


Abstract
Metabotropic glutamate receptor subtype 5 (mGlu5) has been demonstrated to play a role in the modulation of numerous nociceptive modalities. When administered via peripheral, intrathecal, or systemic routes, mGlu5 antagonists have analgesic properties in a variety of preclinical pain models. Despite a wealth of data supporting the use of mGlu5 antagonists to treat pain, studies have been limited to preclinical animal models due to a lack of mGlu5 antagonists that are approved for use in humans. It has been demonstrated previously that fenobam [N-(3-chlorophenyl)-N′-(4,5-dihydro-1-methyl-4-oxo-1H-imidazole-2-yl)urea], an anxiolytic shown to be safe and effective in human trials, is a selective and potent noncompetitive antagonist of mGlu5 (J Pharmacol Exp Ther 315:711–721, 2005). Here, we report a series of studies aimed at testing whether fenobam, similar to the prototypical mGlu5 antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP), has analgesic properties in mice. We show that fenobam reduces formalin-induced pain behaviors and relieves established inflammation-induced thermal hypersensitivity in mice. Similar results were seen with MPEP. Administration of fenobam resulted in an increase in locomotor activity in the open-field task but did not impair performance on the accelerating Rotarod. Analysis of brain and plasma fenobam levels indicated that fenobam is rapidly concentrated in brain after intraperitoneal administration in mice but is essentially cleared from circulation within 1 h after injection. Fenobam had no analgesic effect in mGlu5 knockout mice, whereas the prototypical antagonist MPEP retained significant analgesic efficacy in mGlu5 knockouts. These results demonstrate that fenobam is analgesic in mice and has an improved in vivo selectivity for mGlu5 over MPEP.
Pain is one of the principle components of inflammation and serves as a warning to the organism, allowing for the avoidance of or withdrawal from noxious stimuli. Inflammatory pain, however, may evolve from acute to chronic and become a pathological self-maintained phenomenon, thus losing its purpose as a protective response from tissue injury. One of the primary molecules implicated in the changes underlying chronic inflammatory pain is the excitatory neurotransmitter glutamate. Enhanced glutamate release occurs in response to inflammation at multiple levels of the pain neuraxis (Varney and Gereau, 2002), and several compounds that affect glutamate receptors have been implicated as potential therapeutics for pain (Hewitt, 2000; Bhave et al., 2001; Zhu et al., 2004).
Glutamatergic neurotransmission is mediated via ligand-gated ionotropic receptors and G protein-coupled metabotropic receptors (mGluRs). The mGluR family is classified into eight distinct receptor subtypes divided into three major groups based on sequence homology, signal transduction mechanisms, and pharmacological profiles (Conn, 2003). Group I mGluRs (mGlu1 and 5), which couple to the activation of phospholipase C and the release of intracellular calcium, are expressed at synapses throughout the pain neuraxis (Varney and Gereau, 2002). Both the activation and pharmacological blockade of group I mGluRs, and in particular mGlu5, have been demonstrated to play a role in the modulation of numerous nociceptive modalities (Bhave et al., 2001; Karim et al., 2001; Hu et al., 2002; Zhu et al., 2004). Subcutaneous administration of the selective group I mGlu agonist (R,S)-3,5-dihydroxy phenylglycine into the rodent hind paw results in enhanced sensitivity to thermal stimuli (Bhave et al., 2001) and intrathecal (R,S)-3,5-dihydroxy phenylglycine administration results in immediate spontaneous nociceptive behaviors in rodents. In addition, peripherally (Bhave et al., 2001; Walker et al., 2001; Zhu et al., 2005), intrathecally (Karim et al., 2001), and systemically (Zhu et al., 2004) administered antagonists of mGlu5 have been demonstrated to have analgesic properties in a variety of preclinical pain models.
Past studies of mGlu5 antagonists have been primarily limited to these preclinical models. Research into the efficacy of mGlu5 antagonists in the treatment of human pain conditions would be greatly enhanced by an antagonist that has been shown to be safe and pharmacologically active in humans. Recently, researchers at Hoffmann-La Roche demonstrated that the clinically validated nonbenzodiazepine anxiolytic fenobam [N-(3-chlorophenyl)-N′-(4,5-dihydro-1-methyl-4-oxo-1H-imidazole-2-yl)urea] is a highly potent and selective antagonist of mGlu5 (Porter et al., 2005). McNeil Laboratories originally developed fenobam as a potential anxiolytic in the 1970s and 1980s but with a then unknown molecular target. It was found to be effective at treating anxiety in a double-blinded placebo-controlled clinical trial (Pecknold et al., 1982). In that trial, and two additional trials (Pecknold et al., 1980; Lapierre and Oyewumi, 1982), fenobam was reported to have a good safety profile with no oversedation, no muscle relaxation, and no interaction with ethanol. However, in a different phase II clinical trial (Friedmann et al., 1980), fenobam was reported to have both psychostimulant side effects and to be ineffective as an anxiolytic. In the early 1980s, further development of the molecule as an anxiolytic by McNeil Laboratories was discontinued. In 2005, a functional high-throughput screen and subsequent characterization by Porter et al. (2005) identified fenobam as a potent, noncompetitive, mGlu5-selective antagonist, acting at an allosteric modulatory site shared with the prototypical mGlu5 antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) (Gasparini et al., 1999). It is interesting that, in addition to its anxiolytic efficacy (Ballard et al., 2005), MPEP has also been reported effective in the treatment of animal models of both fragile X syndrome (Yan et al., 2005) and chronic pain (Zhu et al., 2004). Fenobam was recently tested as a potential clinical therapeutic for fragile X syndrome in humans, where it was shown to possess beneficial clinical effect without significant adverse effects (Berry-Kravis et al., 2009). However, the efficacy of fenobam in the treatment of pain remains untested.
Given the interest in mGlu5 as a target for inflammatory pain and the recent demonstration of the efficacy of fenobam as a clinically validated mGlu5 antagonist with a good safety profile, we sought to compare the analgesic efficacy of fenobam with that of MPEP in two different in vivo models of inflammatory pain. Here, we show that fenobam, like MPEP, can prevent formalin-induced spontaneous pain-related behaviors and reduce Complete Freund's adjuvant (CFA)-induced thermal hypersensitivity. Previous pharmacokinetic analysis of fenobam in humans has indicated variable plasma levels (Berry-Kravis et al., 2009); thus, we also sought to characterize the bioavailability of fenobam in both mouse plasma and brain tissue. In addition, we assessed the effects of fenobam on locomotor activity and motor coordination. Finally, to establish selectivity for its analgesic effects on mGlu5, we assessed the pain behaviors of mGlu5 KO mice that were treated with either fenobam or MPEP.
Materials and Methods
Subjects
Experiments were performed in accordance with the Guidelines of the National Institutes of Health and were approved by the Animal Care and Use Committee of Washington University School of Medicine (St. Louis, MO). Male Swiss-Webster mice (29.5–43 g) were purchased from Taconic Farms (Germantown, NY). Male C57BL/6 mice used in metabolism experiments (20.0–29.5 g) were purchased from The Jackson Laboratory (Bar Harbor, ME). For experiments involving mice lacking mGlu5 (mGlu5 KO; 16.5–22.5 g), animals were bred in-house on a C57BL/6 background and compared with WT littermates (Lu et al., 1997). For experiments involving KO animals, the experimenter was blinded to genotype. All other C57BL/6 WT mice used in behavioral experiments were also derived from this colony. Genotyping of mice bred in-house was performed using standard polymerase chain reaction techniques. All mice were group housed on a 12:12-h light/dark schedule with ad libitum access to food and water.
Chemicals and Reagents
Fenobam and MPEP were purchased from Tocris Bioscience (Ellisville, MO). Both compounds were dissolved in 100% dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO) on the day of the experiment. All intraperitoneal injection volumes were 20 μl. Throughout all experiments, the investigator was blinded to pharmacological treatment. Midazolam was purchased from Cerilliant Corporation (Round Rock, TX) and used as the HPLC/MS internal standard. All other reagents were HPLC grade and purchased from Sigma-Aldrich.
Behavioral Assays
Formalin Test. The formalin test was performed as described previously (Karim et al., 2001). In brief, mice were acclimated in a transparent Plexiglas box (25 × 25 × 12.5 cm, length × width × height) for 2 h before any drug injection. Animals were then pretreated by intraperitoneal injection with vehicle, MPEP, or fenobam. All Swiss-Webster mice were pretreated 30 min before formalin injection. Strain differences in responsiveness to fenobam were noted in pilot studies and thus C57BL/6 mice were initially pretreated either 5 or 30 min before formalin injection to determine the optimal time for pretreatment in this strain (n = 4–5 mice/group). In all subsequent formalin tests involving C57BL/6 mice, including those performed with mGlu5 KO mice, vehicle or drugs were injected 5 min before formalin injection. Ten microliters of dilute formalin solution (Sigma-Aldrich) was injected subcutaneously into the plantar surface of the right hind paw. Due to strain differences in formalin sensitivity (Mogil et al., 1999), the concentration of formalin injected into Swiss-Webster mice and C57BL/6 mice was 5 and 2%, respectively. The time spent in nociceptive behavior, defined as licking, lifting, or flicking of the injected paw, was scored in 5-min intervals for 1 h beginning immediately after paw injection. For experiments comparing fenobam with MPEP in Swiss-Webster mice, 30 mg/kg of each drug was administered (n = 7 mice/group). For experiments intended to develop a dose-response curve for fenobam in Swiss-Webster mice, 3, 10, 30, or 100 mg/kg fenobam was injected (n = 7–11 mice/group). For experiments involving the testing of fenobam in C57BL/6 WT and mGlu5 knockout mice, 30 mg/kg fenobam was injected (n = 4–6 mice/group). For experiments involving the testing of MPEP in mGlu5 KO mice, 30 mg/kg MPEP was injected (n = 6–9 mice/group).
CFA-Induced Thermal Hypersensitivity. Thermal hypersensitivity was measured using a modified version of the Hargreaves test (Hargreaves et al., 1988). Swiss-Webster mice (n = 7–8 mice/group) were placed in individual Plexiglas containers (12.5 × 12.5 × 15 cm, length × width × height) on an elevated glass platform. A continuous radiant heat source was delivered through the glass onto the surface of each hind paw (IITC Life Sciences, Woodland Hills, CA), and the latency for animals to withdrawal their paws was measured. The active intensity of the heat source was set to 21%. Five baseline measurements were obtained 15 min apart and averaged. Immediately after baseline CFA (10 μl; 1 mg/ml) was injected into the plantar surface of the right hind paw. Paw withdrawal latency to the thermal stimulus (PWLThermal) was assessed 48 h after CFA injection. One hour after assessment of post-CFA PWLThermal, animals were injected intraperitoneally with fenobam (30 mg/kg), MPEP (30 mg/kg), or vehicle. PWLThermal was then assessed 30 and 60 min after drug injection.
Open-Field Locomotor Test. Locomotor activity was measured in an open-field using a VersaMax Animal Activity Monitoring System (AccuScan Instruments, Inc., Columbus, OH). Swiss-Webster mice (n = 10–11 mice/group) were habituated to the test room individually in Plexiglas boxes (25 × 25 × 12.5 cm, length × width × height) for 2 h. Thirty minutes before the assessment of locomotor activity, mice were injected intraperitoneally with fenobam (3, 10, or 30 mg/kg) or vehicle and returned to their habituation chamber. Locomotor activity was assessed by recording photobeam breaks in a chamber (42 × 42 × 30 cm, length × width × height) for 60 min. Total distance traveled, time spent moving, and the number of beam breaks (horizontal activity) were calculated for the entire chamber as well as a perimeter (outer 8-cm ring) and center (inner 26- × 26-cm square) region. The percentage of time spent in the perimeter was also calculated as (minutes spent in perimeter/60 min × 100%).
Rotarod. An accelerating Rotarod (Ugo Basile, Comerio, Italy) was used to assess motor coordination. Naive Swiss-Webster mice (n = 10–12 mice/group) received two training sessions separated by 1 h. The first training session consisted of two trials of 120 s spent walking on the Rotarod at a fixed speed of 4 rpm. The second training session consisted of one trial of 120 s at 4 rpm. All mice completed the first training session without falling in five attempts or less; all mice completed the second training session in two attempts or less without a fall. One hour after the second training session, mice were injected intraperitoneally with fenobam (3, 10, or 30 mg/kg), pentobarbital (25 mg/kg), or vehicle. Latency to fall as the Rotarod accelerated from 4 to 40 rpm over 5 min was assessed 30 min after injection. Five consecutive acceleration trials were performed, with 10 min between each trial.
Behavioral Data Analysis. All data collected over multiple time points from the formalin, open-field, and Rotarod tests were statistically analyzed using a Bonferroni multiple comparison test after a repeated two-way analysis of variance. Summed data from these tests were analyzed using a two-tailed t test when comparisons were made between two groups or a Bonferroni multiple comparisons test after a one-way analysis of variance when comparisons were made between more than two groups. All data were analyzed using Prism 5.0 (GraphPad Software Inc., San Diego, CA). In all studies, the accepted level of significance was p < 0.05. In all figures, data are reported as mean ± S.E.M.
Method for Quantification of Fenobam in Mouse Brain and Plasma
Plasma and Brain Tissue Collection. Wild-type Swiss-Webster and C57BL/6 mice (n = 4 mice/group) were injected with fenobam (3, 10, or 30 mg/kg i.p.) and administered an overdose of sodium pentobarbital (75 mg/kg) 5, 30, or 55 min after fenobam injection. Whole blood was obtained by transcutaneous cardiac puncture, and plasma was separated via centrifugation (14,000g for 5 min at 4°C) in plasma separator tubes with lithium heparin (BD Microtainer; BD Biosciences, San Jose, CA). Immediately after centrifugation, plasma was frozen using liquid nitrogen and stored at -80°C. Brains were dissected and immediately frozen in liquid nitrogen and stored at -80°C. Whole-brain homogenates were obtained by adding Milli-Q water (Millipore, Billerica, MA) to brain tissue in a 2:1 (v/w) ratio and sonicating using a Sonifier 150 (Branson Ultrasonics Corporation, Danbury, CT). Brain homogenates were immediately refrozen on dry ice.
General Instrumentation. Fenobam was quantitated using liquid chromatography-MS/MS. Calibrators were prepared in a matrix matching the samples (brain homogenates or plasma). Midazolam was used as the internal standard. Instrumental analysis was performed on an API 4000QTRAP triple-quadrupole mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA), equipped with a TurboIonSpray source. The Agilent 1100 HPLC system (Agilent Technologies, Waldbronn, Germany) included a binary pump, a thermostated well plate autosampler, and a column thermostat. An external two-way Valco valve was used to direct HPLC flow to waste before and after column elution of analytes of interest. Chromatographic separation was performed on a SymmetryShield RP18 analytical (3.5 μm; 2.1 × 30 mm) column (Waters, Milford, MA) with a C18 guard (5 μm; 2 × 10 mm) column (Varian, Inc., Lake Forest, CA) at 30°C. Before each injection, the needle was washed with methanol. Separate methods were used for determining fenobam in brain tissue and plasma.
HPLC and Mass Spectrometric Conditions for Quantification of Fenobam in Brain Tissue. Mobile phase A was 20 mM ammonium formate buffer, pH 5.7, and mobile phase B was methanol. Mobile phase was delivered at an initial condition of 2% B and a flow rate of 0.5 ml/min with the following time program: 2% B is held for 0.1 min, followed by a linear gradient to 100% B between 0.1 and 1.0 min. B is held at 100% for 0.1 min and then brought back down to initial condition of 2% between 1.1 and 2.1 min. The column is re-equilibrated with 2% B from 2.1 to 5.0 min. Under these conditions, the retention time for fenobam was 3.3 min and, for midazolam, it was 3.4 min. The injection volume was 10 μl. Both Q1 and Q3 quadrupoles of the mass spectrometer were optimized to unit mass resolution, and the conditions were optimized for each analyte. The instrument was operated in positive-ion mode, with an ion spray voltage of 5500 V. The curtain gas was set at 20 psi, ion source gas 1 and 2 at 20 psi, and the collision gas on “high.” The transitions monitored for each analyte, along with the analyte specific parameters are listed in Table 1.
HPLC-mass spectrometric acquisition parameters for brain tissue
Calibration and Sample Preparation. Each homogenized sample (50 μl) was pipetted into discrete wells of a 96-well 2.2-ml plate. Twenty-five microliters of internal standard (12.5 ng of midazolam) was added to each sample, followed by 200 μl of acetonitrile. The plate was capped and vortexed and then centrifuged at 3000 rpm for 10 min. The supernatants were transferred to a 96-well autosampler plate, and 10 μl was injected. Calibrators and quality control samples were prepared along with experimental samples.
Calibrators, Quality Controls, and Internal Standard Samples. A methanolic solution of fenobam was prepared at 1 mg/ml. Dilutions from this stock standard were prepared and used to make calibrator and quality control (QC) samples in brain homogenate. Brain homogenate calibrators contained 0.5, 2.5, 5, 10, 15, 20, 30, and 40 μg/g. Brain homogenate QCs were made at 2.5 and 20 μg/g.
HPLC and Mass Spectrometric Conditions for Quantification of Fenobam in Plasma. Mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in acetonitrile. Mobile phase was delivered at an initial condition of 5% B and a flow rate of 0.4 ml/min with the following time program: linear gradient between 5 and 60% B for 1.0 min followed by a sharp gradient to 100% B for 0.2 min and hold at 100% B for 0.4 min; mobile phase composition is then brought back down to initial condition of 5% between 1.6 and 2.1 min. The column is re-equilibrated with 5% B from 2.1 to 5.5 min. Under these conditions, the retention time for fenobam was 3.4 min, and for midazolam, it was 3.0 min. The injection volume was 20 μl. Both Q1 and Q3 quadrupoles of the mass spectrometer were optimized to low and unit mass resolution, respectively. The instrument was operated in positive-ion mode, with an ion spray voltage of 5100 V. The curtain gas was set at 20 psi; ion source gas 1 and 2 at 40 and 50 psi, respectively; and the collision gas on high. The transitions monitored for each analyte, along with the analyte-specific parameters are listed in Table 2.
HPLC-mass spectrometric acquisition parameters for plasma
Calibration and Sample Preparation. Mouse plasma samples were thawed, homogenized, and aliquots of 25 μl were transferred into a 96-well plate. Precipitation was performed using 100 μl of acetonitrile, which contained 50 ng/ml midazolam (internal standard). The plate was capped and vortexed and then centrifuged at 3000 rpm for 10 min. The supernatants were transferred to a 96-well autosampler plate, and 20 μl was injected for analysis. Calibrators and quality control samples were prepared along with experimental samples.
Calibrators, Quality Controls, and Internal Standard Samples. A methanolic solution of fenobam was prepared at 1 mg/ml. Dilutions from this stock standard were prepared and used to make calibrator (6–16,000 ng/ml; 10 concentrations) and QC samples (two concentrations) in human plasma. Preliminary experiments using mouse and human plasma had similar liquid chromatography-MS/MS results.
Results
Effects of Fenobam and MPEP on Spontaneous Formalin Behavior in WT Mice. We compared the effects of fenobam to MPEP, an mGlu5 antagonist that has been previously demonstrated to reduce spontaneous behavior during the formalin test. Right hind paw injection of formalin resulted in a characteristic biphasic response. Pretreatment with both fenobam (30 mg/kg i.p.) and MPEP (30 mg/kg i.p.) significantly reduced the time Swiss-Webster mice spent licking or lifting the formalin injected paw during the second phase (p < 0.05 and p < 0.01, respectively) (Fig. 1). In addition, pretreatment with fenobam resulted in a reduction in the time spent licking or lifting during the first phase (p < 0.001).
Next, we examined the effects of four different doses of fenobam (3, 10, 30, and 100 mg/kg i.p.) on nociceptive scores during the formalin test. Pretreatment of Swiss-Webster mice with 30 and 100 mg/kg fenobam 30 min before intraplantar formalin injection significantly reduced the time mice spent licking or lifting the injected paw during both the first (p < 0.05 and p < 0.001, respectively) (Fig. 2B) and second phase (p < 0.01 and p < 0.001, respectively) (Fig. 2C).
The C57BL/6 strain of mice is often used in the generation of KO animals. We therefore examined the effects of fenobam on spontaneous formalin behavior in this strain. In Swiss-Webster mice, pretreatment with fenobam was performed 30 min before formalin injection. When C57BL/6 mice were pretreated with fenobam (30 mg/kg) 30 min before formalin injection, there was a reduction in formalin-induced spontaneous behavior 20 min after formalin injection compared with vehicle (p < 0.05) (Fig. 3); however, the total time in the second phase was not found to be different. When C57BL/6 mice were instead pretreated with fenobam 5 min before formalin injection a significant reduction in the second phase was observed compared with both vehicle-injected mice and mice injected with fenobam 30 min before formalin injection (Fig. 3).
Fenobam Disposition in Plasma and Brain Tissue. Fenobam disposition was examined in both brain and plasma from C57BL/6 and Swiss-Webster mice. Fenobam was readily detectable in plasma and brain tissue 5 min after intraperitoneal injection in both strains (Fig. 4, A and B), and concentrations decreased thereafter. Fenobam (3 and 10 mg/kg) was largely cleared from both plasma and brain tissue after 30 min, whereas the highest dose (30 mg/kg) remained detectable 30 min after injection but was largely cleared after 55 min. A brain/plasma ratio (grams per grams per grams per milliliter) was calculated for the 5-min time point for all three doses (Fig. 4C) and was found to be significantly different in both strains when the 3 and 30 mg/kg doses were compared.
Effects of Fenobam and MPEP on CFA-Induced Thermal Hypersensitivity in WT Mice. Next, we compared the effects of fenobam, MPEP, and vehicle on CFA-induced thermal hypersensitivity. Forty-eight hours after injection of CFA into the right hind paw, PWLThermal relative to baseline was significantly reduced ipsilateral to the injection in all groups (Fig. 5A). No reduction in PWLThermal was observed in the contralateral paw in any group (Fig. 5B). One hour after assessment of the 48-h post-CFA PWLThermal, mice were injected intraperitoneally with vehicle, fenobam (30 mg/kg), or MPEP (30 mg/kg). Both fenobam (p < 0.05) and MPEP (p < 0.05), but not vehicle, significantly increased PWLThermal measured 30 and 60 min after drug injection compared with their respective 48-h post-CFA time point (Fig. 5A). In both the fenobam- and MPEP-treated animals the postdrug PWLThermal returned to a level that was not significantly different from baseline; however, the PWLThermal of the vehicle-treated animals remained significantly different from baseline 30 min after vehicle injection (p < 0.05) (Fig. 5A). No differences were observed in the contralateral paw of any group between baseline, predrug, or postdrug PTWLThermal (Fig. 5B).

Spontaneous formalin behavior is reduced after pretreatment with fenobam or MPEP in Swiss-Webster mice. A, when administered 30 min before intraplantar formalin injection, both fenobam (30 mg/kg i.p.) and MPEP (30 mg/kg i.p.) significantly decrease the time spent licking or lifting the injected paw. Asterisks indicate time points where both drug treatments are significantly different from vehicle (n = 7 mice/group; *, **, and ***, p < 0.05, 0.01, and 0.001, respectively). Fenobam-treated animals were significantly different from MPEP at the 5-min time point (†, p < 0.05). B and C, total time spent licking or lifting in the first phase (<10 min) and second phase (>10 min), respectively.
Open-Field Task as a Measure of Locomotor Activity. The open-field task was used to assess the effects of fenobam (3, 10, and 30 mg/kg i.p. 30 min before placement in the field) on locomotor activity in Swiss-Webster mice. Compared with vehicle-treated animals, fenobam produced no significant effect on spontaneous exploratory behavior at 3 and 10 mg/kg; however, at 30 mg/kg, a significant increase in exploratory behavior was observed in the total distance mice traveled over 60 min (p < 0.05) (Fig. 6, A and B). No significant effects on behaviors in the center (26 × 26 cm) of the open-field apparatus were observed between the different doses. No dose of fenobam was found to alter the percentage of time mice spent in the perimeter versus the center (Fig. 6, C and D).

Effect of fenobam (3, 10, 30, or 100 mg/kg i.p.) on spontaneous formalin behavior in Swiss-Webster mice. A, when fenobam is administered 30 min before formalin, a minimal dose of 30 mg/kg i.p. is required to see a significant effect (n = 7–11 mice/group; ***, p < 0.001 for vehicle compared with 30 and 100 mg/kg; † and ††, p < 0.05 and 0.01, respectively, for vehicle compared with 100 mg/kg. B and C, total time spent licking or lifting in the first and second phase, respectively (*, **, and ***, p < 0.05, 0.01, and 0.001, respectively, compared with vehicle).
Accelerating Rotarod Task as a Measure of Motor Coordination. Compared with vehicle, fenobam injection resulted in no significant differences in latency to fall from an accelerating Rotarod at any dose tested, up to 30 mg/kg (Fig. 7). No significant difference between vehicle-injected and fenobam-injected Swiss-Webster mice was observed over five consecutive trials. To demonstrate that a reduction in fall latency could be induced, one group of mice was injected with 25 mg/kg pentobarbital, which resulted in a significant reduction in the fall latency in both the first (p < 0.01) and second (p < 0.001) trials, compared with vehicle. All groups showed significant improvement in fall latency from the first to the fifth trial (p < 0.001).

Spontaneous formalin behavior is reduced after pretreatment with fenobam in WT C57BL/6 mice. A, when administered 30 min before intraplantar formalin injection, fenobam (30 mg/kg i.p.) significantly decreased the time spent licking or lifting the injected paw compared with vehicle at 20 min after injection (†, p < 0.05). The overall time spent licking or lifting in the second phase was not reduced. When fenobam (30 mg/kg i.p.) was injected 5 min before formalin hind paw injection, there was a significant reduction at multiple time points (A) (* and ***, p < 0.05 and 0.001, respectively) and in the total time spent licking or lifting in the second phase (C) (p < 0.01) compared with both vehicle and animals injected with fenobam 30 min before formalin injection (n = 4–5 mice/group).
Effects of Fenobam and MPEP on Spontaneous Formalin Behavior in mGlu5 KO Mice. The effects of fenobam and MPEP on spontaneous formalin behavior were assessed in mGlu5 knockout mice in the same manner as described for Swiss-Webster WT mice. Because KO mice were bred on a C57BL/6 background, the effects of fenobam (30 mg/kg) were also assessed on littermate WT mice. Pretreatment with fenobam (30 mg/kg i.p. 5 min before formalin injection) significantly reduced the time WT littermates spent licking or lifting the formalin-injected paw in both the first phase (p < 0.01) and second phase (p < 0.05) (Fig. 8, A–C). However, pretreatment with fenobam (30 mg/kg i.p.) did not alter spontaneous nociceptive behavior after formalin injection in mGlu5 KO mice at any time point (Fig. 8, D–F). When mGlu5 KO mice were pretreated with MPEP (30 mg/kg i.p.), there was a significant reduction in the time spent licking or lifting the formalin-injected paw in the second phase (p < 0.001) (Fig. 9) but not the first phase.
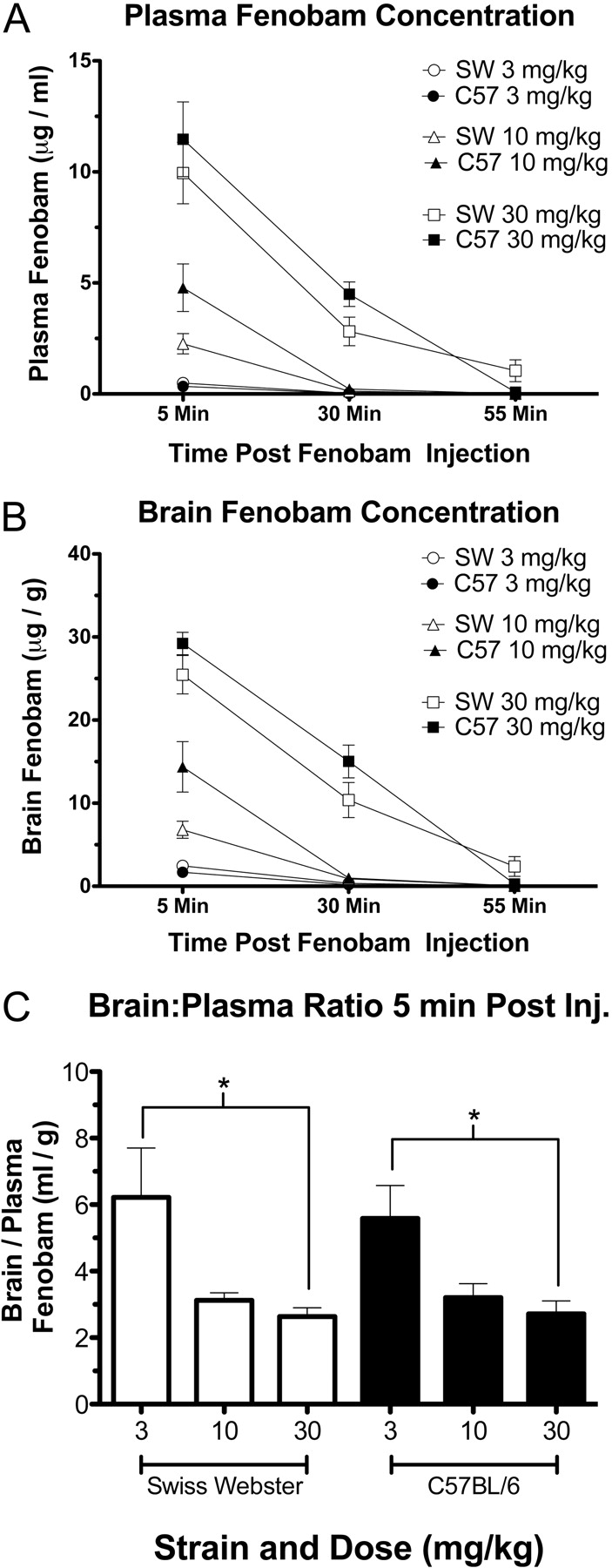
Fenobam brain and plasma concentrations. Swiss-Webster and C57BL/6 mice were injected with either 3, 10, or 30 mg/kg i.p. fenobam and then sacrificed 5, 30, or 55 min later. Plasma (A) and brain (B) fenobam concentrations are presented as mean ± S.E.M. for each animal, dose, and time point. C, a brain/plasma ratio (milliliters per gram) was calculated for each animal 5 min after injection with fenobam. In both strains, the brain/plasma ratio between the lowest dose (3 mg/kg) and the highest dose (30 mg/kg) was significantly different (*, p < 0.05; n = 4 mice/group).

Fenobam or MPEP relieves established CFA-induced thermal hypersensitivity. A, in all groups, paw withdrawal latency to a thermal stimulus was reduced 48 h after an ipsilateral CFA injection, compared with baseline. Both fenobam and MPEP, but not vehicle, significantly increased thermal withdrawal latency 30 and 60 min after treatment in the ipsilateral paw. B, no significant changes in withdrawal latency were observed in the contralateral paw (*, p < 0.05 compared with baseline; †, p < 0.05 compared with 48 h after CFA; n = 7–8 mice/group).

Open-field locomotor activity 30 min after pretreatment with fenobam in WT mice. When administered 30 min before placement in a novel open-field environment, fenobam (30 mg/kg i.p.) significantly increased the total distance mice traveled compared with vehicle between 35 and 40 min (A) (p < 0.05) and as a sum total of distance traveled in 60 min (B) (p < 0.05). C and D, fenobam did not decrease the amount of time mice spent in the perimeter (outer 8-cm ring) compared with the center (inner 26- × 26-cm square) at any dose tested (n = 10–11 mice/group).

Accelerating Rotarod performance is unaffected after pretreatment with fenobam in WT mice. Fenobam (3, 10, or 30 mg/kg i.p.) administered 30 min before placement on an accelerating Rotarod (4–40 rpm increase over 5 min) did not affect latency to fall at any dose tested compared with vehicle. Pentobarbital (25 mg/kg i.p.) significantly decreased the latency to fall on both the first (**, p < 0.05) and second trial (***, p < 0.01) compared with vehicle. All groups showed significant improvement from the first to fifth trial (†††, p < 0.001). Trials were separated by 10-min intervals. Animals that did not fall were allowed to continue walking for a maximum of 30 s after achieving the maximum rpm (330 s, arrow) (n = 10–12 mice/group).
Discussion
Fenobam is a potent and selective mGlu5 antagonist that has anxiolytic properties in both rodents (Porter et al., 2005) and humans (Pecknold et al., 1980, 1982). Multiple groups have reported that pharmacological blockade of mGlu5 is effective at reducing nociception in several rodent pain models (Bhave et al., 2001; Karim et al., 2001; Walker et al., 2001; Zhu et al., 2004); however, until now the analgesic properties of fenobam have not been reported. In the present study, we report that intraperitoneal administration of fenobam (30 mg/kg) is analgesic in two mouse models of inflammatory pain, the formalin test and the CFA-induced thermal hypersensitivity test. Pretreatment with fenobam reduced the time that mice from two different strains (Swiss-Webster and C57BL/6) spent in spontaneous nocifensive behavior during the first and second phase of the formalin test. The effects of fenobam were dose-dependent; 30 mg/kg was the lowest effective dose tested. As reported here, these findings are similar to those seen when mice are pretreated with the prototypical mGlu5 antagonist MPEP (30 mg/kg). MPEP has been shown previously to reduce spontaneous nocifensive behaviors in the formalin test by several groups, with minimal effective doses in mice ranging from 10 mg/kg (Varty et al., 2005) to 30 mg/kg (Satow et al., 2008). We chose to make our comparison between fenobam and MPEP at a dose of 30 mg/kg because MPEP has been shown to be effective at 30 mg/kg in reducing formalin-induced nocifensive behaviors by multiple groups. Our findings suggest that fenobam is capable of reducing both the neurogenic (phase I) and inflammatory (phase II) components of the formalin test. In addition, fenobam (30 mg/kg) was found to relieve established thermal hypersensitivity induced via CFA injection. To our knowledge, this is the first report of the analgesic efficacy of an mGlu5 antagonist that has also been shown to be pharmacologically active and safe in humans.

Spontaneous formalin behavior is not reduced in mGlu5 KO mice after pretreatment with fenobam. In C57 WT mice (A–C), but not mGlu5 KO littermates (D–F), fenobam (30 mg/kg i.p.) administered 5 min before intraplantar formalin injection significantly decreased the time spent licking of lifting the injected paw. Pretreatment with fenobam reduced both the first (p < 0.01) (B) and second (p < 0.05) (C) phases in WT mice (n = 4–6 mice/group).

Spontaneous formalin behavior is reduced in mGlu5 KO mice after pretreatment with MPEP but not with Fenobam. A, when administered 30 min before intraplantar formalin injection, MPEP (30 mg/kg i.p.) significantly decreased the time spent licking or lifting the injected paw. Asterisks indicate time point where MPEP is significantly different from vehicle (n = 6–9 mice/group). Pretreatment with MPEP reduced the second phase (p < 0.001) (C) but not the first phase (B).
These initial tests were performed in outbred Swiss-Webster mice. When attempts were made to begin testing in C57BL/6 mice, a common strain used as a background for KO animals, including the mGlu5 KO mice described here, we noted a difference in sensitivity to the drug compared with Swiss-Webster mice. Interstrain variability in response to both opiate (Pick et al., 1991) and nonopiate analgesics (Wilson et al., 2003) has been reported previously, and our findings suggest that different strains of mice will also exhibit variable sensitivity to fenobam. We hypothesized that a difference in fenobam concentration in brain tissue or plasma might underlie the difference in responsiveness between Swiss-Webster and C57BL/6 mice; however, both strains exhibited nearly identical uptake of fenobam into brain and clearance of fenobam from both brain tissue and plasma at all doses and time points tested. It should be noted that mGlu5 is also expressed in the periphery (Bhave et al., 2001) and the spinal cord (Varney and Gereau, 2002), where potential differences in fenobam concentration were not assessed. Although the brain/plasma ratio of fenobam 5 min after intraperitoneal injection was found to vary depending on dose, this variance was noted in both strains and thus cannot account for any observed differences. Finally, disposition studies also demonstrated that fenobam is rapidly cleared from both brain tissue and plasma, such that when administered at 3 or 10 mg/kg the drug is nearly absent 30 min after injection. This rapid clearance may explain why the 30 mg/kg dose was required to see analgesic effect in the formalin test.
Previous studies of MPEP and the related mGlu5 antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine have reported locomotor side effects at analgesic doses (Zhu et al., 2004). In the present study, administration of fenobam at the analgesic dose (30 mg/kg) but not at lower doses (3 and 10 mg/kg) resulted in an increase in spontaneous locomotor activity in an open-field assay. The increased locomotor activity is interesting in light of findings reported in one clinical trial (Friedmann et al., 1980) that fenobam may have psychostimulant properties. It is possible that the observed increases in locomotor activity reported here are due to a similar psychostimulant effect. However, based on findings from our drug disposition studies, fenobam clearance is nearly complete after approximately 1 h. This is interesting because the drug was injected 30 min before beginning open-field testing and because the increases in locomotor activity compared with vehicle do not become readily apparent until the 40-min time point (70-min after drug injection), after the drug is mostly cleared. Therefore, it is possible, however speculative, that the observed increase in locomotor activity is due to the accumulation of fenobam metabolites and not the parent compound itself. Alternatively, this could represent a “rebound” effect of unblocking mGlu5. Further study is required to assess these possibilities.
In addition, the findings of increased locomotor activity are in opposition to those seen with MPEP (Spooren et al., 2000; Zhu et al., 2004) and the related mGlu5 antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (Zhu et al., 2004), which have both been shown to reduce locomotor activity at analgesic doses. Because the dose of fenobam that resulted in changes in locomotor activity is the same that reduced spontaneous formalin behavior, it is possible that the analgesic effects of fenobam will prove inseparable from some off-target effects. However, motor coordination as measured on an accelerating Rotarod was not affected by fenobam at doses up to 30 mg/kg. Thus, although fenobam may increase locomotor activity, the potentially more deleterious side effect of altered motor coordination seems to be absent at the tested analgesic dose.
We also sought to establish that the in vivo antinociceptive effects of both MPEP and fenobam were specific for mGlu5 by testing these drugs in mGlu5 KO mice bred on a C57BL/6 background (Lu et al., 1997). Pretreatment with fenobam (30 mg/kg i.p.) had no demonstrated effects on mGlu5 KO mice compared with vehicle-treated animals. It is noteworthy that the second phase of the formalin test was reduced in vehicle-treated KO mice (Fig. 8F) to the same extent as in fenobam-treated WT mice (Fig. 8C). This effect is consistent with the importance of mGlu5 in this phase of the formalin test such that pharmacological and genetic knockout of mGlu5 reduces pain related behavior to a similar extent. However, in the first phase of the formalin test spontaneous nociceptive formalin behaviors were reduced in fenobam-administered WT animals (Fig. 8B) but not KO vehicle controls (Fig. 8E), suggesting that fenobam may be capable of blocking nociceptive behaviors in a manner that genetic knockout is not. Therefore, the effects of fenobam on phase 1 of the formalin test may be mediated via a mechanism other than the blockade of mGlu5. However, it is important to note that vehicle-treated KO mice may not have exactly the same behavior as fenobam-treated WT mice due to compensatory changes after the genetic deletion of mGlu5. That fenobam is without effect in mGlu5 KO mice compared with vehicle-treated littermate mGlu5 KO controls is compelling evidence that fenobam is on target for mGlu5.
MPEP (30 mg/kg), in contrast, was found to have robust, residual analgesic effects in mGlu5 KO mice. Questions regarding the selectivity of MPEP for mGlu5 have been noted previously (Lea and Faden, 2006), and here we report definitive evidence that the in vivo analgesic effects of MPEP in an inflammatory pain condition are not exclusively mediated by antagonism of mGlu5. This finding has specific and immediate implications for the pain field, because MPEP has been the primary antagonist used in assessing the role of mGlu5 in pain for more than a decade (Lea and Faden, 2006). Previous results obtained by a multitude of research groups, including our own, must be considered in light of this new finding. However, it is important to note that the data obtained with MPEP in the mGlu5 KO mice should not necessarily be taken to diminish the suggested role for mGlu5 in inflammatory pain. Indeed, as the present study indicates, fenobam is robustly analgesic in inflammatory pain in WT but not mGlu5 KO mice, suggesting its analgesic effects are selective for mGlu5. Rather than detract from a role for mGlu5 in analgesia, these data suggest an additional mechanism for MPEP in analgesia beyond its antagonism of mGlu5.
In conclusion, in the present study we report that the clinically validated mGlu5 antagonist fenobam displays robust analgesic efficacy in two mouse models of inflammatory pain in a manner similar to the prototypical mGlu5 antagonist MPEP. In addition, we report that although exposure to an analgesic dose of fenobam increases spontaneous exploratory behavior, the same dose does not affect motor coordination. Finally, we demonstrate that the in vivo analgesic effects of fenobam, but not MPEP, are selective for mGlu5. These findings, along with previous reports, support a role for mGlu5 antagonists as analgesics. In addition, from previous reports that fenobam has a good safety profile without oversedation, muscle relaxation, and interaction with ethanol, these data suggest that fenobam may represent a reasonable candidate molecule for testing the analgesic efficacy of pharmacological blockade of mGlu5 in human subjects.
Footnotes
-
This work was supported in part by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS42595, NS06462601] (to R.W.G. and to M.C.M., respectively); the National Institutes of Health National Institute on Drug Abuse [Grant K24-DA00417] (to E.D.K.); and the National Institutes of Health [Grant P30-NS057105] (to Washington University, St. Louis).
-
A portion of this work was presented in abstract form: Montana MC, Cavallone LF, and Gereau RW, IV (2008) The metabotropic glutamate receptor 5 (mGlu5) antagonist fenobam is analgesic in rodents; The 2008 Society for Neuroscience Meeting; Dates; Washington, DC. Program 772.5, Society for Neuroscience, Washington, DC.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.109.154138.
-
ABBREVIATIONS: mGlu, metabotropic glutamate receptor; MPEP, 2-methyl-6-(phenylethynyl)-pyridine; CFA, Complete Freund's adjuvant; KO, knockout; WT, wild type; HPLC, high-performance liquid chromatography; MS, mass spectometry; PWLThermal, paw withdrawal latency to the thermal stimulus; QC, quality control.
- Received March 26, 2009.
- Accepted June 9, 2009.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}