


Abstract
Nine membrane-bound adenylyl cyclase (AC) isoforms catalyze the production of the second messenger cyclic AMP (cAMP) in response to various stimuli. Reduction of AC activity has well documented benefits, including benefits for heart disease and pain. These roles have inspired development of isoform-selective AC inhibitors, a lack of which currently limits exploration of functions and/or treatment of dysfunctions involving AC/cAMP signaling. However, inhibitors described as AC5- or AC1-selective have not been screened against the full panel of AC isoforms. We have measured pharmacological inhibitor profiles for all transmembrane AC isoforms. We found that 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (SQ22,536), 2-amino-7-(furanyl)-7,8-dihydro-5(6H)-quinazolinone (NKY80), and adenine 9-β-d-arabinofuranoside (Ara-A), described as supposedly AC5-selective, do not discriminate between AC5 and AC6, whereas the putative AC1-selective inhibitor 5-[[2-(6-amino-9H-purin-9-yl)ethyl]amino]-1-pentanol (NB001) does not directly target AC1 to reduce cAMP levels. A structure-based virtual screen targeting the ATP binding site of AC was used to identify novel chemical structures that show some preference for AC1 or AC2. Mutation of the AC2 forskolin binding pocket does not interfere with inhibition by SQ22,536 or the novel AC2 inhibitor, suggesting binding to the catalytic site. Thus, we show that compounds lacking the adenine chemical signature and targeting the ATP binding site can potentially be used to develop AC isoform–specific inhibitors, and discuss the need to reinterpret literature using AC5/6-selective molecules SQ22,536, NKY80, and Ara-A.
Introduction
The nine membrane-bound mammalian isoforms of adenylyl cyclase (AC) all convert ATP into cAMP, an important second messenger in cell signaling. The structure of the membrane-bound isoforms is well conserved, consisting of a cytosolic N terminus and two other cytosolic domains (C1 and C2) that are separated by a pair of 6-transmembrane helical domains (reviewed in Sadana and Dessauer, 2009). Whereas the N terminus greatly varies across isoforms, the C1/C2 domains that form the catalytic core are well conserved in primary sequence, both to each other and when compared across AC isoforms. Despite this similarity, the C1/C2 domains are a source of differential regulation by heterotrimeric G proteins, kinases, and calcium.
The broad clinical use of Gs-coupled G protein–coupled receptor antagonists or Gi-coupled G protein–coupled receptor agonists that indirectly decrease cAMP synthesis suggests that inhibitors of AC could be highly beneficial (Pierre et al., 2009). Reduction of AC activity, particularly AC5, has well documented benefits in animal models, including increased bone mass and stress resistance in the heart, as well as reduction of aging phenotypes in the heart and bone, decreased pain associated with inflammatory agents, and increased longevity (Sadana and Dessauer, 2009). AC5 knockout models indicate roles for AC5 in mechanical and inflammatory pain (Kim et al., 2007) and with behaviors associated with the use of morphine and related opiates (Kim et al., 2006), indicating a potential role for an AC5 inhibitor in pain relief. AC5 knockout models also show a protective phenotype against chronic heart failure (Okumura et al., 2003, 2007), and increased expression of proteins associated with AC signaling promotes heart failure (Iwase et al., 1997; Engelhardt et al., 1999; Antos et al., 2001). Thus, selective inhibition of AC5 has been proposed as a treatment of chronic heart failure (Pavan et al., 2009; Pierre et al., 2009; Ho et al., 2010). However, this approach is controversial since failing human hearts have reduced amounts of basal cAMP and impaired cAMP generation in response to agonist stimulation (Bristow et al., 1982; Feldman et al., 1987; Phan et al., 2007). Currently, no AC inhibitors are used clinically. Of the nine isoforms, AC5 and AC6 are most closely related (Sadana and Dessauer, 2009). AC5 and AC6 are highly expressed isoforms in the heart but appear to play opposing roles in terms of cardioprotective benefits (Lai et al., 2000, 2004). Deletion of AC6 caused a marked reduction of calcium transients and SERCA2a calcium affinity (Tang et al., 2008) and increased mortality during sustained β-adrenergic receptor stimulation (Tang et al., 2013). Moreover, overexpression of AC6 increases cardiac responsiveness and has advantageous effects on the failing heart (Phan et al., 2007). Thus, an AC inhibitor may need to be truly selective for AC5 over AC6 to be beneficial for chronic heart failure.
There are two clearly distinguished classes of small-molecule inhibitors that target the ATP binding site of AC. The classic P-site inhibitors typically work uncompetitively or noncompetitively by stabilizing a pyrophosphate-bound transition state (Dessauer et al., 1999; Dessauer, 2002). The other class is competitive MANT or ITP containing nucleotide analogs, such as 3′-O-(N-methylanthraniloyl)-guanosine-5′-triphosphate (MANT-GTP) (Gille and Seifert, 2003; Seifert et al., 2012). In both cases, the small-molecule inhibitors are analogs of adenosine, ATP, or other nucleotides and thus have clear potential for off-target effects and/or issues with cell permeability due to the anionic phosphate group. Additionally, these inhibitors have not been well characterized in terms of selectivity, particularly the P-site inhibitors, which have been used in the design of alleged AC5-selective inhibitors.
We have characterized known adenosine-like inhibitors and used structure-based virtual screening targeting the ATP binding site of AC to identify novel AC inhibitors. We found that 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (SQ22,536), 2-amino-7-(furanyl)-7,8-dihydro-5(6H)-quinazolinone (NKY80), and adenine 9-β-d-arabinofuranoside (Ara-A), each previously described as supposed AC5-selective, do not discriminate between AC5 and AC6. We also found that the AC1-selective inhibitor 5-[[2-(6-amino-9H-purin-9-yl)ethyl]amino]-1-pentanol (NB001) does not directly target AC1 to reduce cAMP levels. We identified three novel chemical scaffolds that demonstrate some preference for AC1 or AC2 and appear to bind to the catalytic site. Thus, we show that non–adenine-based chemical structures, targeting the ATP binding site, can potentially be used to develop AC isoform-specific inhibitors, and we discuss the need to reinterpret literature using AC5/6-selective molecules SQ22,536, NKY80, and Ara-A.
Materials and Methods
Plasmids and Viruses.
Rat AC1–4, human AC5, human AC6, and rat AC7 baculoviruses were described previously (Taussig et al., 1994; Yan et al., 2001; Chen-Goodspeed et al., 2005). Eukaryotic expression vectors for rat AC2, rat AC8, and human AC9 in pcDNA3.1 were described previously (Fagan et al., 1996; Piggott et al., 2008). Hexahistidine-tagged Gαs was purified from Escherichia coli and activated with guanosine 5′-3-O-(thio)triphosphate (Chen-Goodspeed et al., 2005).
Small Molecules.
Forskolin and dimethylsulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO). The AC inhibitors SQ22,536 (Enzo Life Sciences, Ann Arbor, MI), NKY80 (Calbiochem, Billerica, MA), and Ara-A under the trade name Vidarabine (Tokyo Chemical Industry, Tokyo, Japan) and NB001 (Sigma-Aldrich) were purchased from the indicated vendor. The following drug-like molecules were purchased from ChemBridge (San Diego, CA): 5-(1'-phenyl-1H,1'H-4,4'-bipyrazol-3-yl)-1,3-benzodioxol-4-ol (no. 6673567), 6-[2-(4-aminophenyl)vinyl]-2-pyridinecarboxylic acid (no. 7921220), and 2-[(4-amino-6-oxo-1,6-dihydro-2-pyrimidinyl)thio]-N-(3-chlorophenyl)propanamide (no. 7833407). All small molecules were purchased in solid form and suspended in 100% DMSO. All inhibitor dilutions were made in DMSO and subsequently added directly to AC membranes; the final concentration of DMSO in adenylyl cyclase assays was less than 5%.
In Silico Structure-Based Screening and Docking.
Screening for novel AC inhibitors was performed using Glide docking software (Schrödinger, LLC, Portland, OR). We screened a generic ChemBridge library of approximately 35,000 small drug-like molecules (downloaded June 2011) that satisfy Lipinski’s rule of five. These ligands were docked to a model of the AC catalytic (i.e., ATP-binding) site derived from the crystal structure of the AC C1/C2 domains bound with either l-2′,3′-dideoxyadenosine-5′-triphosphate [2′3′ddATP; Protein Data Bank (PDB) code 1CJT] or MANT-GTP (PDB code 1TL7). We removed waters, forskolin, Gαs protein, the “A” site metal ion, and buffer molecules from the crystal structures, leaving only the AC C1/C2 domains and the “B” site metal ion. The “A” site metal ion was removed to minimize false-positive hits due to strong electrostatic interaction with the metal ions. The protein preparation and docking protocols used have been reported (Gatica and Cavasotto, 2012). The LigPrep tool (Schrödinger, LLC) was used to prepare the molecules for docking by assigning their protonation state at pH 7, tautomer states, and generating all possible stereoisomers. Docking was restricted to a cubic grid of sides 20 Å encompassing the ATP binding site and centered on the center of the crystallized ligand. This box was large enough to include all the ATP binding site residues with which candidate ligands may interact. To further minimize false positives, only ligand poses that allow for the formation of at least one hydrogen bond with AC residues Lys938, Asp1018, or Ile1019 were considered. Hydrogen bonding was defined by a donor-acceptor distance cutoff of 3.5 Å and a donor-hydrogen-acceptor angle between 150° and 180°. Docked molecules were prioritized by ranking them according to Glide's standard precision (SP) docking score.
In addition to virtual screening with Glide, global and site-directed docking of selected ligands was also performed using AutoDock, version 4.2 (Scripps, La Jolla, CA). Here, known AC5 inhibitors SQ22,536, NKY80, and Ara-A were docked to either the entire surface of AC structures (PDB code 1CJT or 1TL7) or just the ATP binding site. In the former “blind docking” procedure (Grant et al., 2011), the defined search area was a cubic box of 60 Å (with a grid spacing of 0.375 Å) centered on the C1/C2 domains, whereas the second (i.e., site-directed docking) procedure was the same as that used for virtual screening with Glide. Prior to AutoDock docking, ligands were preprocessed with the AutoDock Tools package to assign Gasteiger atomic charges and torsions (Huey et al., 2007; Morris et al., 2009). For both site-directed and global docking experiments, we used a population size of 150, and performed 256 hybrid Lamarckian genetic algorithm runs with a stopping criterion of 10,000 generations. For the site-directed docking, the population size was set to 150. The resulting poses were clustered using a root mean square deviation of cutoff value of 2 Å. The docked poses were then ranked based on the predicted affinity docking scores and cluster size. Protein-ligand interactions were evaluated in terms of hydrogen bonding (using the criteria mentioned in the previous paragraph) and van der Waals contacts (defined by a carbon-carbon distance cutoff of 4.0 Å). We used a combination of the ICM-browser (Molsoft LLC, San Diego, CA) and VMD 1.9 (Humphrey et al., 1996 [NIH Center for Macromolecular Modeling and Bioinformatics, Urbana-Champaign, IL]), both freely available, for visual inspection and image rendering.
Sf9 AC Expression and Membrane Preparation.
A clonal isolate of Spodoptera frugiperda (Sf9) insect cells were maintained in cell suspension with SF-900 II SFM media at 27°C. Baculoviruses encoding AC isoforms 1–7 were amplified for 5–7 days and used to infect Sf9 cells (multiplicity of infection = 1.5). After 48 hours of infection, Sf9 cells were harvested by centrifugation at 1000g for 10 minutes at 4°C. Cells were suspended in ice-cold lysis buffer consisting of 20 mM HEPES (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, 2 mM DTT, and protease inhibitors phenylmethylsulfonyl fluoride, tosyl phenylalanyl chloromethyl ketone, leupeptin, lima bean trypsin inhibitor, and aprotinin. Cells were lysed by nitrogen cavitation at 500 psi for 30 minutes at 4°C. Cell lysates were centrifuged at 750g for 10 minutes to remove intact cells and nuclei. The supernatants were centrifuged at 100,000g for 30 minutes, and the resulting pellets were resuspended and washed in buffer consisting of 20 mM HEPES (pH 8.0), 2 mM DTT, 200 mM sucrose, and protease inhibitors as previously described. Membranes were homogenized with a Dounce homogenizer and centrifuged again at 100,000g for 30 minutes. Protein concentrations of resuspended Sf9 membranes were determined by Bradford assays. Membranes were frozen in liquid nitrogen and stored at −80°C in one-use aliquots for future adenylyl cyclase activity assays.
HEK293 and COS-7 Cell Transfections and Membrane Preparations.
Membranes were prepared from human embryonic kidney 293 (HEK293) cells expressing AC8 or AC9 for all inhibitor studies. COS-7 cells were used to express AC2 wild type and S942P mutant for experiments shown in Fig. 7. HEK293 or COS-7 cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C with 5% CO2. All volumes and amounts are for transfection in 10-cm plates for membrane preparations. HEK293 (3–3.5 × 106 cells) and COS-7 cells (2.5–3 × 106 cells) were seeded 24 hours prior to transfection. Medium was replaced the next day with fresh Dulbecco’s modified Eagle’s medium (no penicillin/streptomycin), and cells were transfected with the appropriate plasmids (10 µg of DNA total per 10-cm plate) using Lipofectamine 2000. Cells were incubated at 37°C for 4–6 hours, the medium was replaced, and membranes were prepared approximately 42 hours (COS-7 cells) or 48 hours (HEK293 cells) after transfection.
Following transfections, cells were rinsed and harvested in cold phosphate-buffered saline and then pelleted by centrifugation at 3000g, 4°C for 5 minutes. Cell pellets were aspirated and resuspended in 20 mM HEPES, 1 mM EDTA, 2 mM MgCl2, 1 mM DTT, 250 mM sucrose, and protease inhibitors. Cells were incubated on ice for 10–30 minutes, subjected to Dounce homogenization, and centrifuged at 1800g for 5 minutes at 4°C to pellet nuclei. The supernatants were centrifuged at 60,000g, 4°C for 20 minutes. The resulting membrane pellet was resuspended in 20 mM HEPES, 1 mM EDTA, 2 mM MgCl2, 1 mM DTT, 250 mM sucrose, and protease inhibitors. HEK293/COS-7 membrane concentrations were determined by Bradford assay. Membranes were immediately used in adenylyl cyclase activity assays or frozen in liquid nitrogen and stored at −80°C in one-use aliquots for future assays.
Adenylyl Cyclase Membrane Assays.
Membrane assays were performed essentially as described previously (Dessauer, 2002). Inhibitors in DMSO (1 µl) were added on ice to Sf9, HEK293, or COS-7 membrane preparations (24 µl). Reactions were initiated with a reaction mix containing [α−32P]ATP, 10 mM MgCl2, and appropriate activators and incubated for 10 minutes at 30°C in a final reaction volume of 50 µl. Unless explicitly stated otherwise, activators for each AC isoform consisted of forskolin (50 µM) for Sf9 membranes expressing AC 1–7, calcium (100 µM) and calmodulin (300 µM) for membranes from HEK293 cells expressing AC8, and guanosine 5′-3-O-(thio)triphosphate–Gαs (300 nM) for membranes from HEK293 cells expressing AC9. Reactions were stopped with a solution of 2.5% SDS, 50 mM ATP, and 1.75 mM cAMP. Nucleotides in each reaction sample were then separated by sequential column chromatography on Dowex and Alumina resin to isolate [32P]cAMP product, using [3H]cAMP to monitor column recovery rates by scintillation counting.
Intact cAMP Accumulation Assays.
HEK293 cells were transfected with either AC1 or a pcDNA vector control in polylysine-coated six-well plates. Forty-eight hours post-transfection, cells were labeled with [3H]adenine for 3–4 hours, washed, and incubated with 1 mM 1-methyl-3-isobutylxanthine (IBMX) and either NB001 or DMSO vehicle control at 37°C for 10 minutes. Forskolin (10 µM) and ionomycin (10 µM) were incubated for an additional 30 minutes. Reactions were stopped upon addition of ice-cold 5% trichloroacetic acid, 1 mM ATP, and 100 µM cAMP. Nucleotides were then separated by sequential column chromatography on Dowex and Alumina resin to isolate [3H]cAMP product, using [32P]cAMP to monitor column recovery rates.
Statistical Analysis.
All biological experiments measuring AC activity or cAMP accumulation were performed in duplicate, with a minimum experiment replication of n = 3. Depending on the experiment, either unpaired or paired t tests were used to determine statistical significance of results. All error bars represent the mean ± SD.
Results
AC5 Inhibitors Are Actually AC5/6-Selective Inhibitors.
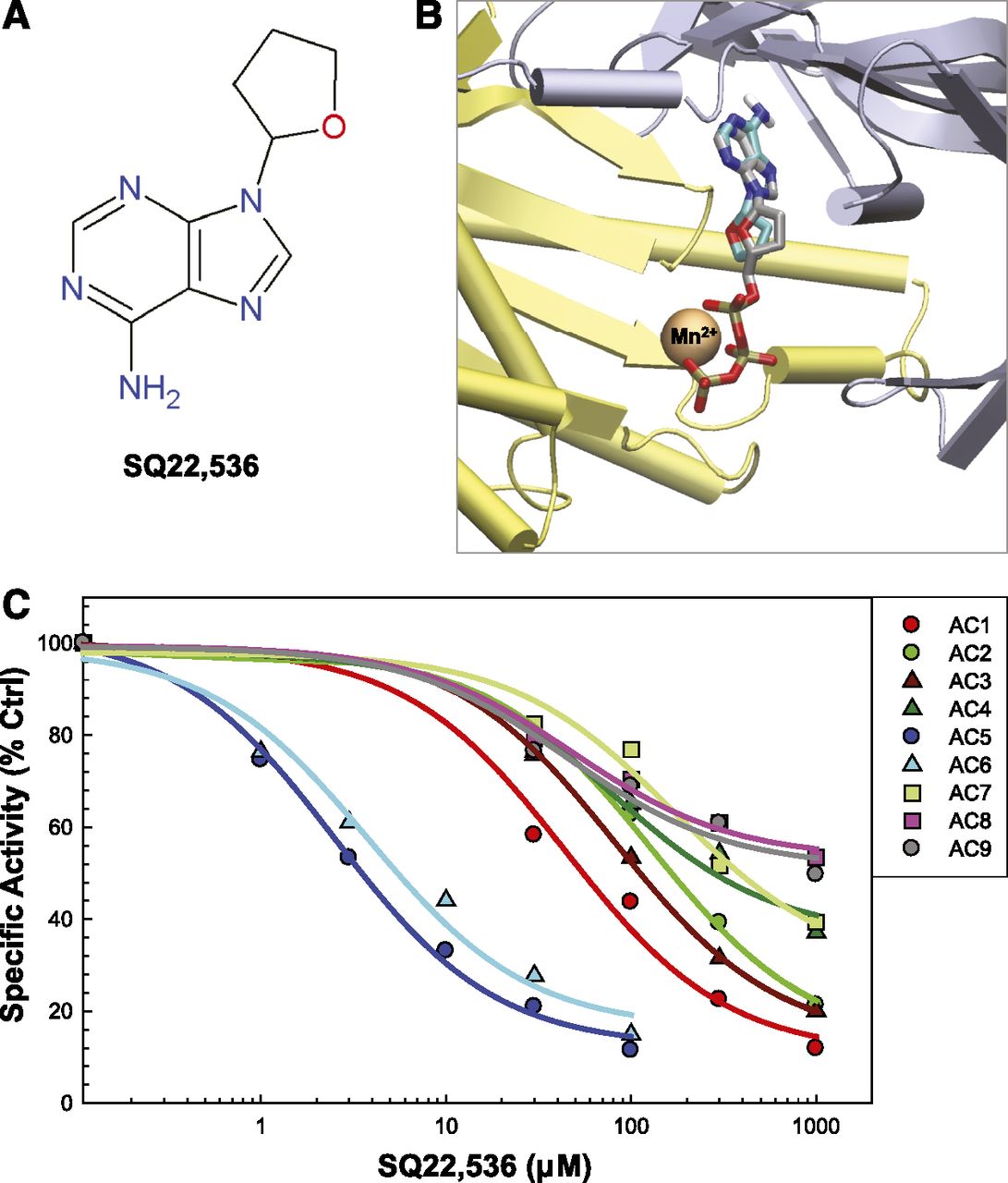
SQ22,536, Ara-A, and NKY80 are three adenine-like inhibitors that are reportedly selective for AC5 (Johnson et al., 1997; Onda et al., 2001; Iwatsubo et al., 2004) (the chemical structures of these ligands are shown in Figs. 1A, 2A, and 3A, and the three-dimensional structures of the ligand-AC complexes obtained from docking are shown in Figs. 1B, 2B, and 3B). However, these inhibitors have never been tested against all nine membrane-bound AC isoforms. Using an in vitro AC activity assay with Sf9 or HEK293 membranes expressing a given AC isoform, we tested these inhibitors for their potency against each transmembrane AC. SQ22,536 was the first of this series of inhibitors developed, and is essentially a dideoxyadenosine analog (Fig. 1A). It has been previously shown to be selective for AC5 over AC2 or AC3 (Johnson et al., 1997; Onda et al., 2001). In addition to confirming these results, we show that SQ22,536 has high potency for AC5 and AC6 with 10- to 15-fold selectivity over the next closest isoform, AC1. Potency and efficacy for AC2, AC3, AC4, AC7, AC8, and AC9 are further reduced, with less than 50% inhibition of AC8 and AC9 at 1 mM. Although inhibition of AC8 and AC9 appears to level off slightly, this is likely only due to our inability to further increase inhibitor concentrations and reach complete saturation. Importantly, inhibition by SQ22,536 is not significantly different for AC5 and AC6 (Fig. 1C; Table 1).

AC inhibition profile of SQ22,536. (A) Chemical structure of SQ22,536. (B) Virtual docking of SQ22,536 to a P-site inhibitor-bound conformation of AC (PDB code 1CJT). SQ22,536 is shown with cyan carbons; the crystallized position of the 2′,3′ddATP ligand is shown with gray carbons for reference. The C1 domain is yellow; the C2 domain is silver. (C) Inhibition profile of SQ22,536 for all nine AC isoforms. Inhibition curves for each AC are shown as a calculated fit to the means of each concentration (indicated by symbols and grouped with colors by AC family; n = 3, performed in duplicate; error bars removed for clarity). Membranes from Sf9 cells expressing AC1–7 were stimulated by 50 µM forskolin. Membranes from HEK293 cells expressing AC8 and AC9 were stimulated by 300 µM calmodulin and 300 nM Gαs, respectively. Ctrl, control.

AC inhibition profile of NKY80. (A) Chemical structure of NKY80. (B) Virtual docking of NKY80 to the 2′,3′ddATP-bound conformation of AC as was described for Fig. 1B. (C) Complete AC isoform inhibition profile of NKY80. Membranes and stimulation conditions were as described in Fig. 1C. Experimental data indicated by symbols (n = 3, performed in duplicate); lines are the calculated fits to experimental means for the dose-response curves. Ctrl, control.

Ara-A does not discriminate between AC5 and AC6. (A) Chemical structure of Ara-A. (B) Virtual docking of Ara-A to the 2′3′ddATP-bound conformation of AC as described for Fig. 1B. (C) Complete AC isoform inhibition profile of Ara-A. Inhibition curves for each AC isoform shown as fit to means of AC activity assays (n = 3). Membranes and stimulation conditions were as described in Fig. 1C (n = 3, performed in duplicate). Ctrl, control.
Pharmacological profile for inhibition of adenylyl cyclase isoforms
Experiments were performed as described in Figs. 1C, 2C, and 3C. Values are reported as pIC50 ± S.E., where pIC50 is calculated as -log(IC50). n = 3, each performed in duplicate. Statistics (t test) were performed on pIC50 of the indicated isoform versus that of AC5.
NKY80 is an AC5 inhibitor derived from SQ22,536, with the adenine group modified to limit potential off-target effects such as inhibition of DNA synthesis (Fig. 2A). It is also reported to be selective for AC5 over AC2 and AC3 (Onda et al., 2001). NKY80 most potently inhibited AC5 and AC6, with only a 2-fold difference in IC50 that did not reach significance. The pattern of isoform selectivity is similar to that of SQ22,536, although AC2 and AC7 are much less potently inhibited. The selectivity of AC5 and AC6 from other AC isoforms, although significant, is less pronounced than it is for SQ22,536.
Ara-A, also known as vidarabine, is an adenosine analog (Fig. 3A) that is used clinically as an antiviral, with a mechanism that is unrelated to inhibition of AC/cAMP signaling (Whitley et al., 1980). Ara-A was shown previously to be selective for AC5 over AC2 and AC3 when expressed in Sf9 membranes, and has been suggested to inhibit AC5 more potently than AC6 in cardiomyocytes (Iwatsubo et al., 2012). We show that, similar to SQ22,536 and NKY80, Ara-A most potently inhibits AC5 and AC6 (Fig. 3C). Additionally, for any given concentration of Ara-A, we observe less than a 2- to 3-fold difference in the inhibition of AC5 versus AC6. Therefore, SQ22,536, Ara-A, and NKY80 are actually AC5/6-selective inhibitors.
AC1 Inhibitor NB001 Does Not Directly Inhibit AC1 Activity.
To gain further insight into the selectivity of different AC inhibitors, we also examined NB001, which is reported to be an AC1-selective ligand with therapeutic potential as an analgesic (Zhou, 2012). In HEK cells stably expressing AC1, NB001 inhibited AC activity when stimulated with forskolin and the calcium ionophore calimycin (Wang et al., 2011). We also observed NB001 inhibition of cAMP accumulation in HEK cells transiently expressing AC1 when stimulated with ionomycin and forskolin (Fig. 4B). However, this experiment, performed both in the present study and by Wang et al., (2011), detects cAMP accumulation within the cell and does not necessarily solely reflect AC1 activity. We were unable to inhibit AC1 activity in vitro by NB001 when assayed in membranes prepared from HEK293 cells expressing AC1, as used in the cAMP accumulation assays (Fig. 4C). This lack of direct action of NB001 on AC1 was irrespective of activation conditions, including stimulation by forskolin, calmodulin, or the combination of forskolin and calmodulin to mimic the effects of forskolin and ionomycin in intact cells. This suggests that, although NB001 can reduce cAMP accumulation in cells in an AC1-dependent manner, the ligand does not act through direct inhibition of AC1 activity.

NB001 decreases AC1-dependent cAMP accumulation but does not directly inhibit AC1. (A) Chemical structure of NB001. (B) HEK293 cells expressing AC1 or pcDNA control were incubated with NB001 (10 or 100 µM) or vehicle prior to stimulation with 10 µM forskolin and 10 µM ionomycin. Statistics (t test) for NB001 inhibition: *P < 0.05. (C) NB001 (100 µM) or vehicle control were incubated with membranes from HEK293 cells expressing AC1 and stimulated as indicated. Experiments (n = 3) for (B) and (C) were performed in duplicate. Representative experiments with SD are shown.
Structure-Based Virtual Screening Identifies Isoform-Selective AC Inhibitors with Novel Chemical Signatures.
The selective AC inhibitors characterized herein are all chemically related to adenine and/or adenosine. Thus, we screened for novel isoform-selective inhibitors of AC that target the ATP binding site but were structurally unrelated to adenine nucleotides. A library of ∼35,000 small drug-like molecules from ChemBridge were tested in two separate high-throughput site-directed virtual screens against an AC structure solved with a bound 2′3′ddATP ligand (P-site inhibitor; PDB code 1CJT), or with a bound MANT-GTP ligand (PDB code 1TL7) (see Materials and Methods). This “receptor ensemble docking” is a way to implicitly incorporate protein flexibility in high-throughput docking (Cavasotto and Singh, 2008). A final list of 100 distinct ligands was compiled from the ∼3000 molecules that scored in the top 10% in the two screens. Additional evaluations involved visual inspection of binding poses taking into account a previous suggestion that the adenine binding subregion is less important for the affinity of known inhibitors (Mou et al., 2006), and the notion that candidate ligands with multiple functional groups are less desirable. Finally, candidate molecules were clustered into chemically related groups to obtain the most structurally varied list of ligands possible. Analyzing hits by a combination of expected ligand-protein interactions, chemical clustering, and visual inspection can be a successful postprocessing strategy (Cavasotto et al., 2008).
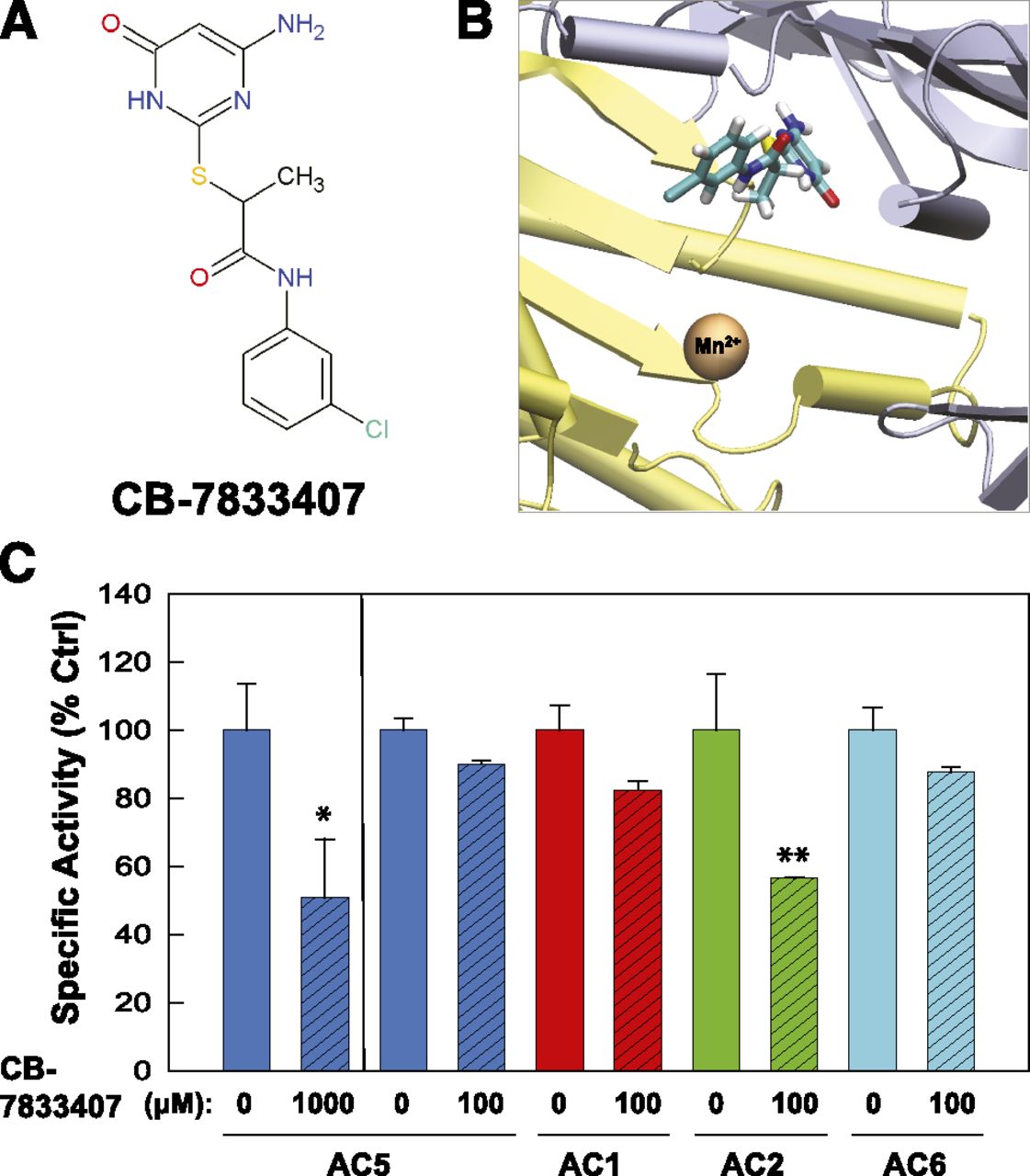
The resulting ligands (a total of 100) were examined in vitro for their ability to inhibit AC5 expressed in Sf9 membranes. For candidates that showed inhibition at 1 mM, a lower concentration of the inhibitor was then tested against AC isoforms 1, 2, 5, and 6 to obtain a limited profile of the candidates’ isoform selectivity. Three novel inhibitors were identified that had chemical structures distinct from adenosine or any other currently known small-molecule AC inhibitors (Figs. 5, A and D, and 6A), supporting the idea that AC inhibitors designed to target the ATP binding site do not necessarily have to resemble ATP. A close examination of the docked poses suggests that these inhibitors show wide variation in their ability to bind in a manner that would block the adenine-binding region (Figs. 5, B and E, and 6B). Surprisingly, all three inhibitors showed a degree of isoform selectivity for AC1 [5-(1'-phenyl-1H,1'H-4,4'-bipyrazol-3-yl)-1,3-benzodioxol-4-ol (CB-6673567) and 6-[2-(4-aminophenyl)vinyl]-2-pyridinecarboxylic acid (CB-7921220); Fig. 5, C and F] or AC2 [2-[(4-amino-6-oxo-1,6-dihydro-2-pyrimidinyl)thio]-N-(3- chlorophenyl)propanamide (CB-7833407); Fig. 6C) over AC5. The IC50 values for CB-6673567 inhibition of AC1 and CB-7833407 inhibition of AC2 are 77 and 147 µM, respectively. CB-6673567 and CB-7833407 display a 2- to 4-fold isoform selectivity over AC6, the next most sensitive AC isoform tested; CB-7921220 cannot distinguish between AC1 and AC6 (Fig. 5; Supplemental Fig. 1).

Identification of novel AC1 inhibitors. Chemical structure of CB-6673567 (A) and CB-7921220 (D). Virtual local docking of CB-6673567 (B) and CB-7921220 (E) to the catalytic site of 2′3′ddATP-bound AC. Inhibitor carbons are cyan, C1 domain is yellow, and C2 domain is silver. Inhibition of AC5 (blue) at 1 mM CB-6673567 (C) and CB-7921220 (F) and inhibition profile at 100 µM for AC5, AC1 (red), AC2 (green), and AC6 (cyan). All Sf9 membranes were stimulated by 50 µM forskolin. Statistics (paired t test) for inhibition of indicated AC isoform (n = 3–4, performed in duplicate): *P < 0.05; **P < 0.01; ***P < 0.001. Ctrl, control.

Identification of a novel AC2 inhibitor, CB-7833407. (A) Chemical structure of CB-7833407. (B) Virtual local docking of CB-7833407 to the catalytic site of 2′3′ddATP-bound AC. (C) Inhibition of AC5 (blue) at 1 mM CB-7833407 and inhibitory profile at 100 µM for AC5, AC1 (red), AC2 (green), and AC6 (cyan). All Sf9 membranes were stimulated by 50 µM forskolin. Statistics (paired t test) for inhibition of indicated AC isoform (n = 3): *P < 0.05; **P < 0.01. Ctrl, control.
Mutation of Serine 942 in AC2 Forskolin Pocket Does Not Alter Inhibition.
Global docking of SQ22,536 and Ara-A predicted that these inhibitors frequently (∼70 and ∼50%, respectively; see Table 2) target the ATP binding site of the AC C1/C2 domains (PDB code 1CJT). Global docking of NKY80 in the absence and presence of forskolin yielded more frequent hits at the forskolin and ATP binding sites, respectively. Similar docking to the MANT-GTP–bound conformation of AC (PDB code 1TL7) was less predictive, suggesting that these molecules bind to a similar AC conformation as their classic P-site predecessors.
Global docking of small-molecule inhibitors to AC catalytic domains
Global docking with AutoDock performed as described in Materials and Methods using the 2′3′ddATP-bound conformation of AC (1CJT). Percentages are based upon 256 independent docking runs. Values are the percentage of total docks.
The catalytic and forskolin sites are pseudosymmetrically related. Although forskolin is an AC activator, inhibitors that target the forskolin binding site have been identified (Pinto et al., 2009; Erdorf et al., 2011). Since our virtual screening focused only on the ATP site, we wanted to rule out that SQ22,536 and the other molecules that inhibited AC activity do not exert their actions through interactions with the forskolin pocket. To this end, we mutated residues within the forskolin-binding pocket. From the crystal structure of AC, Ser942 in the C2 domain of AC2 interacts with forskolin but does not contribute to C1-C2 interaction (Tesmer et al., 1997). Since substitution of this residue by proline was predicted to prevent hydrogen bonding to a nearby water molecule and distort the forskolin binding pocket, we mutated Ser942 to proline in AC2 and prepared membranes from COS-7 cells expressing the wild-type and mutant proteins. S942P activity had decreased sensitivity to stimulation by forskolin compared with wild-type AC2, whereas basal activity and stimulation by Gαs were not significantly altered (Fig. 7, A and B). Synergistic AC2 activation by both forskolin and Gαs was also impaired in the S942P mutant. However, the mutation had no effect on inhibition of Gαs-stimulated AC2 by SQ22,536 or CB-7833407 (Fig. 7C), supporting an interaction of these inhibitors with the ATP binding site.

Mutation of AC2 forskolin binding pocket does not impair inhibition by SQ22,536 or CB-7833407. (A) Basal AC activity of COS-7 membranes expressing AC2 wild type (WT), AC2 S942P mutant, or a pcDNA control. (B) COS-7 membranes expressing AC2 wild type, AC2 S942P mutant, or a pcDNA control were incubated with 10 µM forskolin and/or 50 nM activated Gαs, and AC2 activity was measured. (C) COS-7 membranes expressing AC2 wild type, AC2 S942P mutant, or a pcDNA control were preincubated in the absence or presence of the indicated inhibitor (100 µM SQ22,536 or 500 µM CB-7833407) and then stimulated with 50 nM Gαs. Experiments (n = 3–4) were performed in duplicate; representative experiments with SD are shown. Statistics (paired t test) for inhibition of AC2 WT/S942P by the indicated AC inhibitor: *P < 0.05; **P < 0.01. For differences between inhibition of AC2 WT versus S942P: n.s., not significant.
Discussion
There has been increasing interest in AC5 and other AC isoform–selective inhibitors for treatment of cardiac disorders, pain responses, aging, wound healing, and even jet lag (Seifert et al., 2012). However, we found that SQ22,536, NKY80, and Ara-A all fail to discriminate between AC5 and AC6 isoforms. The IC50 values for the AC5/6 inhibitors obtained in this work (Table 1) are consistent with previously published values (Onda et al., 2001). Ara-A has been reported to be selective for AC5 over AC6 in cardiomyocyte membranes based upon a single concentration point (Iwatsubo et al., 2012). One potential reason for this discrepancy is that prior assays with Ara-A used MgCl2 for AC5 and MnCl2 for AC6 membrane assays. These metals have different properties with regards to the affinity of P-site inhibitors and the Km for ATP. For better direct comparisons, we used MgCl2 for all AC activity assays reported herein. It is also possible that A-kinase anchoring protein (AKAP) signaling complexes that scaffold AC isoforms in cardiomyocytes alter regulatory and/or pharmacological properties of AC (Bauman et al., 2006; Kapiloff et al., 2009; Efendiev and Dessauer, 2011; Li et al., 2012). This has been observed previously for protein kinase C, where anchoring to AKAP complexes changes the pharmacological profile of the enzyme (Hoshi et al., 2010).
The lack of selectivity of SQ22,536, NKY80, and Ara-A that we observed between AC5 and AC6 is consistent with recent studies showing identical inhibition of cardiac AC from wild-type and AC5 knockout animals (Braeunig et al., 2013). SQ22,536 and Ara-A have been used in numerous intact cell assays and are cell permeable (Seifert et al., 2012); the cell permeability of NKY80 is unclear (Iwatsubo et al., 2004). Since these inhibitors have been used to define physiological roles for AC5, re-examination of such studies is in order. For example, SQ22,536 was used to suggest that AC5 was primarily involved in renin secretion (Ortiz-Capisano et al., 2007), but knockout models later clarified that both AC5 and AC6 were important (Aldehni et al., 2011). A similar reinterpretation of results is necessary for animal studies of heart failure (Iwatsubo et al., 2012) and autonomic control of heart rate using Ara-A (Bai et al., 2012). As these three AC inhibitors are adenine-like molecules, additional off-target effects are also of concern. Ara-A is clinically used under the name vidarabine as an antiviral that inhibits DNA synthesis (Whitley et al., 1980) and AMP-activated protein kinase α-2 (Musi et al., 2001), whereas SQ22,536 displays AC-independent effects on neurogenesis, extracellular signal-regulated kinase phosphorylation, and superoxide production (Brunskole Hummel et al., 2013; Emery et al., 2013).
One of the AC inhibitors identified by our in silico screening methods showed a preference for AC1 over AC5, whereas another was somewhat AC2 selective. Since the AC crystal structure consists of a C1 domain from AC5 and a C2 domain from AC2, identifying an AC2-selective inhibitor is not particularly surprising. CB-6673567 (AC1) and CB-7833407 (AC2) display different predicted binding orientations within the catalytic site when docked to the 2′3′ddATP-bound or MANT-GTP–bound AC structure. The AC1 inhibitor CB-7921220 has a more consistent predicted binding position in the two virtual docking screens, and has a binding conformation similar to ATP and P-site inhibitors, which may explain its lack of selectivity between AC1 and AC6.
Global docking predicted that NKY80 and Ara-A would dock to the ATP binding site or the forskolin binding site to varying degrees when forskolin was removed from the structure (Table 2). When forskolin was present, the majority of docked poses predicted binding to the ATP binding site (Table 2). Since mutation of the forskolin binding site did not alter inhibition by SQ22,536 or CB-7833407 (Fig. 7), it is likely that these and the other inhibitors exert their effects through interactions with the ATP binding site. However, local docking predicted no unique interactions consistent with all three AC5/6 inhibitors. The AC isoform selectivity profiles of SQ22,536, NKY80, and Ara-A are similar to the inhibitor MANT-GTP, which shows selectivity for AC 5/6 (and to a lesser extent AC1) over AC2 (Mou et al., 2005, 2006). However, the basis for MANT-GTP isoform selectivity is unclear and could be due to allosteric changes within the binding pocket for the MANT group, which would be difficult to predict from rigid in silico docking (Mou et al., 2006). The MANT binding pocket is bracketed by the α4′ helix in the C2 domain and the α1 helix in the C1 domain (Mou et al., 2005; PDB code 1TL7). Asn1025 is on the α4′ helix and is frequently in the proximity of NKY80 and Ara-A in local docks to the 2′3′ddATP-bound (PDB code 1CJT) conformation of AC. Interestingly, docking did not consistently predict that the novel ChemBridge inhibitors would interact with Asn1025. Therefore, residues that create the MANT pocket may allow for conformations favorable to binding isoform-selective inhibitors. In fact, the α1 helix of the C1 domain that forms the other side of the MANT pocket is poorly conserved across the nine isoforms, although identical in sequence between AC5 and AC6. The isoforms that are the most divergent from AC5 and AC6 in this helix were also the least potently inhibited by the AC5/6 inhibitors, notably AC8 and AC9 (Table 1). Residues adjacent to this helix have been identified as critical for stabilization of interactions between metal ions and triphosphate groups (Tesmer et al., 1999), as well as for inhibition of AC by submicromolar levels of free calcium, a property that is unique to AC5 and AC6 (Mou et al., 2009). Helix α1 may allosterically affect AC-metal interactions in an isoform-dependent manner, and thus may be a novel avenue for designing AC5- and AC6-selective ligands.
Surprisingly, the putative AC1-selective inhibitor NB001 does not appear to directly inhibit AC1 activity. More likely, it interacts with an AC1 regulatory protein or a downstream effector. However, direct interaction with AC does not preclude clinical usefulness. Although the molecular mechanism for NB001 is unclear, it still has important analgesic effects in animals (Wang et al., 2011). AC1 has roles in both pain sensation (Vadakkan et al., 2006; Wang et al., 2007) and memory (Villacres et al., 1998). The ability of NB001 to impair pain sensation, but not memory capability, is consistent with an indirect effect on AC1.
Although “AC5 inhibitors” are in fact AC5/6 inhibitors, these small molecules can be used to potentially uncover roles for additional isoforms. For example, in cardiomyocytes, AC5 and AC6 are the most highly expressed isoforms, but AC4 and AC9 are also expressed in lesser amounts (Espinasse et al., 1995; Li et al., 2012). These isoforms may have important compensatory roles, or even specific roles of their own, explaining the only subtle phenotype of Ara-A on isoproterenol-induced changes in left ventricular ejection fraction in mice (Iwatsubo et al., 2012). Consider the regulation of cardiac repolarization by the KCNQ1 channel. AC9 forms a complex with KCNQ1 and the AKAP Yotiao in the heart and facilitates protein kinase A regulation of the channel (Marx et al., 2002; Li et al., 2012), whereas disruption of the KCNQ1-Yotiao complex results in long QT syndrome (Marx et al., 2002; Chen et al., 2007). Thus, AC9 may be another important source of cAMP in the heart. Effects of cAMP, independent of AC5 and AC6, could be explored using these inhibitors. However, to be clinically useful, inhibitors of AC must show isoform selectivity. Nonselective AC inhibition could have many potential side effects, including deficiencies in spatial and long-term memory (AC1 and AC8), reduced motor coordination (AC5), inability to smell (AC3), obesity (AC3), nephrogenic diabetes insipidus (AC6), reduced renal function (AC3, AC6, AC5), and increased mortality during sustained sympathetic stimulation (AC6) (Pluznick et al., 2009; Sadana and Dessauer, 2009; Rieg et al., 2010; Aldehni et al., 2011; Tang et al., 2013). Clearly there is still a need for more selective tools to unravel cAMP and adenylyl cyclase biology and for therapeutic utility.
Acknowledgments
The authors thank Wei-Jen Tang for the rat AC7 baculovirus.
Authorship Contributions
Participated in research design: Brand, Hocker, Gorfe, Cavasotto, Dessauer.
Conducted experiments: Brand, Hocker.
Performed data analysis: Brand, Hocker, Cavasotto, Dessauer.
Wrote or contributed to the writing of the manuscript: Brand, Hocker, Gorfe, Dessauer.
Footnotes
- Received July 18, 2013.
- Accepted September 3, 2013.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants GM60419 (to C.W.D.); R01GM10078 (to A.A.G.); and T32GM089657 (to C.S.B. and H.J.H.)]; the National Institutes of Health National Institute for Mental Health [Grant MH060397 (to C.W.D.)]; and the Agencia Nacional de Promoción Científica y Tecnológica, Argentina [Grant PICT-2011-2778 (to C.N.C.)].
Portions of this work were previously presented in abstract form: Brand CS, Hocker HJ, Gorfe AA, Cavasotto CN, and Dessauer CW (2013) Isoform selectivity of adenylyl cyclase inhibitors and identification of novel compounds. Experimental Biology; 2013 April 20–25, Boston, MA.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AC
- adenylyl cyclase
- AKAP
- A-kinase anchoring protein
- Ara-A
- adenine 9-β-D-arabinofuranoside
- CB-6673567
- 5-(1′-phenyl-1H,1′H-4,4′-bipyrazol-3-yl)-1,3-benzodioxol-4-ol
- CB-7833407
- 2-[(4-amino-6-oxo-1,6-dihydro-2-pyrimidinyl)thio]-N-(3- chlorophenyl)propanamide
- CB-7921220
- 6-[2-(4-aminophenyl)vinyl]-2-pyridinecarboxylic acid
- 3′ddATP
- l-2′,3′-dideoxyadenosine-5′-triphosphate
- DMSO
- dimethylsulfoxide
- DTT
- dithiothreitol
- HEK293
- human embryonic kidney 293
- MANT-GTP
- 3′-O-(N-methylanthraniloyl)-guanosine-5′-triphosphate
- NB001
- 5-[[2-(6-amino-9H-purin-9-yl)ethyl]amino]-1-pentanol
- NKY80
- 2-amino-7-(furanyl)-7,8-dihydro-5(6H)-quinazolinone
- pcDNA
- XXX
- PDB
- Protein Data Bank
- Sf9
- A clonal isolate of Spodoptera frugiperda (Sf9) insect cells
- SQ22,536
- 9-(tetrahydro-2-furanyl)-9H-purin-6-amine
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}