


Abstract
Morphine administered simultaneously to intracerebroventricular (i.c.v.) and intrathecal (i.t.) sites exhibits synergism, with the antinociceptive potency much greater than would be predicted from a simple addition of the potencies of the same dose administered to either site alone. This synergism was quantified in mice using both a fixed dose method, in which the morphine dose at one site was fixed while the AD50 (antinociceptive dose at 50% effectiveness) of morphine at the other site was determined; and a variable dose method, in which different doses of morphine were administered simultaneously to both sites at a fixed ratio, and the AD50 determined and compared to the AD50 at a single site alone. When animals were made tolerant to morphine by implantation of a 75-mg morphine pellet for 3 days, this synergism was eliminated, so that morphine administered simultaneously to i.c.v. and i.t. sites had an additive effect. However, administration of the peptide DynorphinA-(2–17) i.v. simultaneously to the test doses of morphine in morphine-tolerant animals resulted in a partial restoration of synergism. These results suggest that morphine-induced antinociception is highly dependent on an intact integrated central nervous system system and that the initial tolerance development is the result of a disruption of synergism between the central nervous system sites. Morphine tolerance results not from a reduced sensitivity to morphine at discrete central nervous system sites, but rather from a reduced synergistic interaction of morphine at spinal and supraspinal sites. In support of this conclusion, there was no tolerance observed in morphine-pelleted animals to morphine administered to i.c.v. or i.t. sites alone. DynorphinA-(2–17), a nonopioid peptide has previously been shown to enhance the antinociceptive potency of morphine in morphine-tolerant animals, appears to act by restoring this synergism.
The dynorphins constitute a major family of endogenous opioid peptides. DynorphinA-(1–17) and a synthetic derivative, DynorphinA-(1–13), are unusual in that they have no significant antinociceptive activity of their own in the radiant heat tail flick assay. However, they have the unique property of modulating morphine antinociception (Tulunay et al., 1981). Systemic DynorphinA-(1–13) also suppresses morphine withdrawal signs in mice (Takemori et al., 1992), monkeys (Aceto et al., 1982) as well as humans (Wen and Ho, 1982). Recent studies have demonstrated that DynA-(2–17), which is a nonopioid peptide when given i.v., also retains these modulatory properties in morphine-tolerant mice by shifting morphine AD50 to the left (Takemoriet al., 1993). The mechanism for this phenomenon is not clear; however, because DynorphinA-(2–17) has no appreciable affinity for the known opioid receptors; the peptide is presumed to be exerting its action via a nonopioid system.
A series of studies by Yeung and Rudy (1980) and Fujimoto and colleagues (Roerig et al., 1984; Roerig and Fujimoto, 1988) have demonstrated that morphine induces antinociception by acting synergistically at spinal (i.t.) and supraspinal sites (i.c.v.),i.e., the AD50 for morphine antinociception at one site is greatly reduced when morphine is coadministered even in very low doses at the other site. Further studies of several strains of mice, differing in the degree to which opioid tolerance develops, show that in those strains that developed a high degree of tolerance, the synergism became reduced to an additive effect, although in animals developing relatively little tolerance, the synergism was largely retained (Roerig and Fujimoto, 1988). These observations suggest that the degree of morphine tolerance development seemed to be dependent on the synergism between the neuronal sites in these animals.
Based on these findings, we have hypothesized that the antinociceptive tolerance development to morphine is due to the disruption of the intact integrated central nervous system, i.e., the communication between spinal and supraspinal opioid systems. The suppressant effect of DynA-(2–17) on the antinociceptive tolerance to morphine given systemically may be exerted through a nonopioid system regulating the interaction of morphine between i.t. and i.c.v. sites in the central nervous system. Our study was designed to test this hypothesis.
Materials and Methods
Animals
Male Swiss Webster mice (20–25 g) were purchased from Hilltop Lab Animals, Inc. (Scottsdale, PA). They were housed for at least 24 hr before the experiment in a temperature- and humidity-controlled environment and fed ad libitum. Each animal was used only once.
Chronic Morphine Treatment
Mice were rendered tolerant to morphine by s.c. implantation of one morphine pellet (containing 75 mg morphine free base) for 72 hr. The control group received a placebo pellet for the same length of time.
Antinociception Assay and Measurement of Tolerance
Antinociception was defined as a response latency more than 3 S.D. above the mean of control in the radiant heat tail-flick procedure. The degree of morphine tolerance was measured by the AD50 of morphine in the morphine pelleted mice over that of the naive mice. Thirty min after the injection of morphine solution, the morphine AD50 values were determined from a dose-response curve derived from at least three groups of animals, each consisting of 10 mice. The implanted morphine pellet was left intact during the assessment of antinociception. DynA-(2–17) solution was given i.v. when indicated simultaneously with morphine for a total of 30 min between morphine AD50 values were determined.
Evaluation of Synergism of Spinal and Supraspinal Sites
Spinal/supraspinal synergism was evaluated in two independent ways.
Variable ratio.
The morphine dose was fixed at one site,e.g., i.c.v., and the dose response relation to morphine was determined at the other site, e.g., i.t. The apparent AD50 of morphine at the variable site was then calculated.
Fixed ratio.
The ratio of morphine doses at both i.c.v. or i.t. sites were held constant, and the dose response of this combination and the apparent AD50 were determined. The interaction of morphine between these two sites were analyzed in an isobologram that was constructed by plotting the AD50 for i.c.v. vs. i.t. The straight line connecting these two points was defined as the theoretical additive line, which consists of points for the purely additive effect at all the ratios between i.t. and i.c.v. sites. Those values and their S.E. were calculated according to the method described by Tallarida (Tallarida et al., 1989; Finney, 1971). The experimentally derived apparent AD50 values were compared with theoretical additive values and if statistically significant by Student’s t test, this was taken as indicative of a synergistic effect.
Statistics.
Morphine AD50 values and their 95% confidence limits were calculated by the method of Litchfield and Wilcoxon (1949). The interactions of morphine between i.t. and i.c.v. sites were analyzed in the isobolograms (Tallarida et al., 1989) and assessed statistically by Student’s t test. The linear regression analysis was used to evaluate the effect of different doses of DynA-(2–17) on the modulation of morphine synergism between spinal and supraspinal sites in the tolerant mice.
Drugs.
Morphine base pellets were provided by the National Institute on Drug Abuse (Rockville, MD). Morphine sulfate was purchased from Mallinckrodt, Inc. (St. Louis, MO). DynA-(2–17) was purchased from Phoenix Co.(Mountain View, CA). Aqueous solutions of DynA-(2–17) and morphine were administered i.v. and s.c., respectively, in a volume of 10 ml/kg and administered i.t. (Hylden and Wilcox, 1980) or i.c.v. (Haley and McCormick, 1957) in a volume of 5 μl/mouse. The sequence of administrative routes was usually in the order of i.t., i.c.v. and i.v.
Results
Effect of mode of administration on the morphine sensitivity in mice rendered tolerant to morphine by pellet implantation.
To provide a baseline against which to compare synergistic effects, the AD50 of morphine was determined separately at i.t. and i.c.v. sites, in both naive and morphine-pelleted animals. In agreement with many others, we found that in the morphine-pelleted mice there was a 8- to 10-fold increase in the morphine AD50 by morphine s.c.; however, it appears no significant tolerance development was demonstrable when morphine was given i.t. or i.c.v. alone. As shown in table1, The AD50 of morphine given i.t. or i.c.v. were essentially the same in the placebo- and morphine-implanted animals. These findings contrast with the high degree of morphine tolerance observed in animals challenged by the systemic route.
The AD50 for morphine by different routes of administration in mice rendered tolerant to morphine by pellet implantation
Interaction of morphine between i.t. and i.c.v. sites in naive mice.
To assess the synergism between morphine administered simultaneously at i.c.v. and i.t. sites, we first used a variable ratio approach. The morphine i.c.v. dose response curves were shifted left with increasing concentrations of morphine given i.t. In the presence of 0.05 nmol/mouse i.t., a dose that was virtually ineffective when given to this site alone, the apparent AD50 at the i.c.v. site was significantly reduced from 5.5 nmol/mouse to 1.1 nmol/mouse. The apparent AD50 values at the i.c.v. sites were further reduced to 0.52 and 0.3 nmol/mouse in the presence of 0.1 and 0.2 nmol/mouse, respectively, of morphine given i.t. (fig. 1).

Antinociceptive effect of morphine in naive mice using the variable ratio method as described in “Materials and Methods.” The i.c.v. morphine AD50 was determined in the presence of increasing fixed dose of morphine i.t. Doses of fixed i.t. dose were as follows, (○) saline, i.t.; (•]) 0.05 nmol, i.t.; (□) 0.1 nmol, i.t.; (▪) 0.2. nmol, i.t. The apparent AD50 values with 95% confidence limits are indicated.
Similarly, the apparent AD50 for i.t. morphine could also be shifted leftward in the presence of increasing concentrations of morphine given i.c.v. The apparent AD50 was 0.9 nmol/mouse in the absence of i.c.v. morphine and was significantly reduced to 0.3 and 0.1 nmol/mouse in the presence of 0.2 and 1.0 nmol/mouse, respectively, of i.c.v. morphine (fig. 2).

Antinociceptive effect of morphine in naive mice using the variable method as described in the method section. The i.t. morphine AD50 was determined in the presence of increasing fixed dose of morphine i.c.v. Doses of fixed i.c.v. dose were as follows, (○) saline, i.c.v.; (•) 0.1 nmol, i.c.v.; (□) 0.2 nmol, i.c.v.; (▪) 1.0 nmol, i.c.v.. The apparent AD50 values with 95% confidence limits are indicated.
The synergistic effect of morphine at supraspinal and spinal sites was further confirmed in the fixed ratio protocol. The ratio between i.t. and i.c.v. morphine doses was fixed at 1:5, 1:10 or 1:20. The apparent AD50 values of each fixed ratio were calculated and compared with the theoretical additive values. The experimentally obtained apparent AD50 values for fixed ratios were statistically significantly lower than those of the theoretical additive AD50 (fig. 3; P < .05). These results were thus consistent with those of the variable ratio method, again suggesting that there is a marked degree of synergism for morphine at spinal and supraspinal sites, and the effects of morphine are generally enhanced when coadministered at both sites.

Isobolographic plot for the interactive effect of morphine between i.t. and i.c.v. sites in naive mice. The apparent AD50 values for individual administration at two sites are plotted on the x- and y-axes, respectively. The line connecting these points is the theoretical additive line. The open square, circle and triangle represent the theoretical additive points at 1:5, 1:10 and 1:20 ratios and closed square, circle and triangle represent the experimental AD50 values at 1:5, 1:10 and 1:20 ratios, respectively. The vertical bars crossing the points represent S.E. at i.c.v. sites whereas, horizontal bars represent S.E. at the i.t. sites. The statistical significance was obtained using t test (*P < .05), indicating a synergistic effect at all three ratios.
Interaction of morphine between i.t. and i.c.v. sites in morphine-tolerant mice.
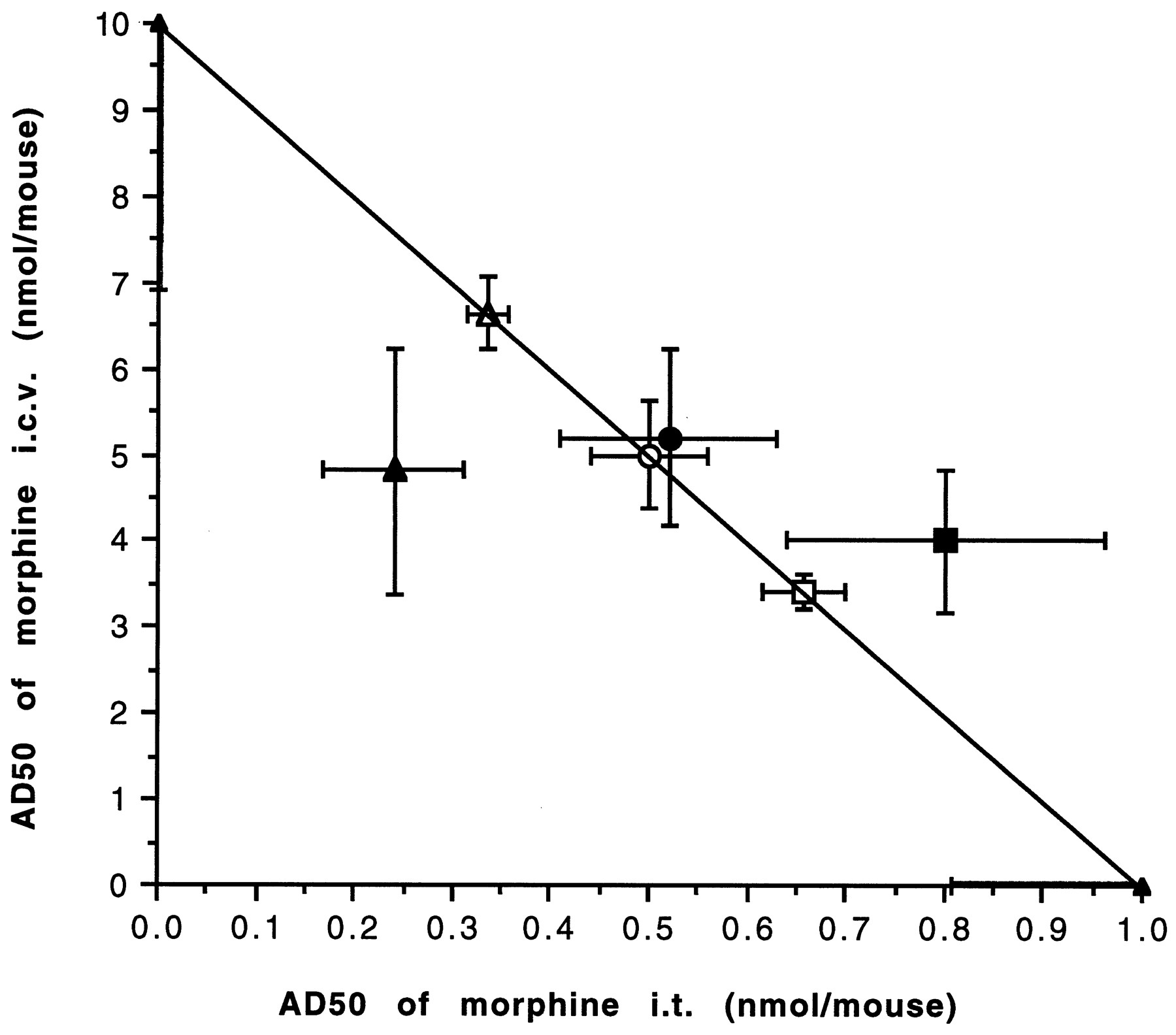
The morphine synergism at i.t. and i.c.v. sites in naive mice was completely eliminated when mice were rendered tolerant with a 3-day morphine pellet treatment. Using the fixed ratio method, we observed that morphine given either i.c.v or i.t. was unable to synergize the effects of morphine at the other site (fig.4). The apparent AD50 derived from 1:5, 1:10 and 1:20 ratios were nearly identical to the theoretical additive values. This suggests that 3-day morphine pellet treatment results in the loss of synergism between spinal and supraspinal sites.

Isobolographic plot for the interactive effect of morphine between i.t. and i.c.v. sites in morphine tolerant mice. The apparent AD50 values for individual administration at two sites are plotted on the x- and y-axes, respectively. The line connecting these points is the theoretical additive line. The open square, circle and triangle represent the theoretical points at 1:5, 1:10 and 1:20 ratios, whereas close square, circle and triangle represent the experimental AD50 values at 1:5, 1:10 and 1:20 ratios, respectively. The vertical bars crossing the points represent S.E. at i.c.v. sites whereas, horizontal bars represent S.E. at the i.t. sites. No statistical significance was obtained using t test (P > .05), indicating an additive effect.
Effect of Dyn A-(2–17) on the interaction of morphine between i.t. and i.c.v. sites.
The role of DynA-(2–17) in the synergistic interaction of morphine at i.t. and i.c.v. sites in morphine-tolerant mice was investigated by examining whether DynA-(2–17) could restore the synergism that was abolished after the development of s.c. tolerance to morphine. Figure 5 shows that in tolerant animals the AD50 of i.c.v. or i.t. morphine in the presence of 8.4 μmol/kg DynorphinA-(2–17) were significantly different from additive values (P < .05) for 1:5 and 1:10 ratios but not for 1:20 ratio, suggesting that synergism was partially restored. In contrast, when DynorphinA-(2–17), i.v. was administered to naive animals under the same conditions (route and dose), no change in morphine effect was demonstrable.

The effect of DynA(2–17) on restoring the synergism of morphine between i.t. and i.c.v. sites in morphine tolerant mice was displayed with isobolographic plot. The open square, circle and triangle represent the theoretical additive points at 1:5, 1:10 and 1:20 ratios, respectively, and closed square, circle and triangle represent the experimental AD50 values at 1:5, 1:10 and 1:20 ratios in the presence of DynA(2–17). The vertical bars crossing the points represent S.E. at i.c.v. sites whereas, horizontal bars represent S.E. at the i.t. sites. The statistical significance for 1:5 and 1:10 ratios was obtained (*P < .05), indicating a synergistic effect; but no significance was seen for 1:20 ratio (P > .05), indicating an additive effect.
The effect of DynorphinA-(2–17) on the morphine synergism is dose related. The AD50 of morphine decreased as the dose of DynorphinA-(2–17) increased as shown in table 2. The statistical analysis by linear regression of AD50 on dose shows a significant variation in those AD50 values (P < .005), indicating that the effect of dynorphin is highly concentration dependent.
Effect of varying doses of Dyn A-(2-17) i.v. on the sensitivity to morphine coadministered spinally and supraspinally at the fixed ratio of 1:10 in mice rendered tolerant to morphine by pellet implantation
Discussion
Our results indicate that morphine-induced antinociception is highly dependent on an intact integrated central nervous system and that tolerance development is the result of a disruption of synergism between the central nervous system sites. Several laboratories have reported that the potency of morphine in naive mice is greatly enhanced by coadministration of morphine at discrete sites in the spinal cord and the brain (Yeung and Rudy, 1980; Roerig et al., 1984). We have confirmed this synergism, using two independent assays, involving either a variable or fixed ratio of doses. We have further shown that this synergism of concomitant administration of morphine i.t. and i.c.v. is reduced after s.c. morphine pellet implantation. Treatment of these mice with DynorphinA-(2–17), i.v., which we have previously shown to shift morphine AD50 curve to the left in tolerant animals (Takemori et al., 1993), resulted in a partial restoration of this synergism.
These results thus offer support for the hypothesis that Dynorphin’s modulation of morphine antinociception is elicited by altering the synergistic interaction of spinal and supraspinal opioid receptors. Specifically, we suggest that the increase in morphine AD50, characteristic of opioid tolerance, results from a loss of synergism that dynorphin restores at least in part. These results are consistent with earlier studies that failed to demonstrate changes in either the number or affinity of opioid receptors in the development of tolerance to morphine (Smith and Loh, 1981), for we found that after 3-day morphine pellet implantation, the AD50 of morphine administered to either i.c.v. or i.t. sites alone was unchanged.
It appears that the well-documented tolerance to morphine observed when the drug is administered systemically, results from its simultaneous interaction with receptors at both spinal and supraspinal sites. However, we emphasize that tolerance at individual spinal and supraspinal sites does develop over longer periods of exposure to morphine. We have recently observed that when mice are implanted with morphine pellets for a total of 8 days, the AD50 of morphine administered either i.c.v. or i.t. increases; however, the systemic morphine tolerance was the same as the 3-day implanted animal (L. He., X. H. Zhang and N. M. Lee, manuscript in preparation). Thus tolerance development has a different time course at i.c.v. and i.t. sites compared with that of s.c. site. The loss of synergism and resulting tolerance to systemic morphine challenge is largely complete after 3 days, before any tolerance at these individual central sites is detectable.
Dynorphin A-(2–17), unlike the full-length A-(1–17) compound, appears not to interact with opioid receptors. This conclusion is based on several observations, including 1) studies demonstrating the importance of the terminal tyrosine in opioid peptides for receptor binding (Chavkin and Goldstein, 1981); 2) the inability of DynorphinA-(2–17) to induce antinociception in animals as measured by the classic tail flick or hot plate assays, although it is antinociceptive in the writhing test (Hooke, et al., 1995) and 3) the inability of this peptide to compete with opioid ligands in in vitrobinding assays (Walker et al., 1982a). Nevertheless, DynorphinA-(2–17) is able to modulate morphine antinociception in both naive and tolerant animals (Walker et al., 1982b; Takemoriet al., 1993). Our results offer a mechanism where this peptide can have effects on opioid antinociception, by acting through nonopioid receptors. We propose that DynorphinA-(2–17) acts through a distinct nonopioid receptor mechanism to regulate synergistic interaction between spinal and supraspinal opioid receptors.
The nature of the receptor at which DynorphinA-(2–17) acts has not been established. However, several lines of evidence implicate excitatory amino acids, particularly NMDA, in the actions of Dynorphin. NMDA antagonists block the development of opioid tolerance (Tiseo and Inturrisi, 1993; Trujillo and Akil 1991) and DynorphinA increases the release of excitatory amino acids and potentiate the NMDA-induced activity (Skilling et al., 1992). Chen et al.(1995) reported that dynorphin blocks NMDA-activated ion currents through a nonopioid receptor, suggesting a direct action of the peptide on NMDA receptors. A common site of action at the receptor level is also suggested by our observation that NMDA antagonists have activity in the mouse writhing test, an alternative antinociceptive model in which both DynorphinA-(1–17) and DynorphinA-(2–17) are highly potent.
The ability of DynorphinA to modulate opioid tolerance has been known for more than a decade, and may be the basis for novel therapeutic approaches to treating opioid tolerance. Its role in modifying spinal-supraspinal synergism is an important step in identifying its mechanism of action, and should ultimately help in locating the most effective targets for such treatment.
Acknowledgments
The authors thank Drs. E. L. Way and Andy Smith for valuable suggestions.
Footnotes
-
Send reprint requests to: Dr. Nancy M. Lee, Geraldine Brush Cancer Research Institute, California Pacific Medical Center, Research Institute, 2330 Clay St., San Francisco, CA 94115.
-
↵1 This work was supported by National Institute on Drug Abuse Grants DA-02643 and DA-10048.
- Abbreviations:
- i.t.
- intrathecal
- i.c.v.
- intracerebroventricular
- dyn A
- dynorphin A
- NMDA
- N-methyl-D-aspartate
- AD50
- antinociceptive dose at 50% effectiveness
- Received August 12, 1996.
- Accepted November 15, 1996.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}