrageenan (FMC, Rockland, ME) and ketorolac and ELISA reagents (Cayman Chemical Co., Ann Arbor, MI). The monoclonal anti-PGE2 antibody 2B5 was generated as an ascites fluid in syngeneic BALB/c mice, and the selective COX-2 inhibitor SC-58635 (Penning et al., 1997) was synthesized by Searle Chemistry.
Rats.
Male Sprague-Dawley rats obtained from Charles River (Portage, MI) and weighing 180 to 250 g were maintained in temperature- and humidity-controlled quarters with free access to water and food. Rats (five per group) were fasted, with free access to water, for more than 16 hr before testing. All experiments using animals were conducted under protocols approved by the Monsanto Animal Care and Use Committee.
Carrageenan-induced paw inflammation in rats.
The carrageenan-induced paw inflammation model has been described previously (Seibert et al., 1994; Winter et al., 1962; Hargreaves et al., 1988). In brief, inflammatory-grade carrageenan was prepared as a 1% suspension in saline, and a volume of 0.1 ml was injected with a 27-gauge needle into the footpad tissue of the right hind paw; the noninjected contralateral footpad of each animal served as a normal control. Carrageenan caused visible redness and pronounced swelling that was well developed by 3 hr. The average length of time that rats delayed before withdrawing a carrageenan-injected paw from an intense light stimulus was much shorter than for a normal paw (latency = 3 sec vs. 12 sec), which indicated that inflamed paws have increased sensitivity (hyperalgesia) to this thermal stimulus.
Measurement of edema and hyperalgesia in carrageenan-induced paw inflammation.
Paw volumes were determined by water displacement using a Ugo Basile (Comerio, Italy) plethysmometer before carrageenan injection and at selected times after compound dosing. Edema was defined by subtracting the initial volume displacement for each paw from its subsequent postcarrageenan measurements. A hyperalgesic response to heat was determined in the same animals using a University of California, San Diego paw thermal stimulator with bulb intensity adjusted for latency of 10 to 12 sec in normal animals. Rats were individually confined to Plexiglas chambers beneath which the high-intensity projector bulb was positioned to deliver a thermal stimulus to the hind paws. The withdrawal latency period for inflamed and contralateral control paws was determined with an electronic clock.
Compounds were administered therapeutically from 1 to 3 hr after carrageenan injection by i.v. route in saline or by oral gavage in 0.5% methyl cellulose and 0.025% Tween-20 vehicle. Monoclonal anti-PGE2 antibody or isotype-matched MOPC-21 control protein were administered in saline by i.v. injection at 30 mg/kg. In certain experiments, ketorolac (10 mg/kg), dexamethasone (1 mg/kg) or SC-58635 (30 mg/kg) was administered prophylactically by oral gavage 2 hr before carrageenan injection.
Measurement of PG production in rat paw tissue.
At selected times after dosing, rats were euthanized with CO2 and their hind feet removed. A volume of 0.1 ml saline containing 10 μM indomethacin was injected into the paw to aid the removal of eicosanoid-containing fluid and prevent further PG production in paw tissue. Paw pads were incised with a scalpel, suspended off the bottom of polypropylene tubes × with an Eppendorf pipet tip to facilitate fluid exudation and centrifuged at 1800 g for 15 min. The volume of exudate from each paw was measured. PGE2levels in paw exudates were quantitated with a competitive binding ELISA using Dynatech (Chantilly, VA) Immulon-4 microtiter plates coated with donkey anti-mouse antibody from Jackson ImmunoResearch (West Grove, PA) AChE-linked PGE2 tracer from Cayman Chemical Co. (Ann Arbor, MI) and monoclonal anti-PGE2 antibody as previously described (Mnich et al., 1995).
Statistical analysis.
Either a one-tailed Student’st test (when there were only two groups) or a Dunnett’s test (based on a one-way ANOVA, when there were more than two groups) was used to compare control-group means with treatment-group means at each time-point. When equivalent time-points did not occur, the last previously occurring control-group time-point was used.
Results
Rapid reversal of hyperalgesia by ketorolac.
In preliminary studies, we found that the injectable NSAID analgesic ketorolac was a potent inhibitor of COX-1 and COX-2 (IC50 = 20 nM for both enzymes; personal communication, for method see Gierse et al., 1995). To investigate the ability of this nonselective COX inhibitor to affect established inflammatory hyperalgesia, we administered ketorolac i.v. and tracked paw swelling and thermal sensitivity as indicators of inflammation. As expected, carrageenan injection induced substantial paw swelling with rapid increases for the first 2 hr, followed by slower but continuing increases thereafter (fig. 1A). The hyperalgesic response to a thermal stimulus measured in the same animals increased rapidly after carrageenan injection, reaching maximal levels after about 2 hr, and remained elevated for 6 hr. A single i.v. injection of ketorolac (30 mg/kg) at 1 to 3 hr after initiation of inflammation did not reduce swelling over the time period studied but did block further increases in paw volume. In the same animal, ketorolac caused a rapid reduction of hyperalgesia at each of the three times of compound administration (fig. 1B). Maximal inhibition of hyperalgesia was achieved 30 to 60 min after dosing, and the analgesic effect persisted throughout the study period.

Rapid reversal of hyperalgesia by a single ketorolac injection at different times. A volume of 0.1 ml of 1% carrageenan was injected into the footpad of the right hind paw in rats. Paw swelling (panel A) and hyperalgesic activity (panel B) were measured from 5 min to 3 hr after a single i.v. administration of 30 mg/kg ketorolac dosed at 1 hr (•), 2 hr (▴) or 3 hr (⧫) after carrageenan injection; saline control (▪). Data represent the difference in withdrawal latency or paw volume between normal and injected paws (mean ± S.E.M., n = 5 – 15 rats). Figure combines data from seven separate experiments. Error bars are omitted for clarity. Greatest S.E.M. = 9.5% of the mean.
Role of PGE2 in inflammatory hyperalgesia.
To study more directly the role of PGE2 in established inflammation, we compared the hyperalgesic response with PGE2 production at the site of inflammation. Ketorolac was administered i.v. 3 hr after injection of carrageenan into the footpad. The data presented in figures 2A and B demonstrated that ketorolac dramatically reduced pain responses within 1 hr of administration but had little effect on edema within the same time period. Measurement of PGE2 levels from the paw exudate fluid indicated that the reversal of hyperalgesia was accompanied by a concomitant reduction in PGE2 levels in affected paws. In a result consistent with previous studies (Portanovaet al., 1996), we found that paw PGE2 levels rose 4-fold between 1 and 3 hr and remained elevated for several hours. Interestingly, reduction in PGE2 levels in affected paws occurred within 5 min after i.v. dosing with ketorolac (fig. 2C). Furthermore, PGE2 levels dropped below those detected in normal paw tissue (0 time-point).

Effect of ketorolac on carrageenan-induced hyperalgesia and paw PG production. Animals were dosed 3 hr after carrageenan injection. Paw volume (panel A), hyperalgesic activity (panel B) and PGE2 levels in paw tissue (panel C) were measured from 5 min to 1 hr after i.v. dosing with ketorolac (30 mg/kg, •) or saline (▪). Data represent mean ± S.E.M.,n = 5 rats. *indicates group means significantly (P < .05) less than vehicle controls for the same (or the closest) time. Representative data from 1 of 5 experiments are shown.
A neutralizing monoclonal antibody to PGE2 (2B5) was utilized to address specifically the role of PGE2 in sustaining the inflammatory response. Two hours after carrageenan injection, i.v. administration of 30 mg/kg of the 2B5 antibody reduced hyperalgesia rapidly (by 50% in 30 min) and progressively, with near complete reversal by 3 hr (fig. 3). In contrast, MOPC-21 control protein had no effect at an identical dose over this time period. 2B5 antibody was detected at only 50 ng/ml in cerebrospinal fluid, a level 500-fold lower than that in plasma from the same animals (data not shown).

Effect of anti-PGE2 antibody on carrageenan-induced hyperalgesia. Monoclonal anti-PGE2antibody (▴), MOPC21 control antibody (•) or vehicle (▪) was administered i.v. 2 hr after carrageenan injection. The hyperalgesic activity was measured from 15 min to 3 hr after dosing. Data represent the mean ± S.E.M., n = 5 rats. *indicates group means significantly (P < .05) less than vehicle controls for the same time. Representative data from 1 of 2 experiments are shown.
Therapeutic intervention with p.o. administered compounds.
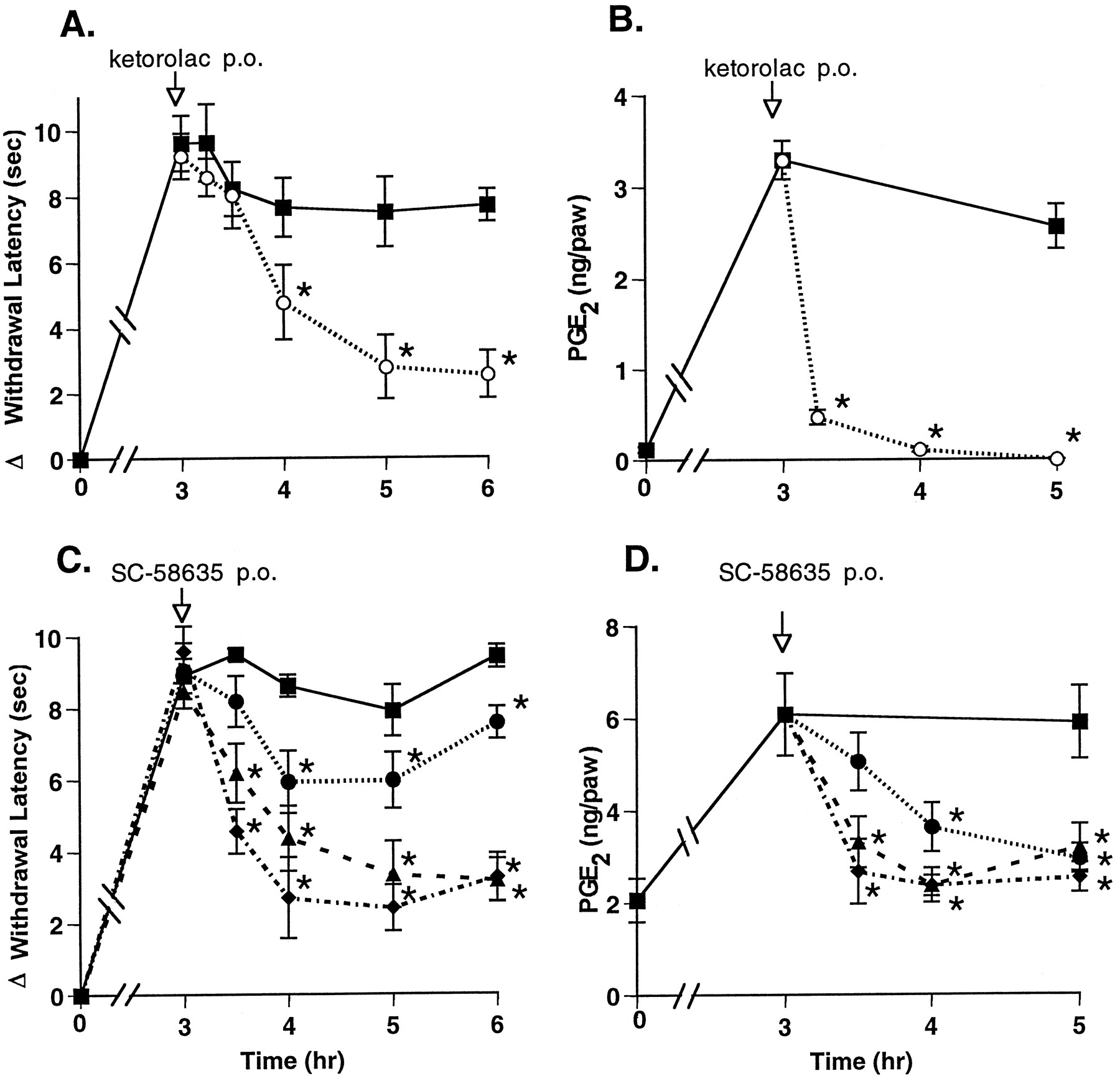
The results described above indicate that a mixed inhibitor of COX-1 and COX-2 could rapidly reverse hyperalgesia. To address whether COX-2-mediated PG production was responsible for this effect, we used a selective inhibitor of COX-2, SC-58635 (Penning et al., 1997). Compounds were administered p.o., and a maximal reduction of hyperalgesia was observed approximately 1 to 2 hr after p.o. administration of 30 mg/kg ketorolac (fig.4A). The selective COX-2 inhibitor SC-58635 (IC50 COX-1 = 15 μM, IC50COX-2 = 0.04 μM) was tested (5, 10 and 50 mg/kg) and found to reduce hyperalgesia in a dose-dependent manner, in a similar time-frame, and as effectively as ketorolac (fig. 4C). SC-58635 administered p.o. also reduced paw exudate PGE2 levels in a dose-dependent manner. All tested doses of SC-58635 reduced PGE2 levels to near baseline (fig. 4D). In contrast, ketorolac reduced paw PGE2 levels to below base-line levels within 1 hr, but it did so without achieving greater analgesic activity than SC-58635 (fig. 4B).
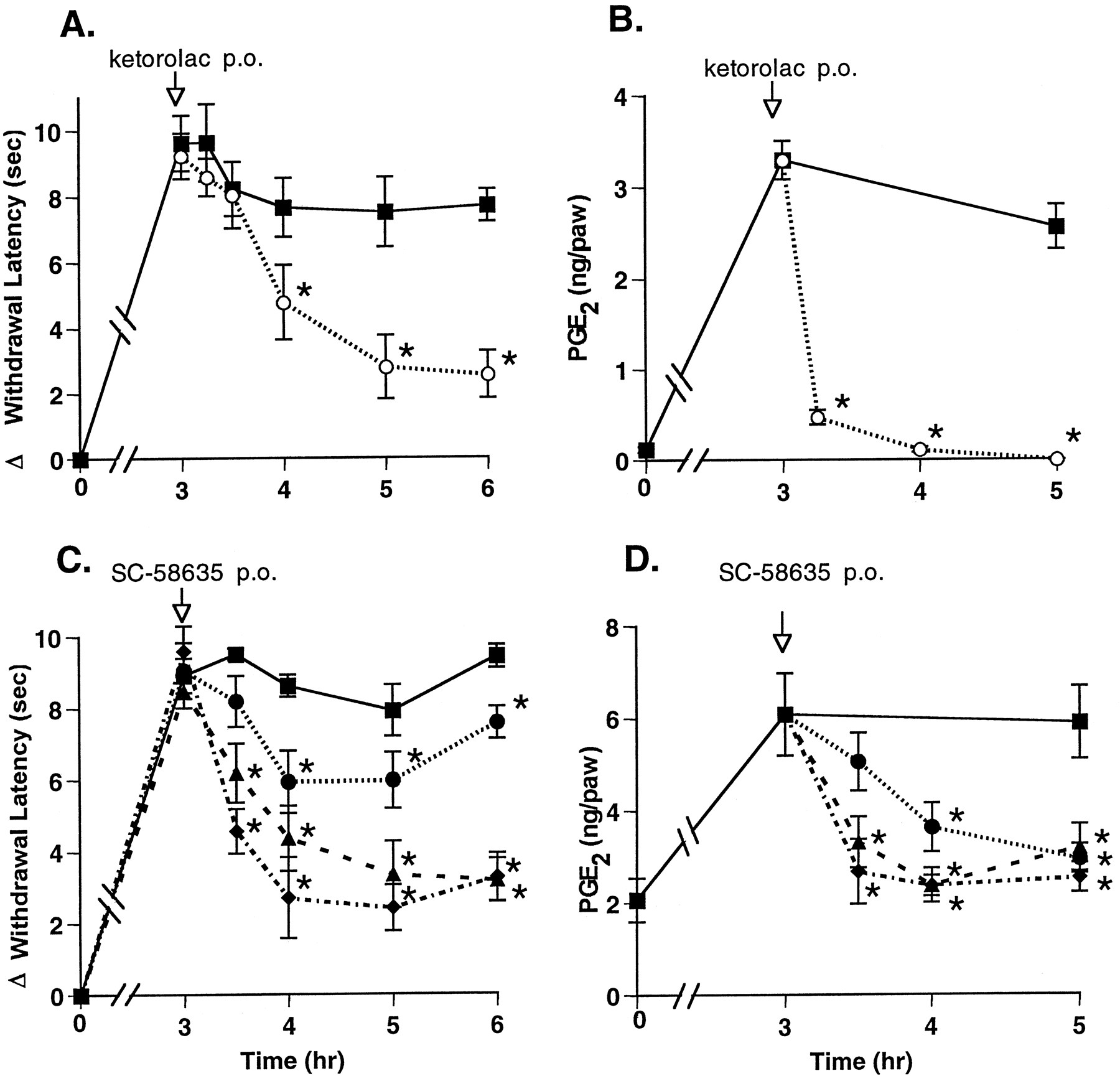
Effect of p.o. administered COX inhibitors on hyperalgesia and PG production. Drug or vehicle was administered p.o. 3 hr after carrageenan footpad injection. Hyperalgesic activity (panels A, C) and PG levels in the paw tissue (panels B, D) were measured from 15 min to 3 hr after dosing with 30 mg/kg ketorolac (○), SC-58635 (•, 5 mg/kg; ▴, 10 mg/kg; ⧫, 50 mg/kg) or vehicle (▪). The data are expressed as mean ± S.E.M., n = 5 rats. *indicates group means significantly (P < .05) less than vehicle controls for the same times and, for PGE2 group means at 3.25, 3.5 and 4 hr, significantly (P < .05) less than vehicle control at the 3-hr time. Shown are representative data from 1 of 5 experiments with ketorolac and from 1 of 4 experiments with COX-2 inhibitors.
Prophylactic treatment: comparison of mechanistically distinct agents.
The results shown in figure 4 suggest that basal levels of PG found in the footpad may derive from constitutively expressed COX-1, because the selective COX-2 inhibitor reduced PG levels to base line, whereas the nonselective inhibitor reduced PG levels to nearly undetectable levels. To assess this possibility, we performed an experiment with the anti-inflammatory glucocorticoid dexamethasone, which inhibits expression of COX-2 but not that of COX-1 (Kujubuet al., 1991; Fu et al., 1990; Masferrer et al., 1992). Edema, hyperalgesia and PG production at the inflammatory site were measured in a pretreatment regimen with compounds administered p.o. 2 hr before the carrageenan footpad challenge. In agreement with previous studies, edema, hyperalgesia and paw PGE2 levels were maximally elevated about 3 hr after administration of carrageenan (Portanova et al., 1996). The increases in edema and hyperalgesia were both prevented to an equal extent by pretreatment with ketorolac 30 mg/kg, dexamethasone 10 mg/kg or SC-58635 30 mg/kg (fig. 5A). The increase in PGE2 levels after carrageenan injection was prevented by preadministration of dexamethasone or the COX-2 inhibitor, but only the nonselective COX-1/COX-2 inhibitor ketorolac reduced PGE2 levels below base line (fig. 5B).

Effect of p.o. pretreatment with ketorolac, dexamethasone or COX-2 inhibitor on edema, hyperalgesia and PG in the paw. Sprague-Dawley rats were administered ketorolac (10 mg/kg), SC-58635 (30 mg/kg) or dexamethasone (1 mg/kg), or vehicle 2 hr before injection of carrageenan into the footpad. Edema and hyperalgesia were measured (panel A) 3 hr after carrageenan injection. Paws were then excised and exudate fluid measured for PGE2 (panel B). The data are expressed as mean ± S.E.M., n = 5 rats. *indicates group means found to be significantly less (P < .05) than the vehicle mean for edema, hyperalgesia and PGE2. Representative data from 1 of 4 experiments are shown.
Discussion
NSAIDs are widely used for the treatment of inflammation and pain, and they are thought to act via inhibition of the synthesis of PGs (Vane, 1971; Smith and Willis, 1971). In typical clinical use, these drugs are administered after pain and inflammation are present. This contrasts with most animal models of inflammatory pain, such as the Hargreaves model (Hargreaves et al., 1988) and the formalin test (Malmberg and Yaksh, 1992a; Malmberg and Yaksh, 1992b;Dubuisson and Dennis, 1977), wherein drugs are administered before the inflammatory stimulus. To understand better the mechanisms involved in acute inflammatory pain, we have modified the Hargreaves model to allow for therapeutic dosing after inflammation is established. One of the important results obtained with this model is its demonstration of the pivotal role of COX-2-derived PGE2 in sustaining a hyperalgesic response at sites of tissue inflammation. Thus blockade of PG synthesis with the mixed COX-1/COX-2 inhibitor ketorolac, 1 to 3 hr after carrageenan injection, rapidly eliminated established hyperalgesia. This result is consistent with those reported by Boyceet al. (1994) who also found that NSAIDs reversed established hyperalgesia. The length of time animals had been in a hyperalgesic state did not appear to influence the subsequent response (fig. 1), at least over this relatively short time course. It would be interesting to determine whether, after longer periods of inflammation, animals become more refractory to analgesic intervention. The prompt reversal of hyperalgesia seen in this model appears to reflect accurately what occurs clinically in humans, where rapidly absorbed NSAIDs such as ibuprofen quickly reverse pain caused by dental extraction (Troullow et al., 1990).
The results obtained with the anti-PGE2 antibody 2B5 are also of considerable interest. The ability of this antibody to reverse established hyperalgesia suggests that continuous production of PGE2 is needed to sustain the hyperalgesic response, and it complements earlier work that showed marked anti-inflammatory and analgesic activity of 2B5 when it was administered in a pretreatment regimen (Portanova et al., 1996). The slower onset of action of the antibody compared with the NSAID probably reflects a difference in the rates at which the antibody protein and the small-molecule inhibitor gain access to the inflamed tissue, but experiments to test this hypothesis directly have not been performed. The large observed difference in antibody concentration between the CNS and the circulation suggests that the site of action of the anti-PGE2 antibody is probably the inflamed tissue. Preliminary results with direct intrathecal administration of 2B5 antibody support this interpretation (data not shown).
The therapeutic model employed here also made it possible to analyze the rate at which PGs are cleared from inflamed tissue after blockade of biosynthesis. A highly water-soluble and potent NSAID, ketorolac, was used for these studies so that drug could be delivered i.v., eliminating absorption time as a variable. Ketorolac reduced PG levels in the footpad very rapidly, effects being apparent 5 min after its administration. This remarkable result suggests that PGs are cleared extremely quickly in inflamed tissue; whether this clearance is due to metabolism or diffusion-related processes cannot be determined from these studies.
Reversal of hyperalgesia by the selective COX-2 inhibitor SC-58635 was equivalent to that obtained with the nonselective COX inhibitor ketorolac. The COX-2 selective inhibitor had pharmacological effects on hyperalgesia in this model that were indistinguishable from those of ketorolac, which suggests that COX-2 is the enzymatic source of pro-inflammatory PGs. This result complements earlier studies showing that selective COX-2 inhibitors are as efficacious as NSAIDs in preventing acute inflammation and pain (Seibert et al., 1994; Futaki et al., 1993; Chan et al., 1995;Boyce et al., 1994). It is noteworthy that SC-58635 appeared to inhibit only the “induced” PGs, whereas ketorolac typically reduced PGs to below control levels (fig. 3). This suggests that the PGE2 that remains after SC-58635 treatment may be the product of constitutively expressed COX-1 enzyme in the paw. This hypothesis was supported by the experiment shown in figure 5, where dexamethasone also reduced PG levels to, but not below, base line. This result is consistent with the known ability of glucocorticoids in other cells and tissues selectively to modulate COX-2 but not COX-1 expression (Kujubu et al., 1991; Fu et al., 1990;Masferrer et al., 1992). As noted above, ketorolac reduced PGE2 to nearly undetectable levels. These results are somewhat paradoxical in that they suggest that quantitative blockade of PG production at the site of inflammation is not required for a complete analgesic effect. Further experiments with selective COX-1 inhibitors may shed light on this issue.
Once established, inflammation (as assessed by paw swelling or edema) is clearly less tractable to PG modulation; it showed little reduction over the time period studied. Mediators other than PG that increase vascular permeability, such as nitric oxide (Salvemini et al., 1996) and serotonin, may persist at the site, or it may be that once fluid has accumulated in the extracellular spaces, the absence of vasoactive agents may not be sufficient to return fluid rapidly to the lymphoid circulation. Production of an analgesic effect at the same time that considerable swelling persists suggests that PG and its associated mediators can achieve their effect by acting directly on the nociceptive pathway without concomitant reduction of inflammation. This is consistent with previous results in the formalin model, where intrathecal administration of NSAIDs prophylactically or therapeutically was effective in reducing hyperalgesia (Malmberg and Yaksh, 1992a). It is known that significant spinal facilitation of the nociceptive pathway occurs in hyperalgesic states (Yaksh, 1993). All of the models in which NSAIDs show activity are characterized by prolonged C fiber barrage that leads to spinal release of excitatory amino acids, substance P and PGs (Ramwell et al., 1966; Coderre et al., 1990; Malmberg and Yaksh, 1995; Yaksh 1993). The mechanisms by which PGs mediate hyperalgesia are unknown, but it seems likely that they help sensitize terminal afferent C fibers in the periphery and that these prolonged discharges release excitatory amino acids and peptides in the spinal cord. Yaksh and co-workers have suggested that spinal PGs play an important role in hyperalgesia; they base this conclusion on results demonstrating analgesic activity of NSAIDs administered intrathecally (Malmberg and Yaksh, 1992a; Malmberg and Yaksh, 1992b), a finding recently confirmed by others (Yamamoto and Nozaki-Taguchi, 1996). The observation that PGs are readily formed by spinal cord either in vitro or in vivo (Ramwellet al., 1996; Yaksh, 1982; Coderre et al., 1990;Malmberg and Yaksh, 1995) supports the hypothesis that central PGs are important in the initiation or maintenance of hyperalgesia. Nevertheless, the results with the 2B5 antibody strongly indicate an important role for peripheral PGs in the induction and maintenance of the hyperalgesic state, because antibodies do not cross the blood-brain barrier. This observation does not, however, rule out additional pharmacological sites of action for COX inhibitors in the CNS.
The evidence presented here for very fast turnover of PG in inflamed tissue and the ability of COX inhibitors to reduce inflammatory PG rapidly may, in part, account for their usefulness as analgesics in the clinic (Insel, 1996). Here we demonstrate the equivalent capacity of selective COX-2 inhibitors such as SC-58635 to reduce PG at the site of inflammation and to alleviate hyperalgesia rapidly. These results lend support to the concept of the clinical utility of this new class of analgesic and anti-inflammatory compounds with a greatly reduced side effect profile.
Acknowledgments
The authors wish to thank W. Smith and C. Smith for critical evaluation of the manuscript and Steve Mnich for production of and assistance with the 2B5 antibody.
Footnotes
-
Send reprint requests to: Peter C. Isakson, Monsanto Company, BB2B, 700 Chesterfield Parkway North, Saint Louis, MO 63198.
- Abbreviations:
- COX-1
- constitutive cyclooxygenase enzyme
- COX-2
- inducible cyclooxygenase enzyme
- NSAID
- nonsteroidal anti-inflammatory drug
- Received June 2, 1997.
- Accepted August 25, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}