


Abstract
The hemagglutinin-tagged human trace amine-associated receptor1 (TAAR1) was stably coexpressed with rat Gαs in the AV12-664 cell line, and receptor activation was measured as the stimulation of cAMP formation. After blockade of endogenously expressed α2- and β-adrenoceptors with 2-[2-(2-methoxy-1,4-benzodioxanyl)]-imidazoline hydrochloride (2-methoxyidazoxan, RX821002) and alprenolol, respectively, the resulting pharmacology was consistent with that of a unique receptor subtype. β-Phenylethylamine (β-PEA), the putative endogenous ligand, gave an EC50 of 106 ± 5 nM in the assay. For a series of β-PEA analogs used to explore the pharmacophore, small substituents at ring positions 3 and/or 4 generally resulted in compounds having lower potency than β-PEA, although several were as potent as β-PEA. However, small substituents at ring position 2 resulted in a number of compounds having potencies as good as or better than β-PEA. A number of nonselective antagonists known to share affinity for multiple monoaminergic receptors were evaluated for their ability to inhibit β-PEA stimulation of the human TAAR1. None had an IC50 <10 μM. For comparison, the rat TAAR1 receptor was expressed in the AV12-664 cell line. A number of agonist compounds had significantly different relative potencies between the rat and human TAAR1, demonstrating a significant species difference between the rat and human TAAR1. The TAAR1 receptor exhibits a pharmacologic profile uniquely different from those of classic monoaminergic receptors, consistent with the structural information that places them in a distinct family of receptors. This unique pharmacologic profile suggests the potential for development of TAAR-selective agonists and antagonists to study their physiologic roles.
The trace amines are congeners of the so-called classic monoamine or biogenic amine neurotransmitters, e.g., dopamine, norepinephrine and serotonin, but are found in the brain in much lower concentrations (nanograms per gram or less) than the classic neurotransmitters (Baldessarini and Fischer, 1978; Philips et al., 1978). Compounds typically discussed under the category of trace amines include (but are not limited to) β-phenylethylamine, m- and p-tyramine, octopamine, and tryptamine. Hypotheses regarding the possible actions of the trace amines in normal physiology and disease states were published as early as the 1970s (Baldessarini and Fischer, 1978; Philips et al., 1978; Boulton, 1980). However, this field of study remained on the fringes of neurotransmitter research because of the lack of tools that would differentiate the actions of trace amines from those of other biogenic amine neurotransmitters. In 2001, cloning studies revealed the existence of a group of receptors described as the trace amine receptor family (Borowsky et al., 2001; Bunzow et al., 2001). These initial reports have been followed by several reviews and additional characterizations of these receptors (Branchek and Blackburn, 2003; Lindemann and Hoener, 2005; Lindemann et al., 2005; Lewin, 2006; Navarro et al., 2006). However, given the time since the first publications, there are surprisingly few actual data published on this receptor family. One reason for this lack of data seems to be that this family of receptors is difficult to express and characterize in recombinant systems. Nomenclature of the trace amine receptors has been somewhat haphazard, as various members of this family, including the putative neurotransmitter receptor (Zeng et al., 1998) and G protein-coupled receptors GPR57 and GPR58 (Lee et al., 2000), were named before it was realized that they were part of a larger related group of receptors described by Borowsky et al. (2001). Recently, Lindemann et al. (2005) proposed a uniform nomenclature, and we have adopted this as the form for use in this article.
With the successful expression of the human trace amine-associated receptor1 (TAAR1) (also known as TA1 in the literature) by our group, the primary goal of the present work was to explore the pharmacologic properties of this receptor for comparison with those of the classic biogenic amine receptors. These studies point to significant differences in the pharmacology of the human TAAR1 receptor compared with those of adrenergic, dopaminergic, and serotonergic receptors. With the recent successful expression of the rat TAAR1, we also provide some initial indications of species differences in the pharmacology of the TAAR1, including a differential in the potency of 3-iodothyronamine, which has previously been described as a potent agonist at the rat TAAR1 receptor (Scanlan et al., 2004).
Materials and Methods
Materials
d-Amphetamine, l-amphetamine, psilocin, and psilocybin were purchased from Alltech-Applied Science Labs (State College, PA). Lysergic acid diethylamide was purchased from Cerilliant Corporation (Round Rock, TX). S-(–)-Lisuride was purchased from MP Biomedicals (Solon, OH). 2-Hydroxy-β-PEA (o-tyramine), 2-methyl-β-PEA, 3-methyl-β-PEA, 2-bromo-β-PEA, 3-bromo-β-PEA, and 3-iodothyronamine were synthesized at the Lilly Research Laboratories (Indianapolis, IN). All other compounds were obtained from Sigma-Aldrich (St. Louis, MO).
Cloning of Human TAAR1 and Rat TAAR1 Full-Length cDNA from Genomic DNA by Use of a PCR Cloning Method
Human and rat genomic DNA was purchased from Clontech (Mountain View, CA). PCR primers were designed based on the hTAAR1 sequence from chromosome 6. The sense primer hTA1S CTCGAGCCACCATGATGCCCTTTTGCCACAATATA contained a Kosak sequence and an XhoI restriction enzyme site. The antisense primer hTA1AS AATGCGGCCGCCTATGAACTCAATTCCAAAAATAAT contained a NotI restriction enzyme site. For rat TAAR1 constructs; the sense primer rTA1S CCGTCTCGAGCCACCATGCATCTTTGCCACAATAGCGCGA also contained a Kosak sequence and an XhoI restriction enzyme site. The antisense primer rTA1AS ATAAGAATGCGGCCGCTTACAAAAATAACTTAGACCTAGA also contained a NotI restriction enzyme site. The same PCR conditions were used for the human and rat constructs. The PCR reactions were carried out in a total volume of 100 μl containing 1 unit of Pfu DNA polymerase (Promega, Madison, WI), 1 μl of 100 ng/μl genomic DNA, 10 μl of 10× Pfu PCR reaction buffer [300 mM Tris-SO4, pH 9.1, 10 mM MgSO4, and 90 mM (NH4)2SO4], 8 μl of 25 μM concentrations of dNTPs, and 4 μl containing 5 pmol of each oligonucleotide. The PCR reaction parameters were 98°C for 1 min for denaturation of genomic DNA and amplification for 20 cycles with each cycle being 95°C of denaturation at 20 s, annealing at 58°C for 30 s, and elongation at 68°C for 3 min, with a final elongation at 68°C for 5 min. PCR products were run on 1% TAE-agarose gels that were stained with ethidium bromide. A single band was excised from the gel, and the DNA was purified using a QIAquick gel extraction kit (QIAGEN, Valencia, CA). Purified DNA was digested with XhoI and NotI for 3 h followed by the use of a QIAquick DNA purification kit (QIAGEN). The human and rat PCR TAAR1 fragments were then cloned into hemagglutinin (HA) epitope-tagged pCMV mammalian expression vector (pCMV-HA; Clontech) according to the manufacturer's protocol. After transformation, single bacterial colonies were picked and grown overnight. Restriction enzyme XhoI and NotI digestions were carried out to confirm that the TAAR1 fragments were inserted into the pcDNA 3.1(–) vector. Clones with inserts were selected for plasmid DNA isolation using a mini-DNA preparation kit (QIAGEN) according to the manufacturer's protocols. Four hTAAR1 and rTAAR1 clones were sequenced for the confirmation of the DNA sequence.
Establishment of Human and Rat TAAR1 Stable Cell Lines
For stable cell clones, cells in six-well plates (6 × 105 cells/well) were cotransfected with 0.5 μg of pcDNA3.1/hygromycin vector (Invitrogen, Carlsbad, CA) and 2 μg of human or rat TAAR1 plasmid (pCMV-HA) using FuGENE 6 Transfection Reagent (Roche Diagnostics, Indianapolis, IN). Cells were then split into 100-mm tissue culture dishes 24 h post-transfection and selected with 200 μg/ml hygromycin (Invitrogen, Carlsbad, CA). At least 100 clones for each transfection were selected and expanded for determination of human TAAR1 function by measuring the accumulation of cAMP using an AlphaScreen cAMP assay kit (PerkinElmer Life and Analytical Sciences, Boston, MA). Stable cell lines coexpressing human TAAR1 and rat Gαs signaling protein were also established by transfection of human TAAR1 stable cell lines with rat Gαs cDNA in the pcDNA 3.1/neomycin vector. An AV12-664 cell line expressing the cloned rat Gαs protein was also established for background studies.
cAMP Formation and Detection
The rat TAAR1, stably expressed in AV12-664 cells (Syrian hamster fibroblast cell line, American Type Culture Collection No. CRL-9595), or the human TAAR1, stably expressed in rGαsAV12-664 cells (AV12-664 cells stably transformed with rat Gαs protein), was suspended in cAMP stimulation buffer (Hanks' balanced salt solution containing bovine serum albumin, pargyline, and HEPES with or without alprenolol and/or RX821002), and the suspension was then incubated at room temperature for 25 min. The cells were centrifuged at 200g for 5 min and resuspended in cAMP stimulation buffer with 3-isobutyl-1-methylxanthine. By using a Tecan Freedom EVO 200 platform (Tecan Schweiz AG, Männedorf, Switzerland), 10 μl of cell suspension was mixed and added to 5 μl of test compound, and the suspension was then incubated for 30 min. All incubations were at room temperature. For antagonist assays, 2 μl of β-PEA (500 nM final concentration) was added after a 30-min preincubation with the antagonist, and the suspension was incubated an additional 30 min. The concentrations of reagents in the cAMP formation incubation were 0.075% bovine serum albumin, 10 μM pargyline, 20 mM HEPES, 125 μM 3-isobutyl-1-methylxanthine, and, when used, 10 μM alprenolol and/or 3 μM RX821002. The HitHunter cAMP XS kit (DiscoveRx, Fremont, CA) was used for the determination of the amount of cAMP formed. The cAMP formation assay was terminated by addition of 10 μl of cAMP XS antibody/lysis mix. After a 1-h incubation, 10 μl of cAMP XS ED reagent was added, followed by an additional 1-h incubation; then 20 μl of cAMP XS EA/CL substrate mix was added, and the plates were covered with sealing tape. The plates were allowed to stand for 1 h and then were centrifuged at 200g for 5 min. The plates were incubated for an additional 14 h in the dark, followed by counting using a PerkinElmer 1450 MicroBeta TriLux counter (PerkinElmer Life and Analytical Sciences) in luminescence mode.
Data Analysis
For all compounds, the luminescence counts per second were converted to amount of cAMP formed by interpolating from the cAMP standard curve, which had been fit with nonlinear regression (GraphPad Prism; GraphPad Software Inc., San Diego CA). The amount of cAMP formed by a β-PEA concentration-response curve, run as a standard on each assay plate, was fit to a four-parameter logistic equation by nonlinear regression analysis. Individual compound data were then transformed to the percentage of maximal response to β-PEA and fit with nonlinear regression to obtain the EC50 and Emax for each test compound.
Results
Blockade of α2- and β-Adrenoceptors
Initially in the course of these studies, a number of the compounds reported to have activity at the cloned rat TAAR1 receptor expressed in HEK-293 cells (Bunzow et al., 2001) were investigated for activity at the cloned human TAAR1. Among these was the β-adrenoceptor agonist fenoterol. Fenoterol (EC50 = 660 nM, data not shown), (–)-isoproterenol, and (–)-norepinephrine were found to have activity in rGαsAV12-664 cells expressing the cloned human TAAR1 (Fig. 1A). However, because of the high potency of (–)-isoproterenol (EC50 = 2.42 ± 0.4 nM), an endogenous β-adrenoceptor was suspected as being responsible for its activity. In addition, the biphasic nature of the (–)-norepinephrine curve (Fig. 1A) suggested that multiple pharmacologies were present in this response, with the stimulation being activation of a β-adrenoceptor positively coupled to adenylyl cyclase and the inhibition being activation of an α2-adrenoceptor negatively coupled to adenylyl cyclase. AV12-664 cells were known to endogenously express an α2-adrenoceptor (Wainscott et al., 1998).

Stimulation of cAMP formation in human TAAR1-expressing rGαsAV12-664 cells in the absence of adrenergic α2- and β-adrenoceptor antagonists (A), in the presence of 3 μM of the α2-adrenoceptor antagonist RX821002 (B), in the presence of 10 μM of the β-adrenoceptor antagonist alprenolol (C), and in the presence of 3 μM RX821002 and 10 μM alprenolol (D). All curves are the means ± SE of at least three separate experiments.
To test the contribution of α- and/or β-adrenoceptors to the cAMP response in human TAAR1-expressing rGαsAV12-664 cells, the α-adrenoceptor antagonist RX821002 at 3 μM (Fig. 1B), the β-adrenoceptor antagonist alprenolol at 10 μM, (Fig. 1C), or 3 μM RX821002 and 10 μM alprenolol (Fig. 1D) were preincubated with the cells for 30 min before testing agonists and were included in the cAMP formation assay. In the absence of adrenergic antagonists (Fig. 1A), β-PEA stimulated cAMP formation with an EC50 of 147 ± 15 nM. The cAMP response to β-PEA (EC50 = 130 ± 13 nM) or (–)-isoproterenol (EC50 = 3.02 ± 0.56 nM) was not affected by 3 μM RX821002. However, RX821002 did unmask the β-adrenoceptor agonist properties of (–)-norepinephrine, changing the bell-shaped dose-response curve to a simple sigmoidally shaped curve (EC50 = 24.6 ± 4.0 nM; Fig. 1B). Alprenolol, 10 μM, also did not affect the β-PEA response (EC50 = 129 ± 15 nM) but caused a marked 1900-fold rightward shift of the (–)-isoproterenol response (EC50 = 4610 ± 1800 nM) and virtually eliminated the response to (–)-norepinephrine (Fig. 1C). The dose shift produced by this single concentration of alprenolol was used to estimate the Kb according to the modification of the Schild equation described by Limbird (1986): Kb = [antagonist]/([A′]/[A] – 1). This gave a Kb = 7.21 ± 2.77 nM for alprenolol inhibition of (–)-isoproterenol cAMP formation at the endogenous β-adrenoceptor. The combination of 3 μM RX821002 and 10 μM alprenolol again did not affect the β-PEA response (EC50 = 142 ± 18 nM) (Fig. 1D) and resulted in the same rightward shift of the (–)-isoproterenol curve (EC50 = 3540 ± 780 nM) as seen with alprenolol alone. However, blockade of the α2-adrenoceptor with RX821002 unmasked a 840-fold rightward shift of the (–)-norepinephrine response (EC50 = 20,700 ± 3900 nM) by alprenolol, resulting in a Kb calculation for alprenolol of 12.0 ± 0.5 nM in the rGαsAV12 cell line expressing the cloned human TAAR1. The in vitro potencies of (–)-isoproterenol and (–)-norepinephrine and the affinity of alprenolol are in agreement with these compounds interacting with an endogenous β-adrenoceptor (Hoffman et al., 2004). Note that alprenolol with or without RX821002 virtually eliminated the response to fenoterol seen in the absence of the adrenoceptor antagonists (data not shown). Because 3 μM RX821002 and 10 μM alprenolol did not affect the β-PEA response, these compounds were used to block any potential interfering α2-or β-adrenoceptor activity in human TAAR1-expressing rGαsAV12-664 cells.

Stimulation of cAMP formation in AV12-664 cells expressing the cloned rat TAAR1 in the absence of adrenergic α2- and β-adrenoceptor antagonists (A), in the presence of a 3 μM concentration of the α2-adrenoceptor antagonist RX821002 (B), in the presence of a 10 μM concentration of the β-adrenoceptor antagonist alprenolol (C), and in the presence of 3 μM RX821002 and 10 μM alprenolol (D). All curves are the means ± SE of at least three separate experiments.
Once it was determined that α2- and β-adrenoceptors could be blocked with RX821002 and alprenolol, the same studies were conducted for rat TAAR1-expressing AV12-664 cells. In the absence of RX821002 and alprenolol, (–)-isoproterenol was again a potent stimulator of cAMP formation (EC50 = 14.6 ± 0.1 nM) and (–)-norepinephrine gave a biphasic response (Fig. 2A). The EC50 for β-PEA was 187 ± 10 nM. When the assay was performed in the presence of 3 μM RX821002 (Fig. 2B), the β-adrenoceptor activity of (–)-norepinephrine was unmasked (EC50 = 130 ± 14 nM). However, the β-PEA curve was unexpectedly shifted to the right (EC50 = 635 ± 45 nM). The EC50 for (–)-isoproterenol (17.8 ± 0.6 nM) was unaffected. Inclusion of 10 μM alprenolol (Fig. 2C) or 3 μM RX821002 and 10 μM alprenolol (Fig. 2D) resulted in a strong rightward shift of the β-PEA response (EC50 = 6090 ± 2750 or 9320 ± 5210 nM, respectively). The responses to (–)-isoproterenol and (–)-norepinephrine were virtually eliminated by the inclusion of 10 μM alprenolol (Fig. 2C) or 3 μM RX821002 and 10 μM alprenolol (Fig. 2D). The Kb for alprenolol at the endogenous β-adrenoceptor in the cell line expressing the rat TAAR1 could not be calculated under the conditions used because the 10 μM concentration of alprenolol completely blocked the isoproterenol response at the concentration tested. The rightward shifts in the β-PEA concentration-response curve produced by RX821002 and alprenolol suggested that the rat TAAR1 had some sensitivity to these adrenergic antagonists. Because the human TAAR1 did not show this sensitivity, a species difference in the pharmacology of the TAAR1 was suggested.

Endogenous monoamine stimulation of cAMP formation in human TAAR1-expressing rGαsAV12-664 cells in the presence of 3 μM RX821002 and 10 μM alprenolol. All curves are the means ± the SE of at least three separate experiments.
Human TAAR1 Agonist Characterization
Examination of putative endogenous trace amines revealed a unique profile at the human TAAR1 receptor. β-PEA showed the highest potency among these compounds, with p-tyramine having the next highest potency (Fig. 3; Table 1). Standard ligands that interact with 5-hydroxytryptamine, noradrenergic, or dopaminergic receptors showed only low potency at the human TAAR1 receptor.
EC50 and Emax values for endogenous monoamine stimulation of cAMP formation in human TAAR1-expressing rGαsAV12-664 cells in the presence of 3 μM RX821002 and 10 μM alprenolol
Examination of a series of simple phenylethylamines revealed that halogen substitution in the 2-position resulted in compounds having greater potency than β-PEA with 2-chloro-β-PEA being ∼3 times more potent than β-PEA (Table 2). For most series, substitutions in the 3- and 4-positions typically reduced potency compared with the 2-substituted compounds. Interestingly, for the hydroxyl-substituted compounds, the 4-position was better tolerated than either the 2- or 3-position. For the two series for which a 2,6-substituted compound was available, the 2,6-substituted compound was nearly as well tolerated as the respective 2 substitution in that series.
EC50 and Emax values for substituted β-phenylethylamine stimulation of cAMP formation in human TAAR1-expressing rGαsAV12-664 cells in the presence of 3 μM RX821002 and 10 μM alprenolol
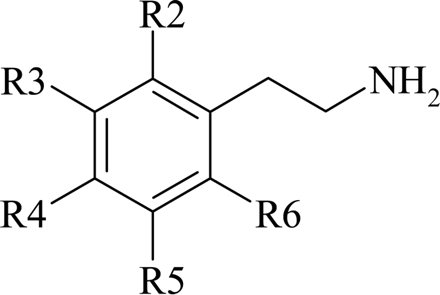
Substitution on the ethylamine side chain produced a variety of effects on potency at the human TAAR1, depending on the nature of the substituent. For example, a β-methyl substituent was well tolerated, being as potent as β-PEA itself (Table 3). However, changing that substitution to a β-hydroxy resulted in a 10-fold reduction in potency (Table 3). In contrast with the effect of a β-methyl substituent, α-methyl-substituted compounds, i.e., both d- and l-amphetamine, had reduced potency compared with β-PEA (Table 4). Interestingly, the d-isomer of amphetamine was more active at the human TAAR1 than the l-isomer, as is the case for amphetamine-induced adrenergic stimulation. The monoamine oxidase inhibitor tranylcypromine, which can be considered as a bridged α/β-methyl substitution, was ∼20-fold less potent than β-PEA (Table 3). Simple monomethylation of the amino function of β-PEA reduced potency by a factor of ∼2-fold (Table 4). However, N,N-dimethylation of β-PEA resulted in a 10-fold reduction in potency (Table 4).
EC50 and Emax values for β-carbon-substituted β-phenylethylamine stimulation of cAMP formation in human TAAR1-expressing rGαsAV12-664 cells in the presence of 3 μM RX821002 and 10 μM alprenolol
EC50 and Emax values for α-carbon-substituted and N-substituted β-phenylethylamine stimulation of cAMP formation in human TAAR1-expressing rGαsAV12-664 cells in the presence of 3 μM RX821002 and 10 μM alprenolol

Monoaminergic antagonist inhibition of 500 nM β-PEA-stimulated cAMP formation in human TAAR1-expressing rGαsAV12-664 cells in the presence of 3 μM RX821002 and 10 μM alprenolol. All curves are the means ± SE of at least three separate experiments. These represent the antagonists from Table 5 that produced significant inhibition of β-PEA stimulation of the human TAAR1 receptor.
A number of nonselective monoaminergic compounds were examined for agonist and/or antagonist activity. None of these compounds was found to have high or even moderate affinity for the cloned human TAAR1 (Table 5). The most potent IC50 values for the inhibition of β-PEA-stimulated cAMP formation were in the range of 10 to 30 μM (Fig. 4). Most compounds produced no significant inhibition up to 10 μM.
Examples of nonselective monoaminergic compounds having no significant interaction with the human TAAR1 receptor
Human Versus Rat TAAR1
Subsequent to the pharmacologic characterization of the human TAAR1 receptor, we identified a stably transfected cell line with the rat TAAR1. Initial characterization of this receptor revealed that, unlike the human TAAR1, the rat form of the receptor was inhibited by the high concentrations of alprenolol and RX821002 used to block the α2- and β-adrenergic receptors endogenously expressed by the AV12-664 cell line (Fig. 2). This inhibition was manifested by a rightward shift in the dose-response curve of β-PEA in the presence of these adrenergic antagonists. Thus, neither alprenolol nor RX821002 could be used in the assay to block the endogenous α- and β-adrenergic receptors in the cell line to characterize the rat TAAR1. Likewise, use of agonists to characterize the rat TAAR1 was limited to those compounds that were shown not to have significant interactions with the α- and β-adrenergic receptors endogenously expressed by the AV12-664 cell line.
Based on the above criteria, a number of agonists were selected for comparison of the rat and human TAAR1 pharmacology (Table 6). Some compounds, including p-tyramine, dopamine, (±)-octopamine, 4-chloro-β-PEA, 3-iodothyronamine, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, had notably higher potency for the rat versus the human TAAR1. For dopamine, the efficacy relative to β-PEA was decreased in the rat compared with the human. Some compounds did appear to have slightly higher potency in the human versus the rat TAAR1, including β-PEA and betahistine. However, more attention should be paid to the relative potencies among compounds, rather than absolute potencies, because we do not know the expression densities of the human and rat TAAR1 and because the backgrounds of the host cells are not exactly the same for the two recombinant systems. The rat TAAR1 also appeared to differ in its response to the pattern of small substituents on the phenyl ring. Whereas the human form clearly showed that substitution at C4 dramatically reduced potency relative to substitution at C2 (Table 2), this effect was much less pronounced on the basis of the comparison of 2-chloro-β-PEA to 4-chloro-β-PEA (Table 6). Other examples of rat/human differences in the effects of substituents at the C4 position were seen. For example, (±)-octopamine was ∼3.6-fold less potent than β-PEA at the rat receptor but 71-fold less potent than β-PEA at the human receptor. 3-Iodothyronamine was actually more potent than β-PEA at the rat TAAR1, but 14-fold less potent than β-PEA at the human receptor.
EC50 and Emax values for stimulation of cAMP formation in human TAAR1-expressing rGαsAV12-664 cells versus rat TAAR1 expressing AV12-664 cells
Species differences were also seen with amphetamine. For example, the two enantiomers of amphetamine have similar efficacy at the human TAAR1, whereas l-amphetamine had decreased efficacy relative to d-amphetamine at the rat TAAR1.
Discussion
Examination of the human TAAR1 in the present work has revealed a pharmacologic profile that clearly distinguishes it from the so-called classic monoamine (dopamine, norepinephrine, serotonin, and histamine) receptor subtypes. However, getting to that conclusion required the use of pharmacologic masks of adrenergic receptors that were endogenously expressed by the cell line (AV12-664) chosen as the host for expression of TAAR1. Previous work in our laboratory had shown radioligand binding to α2-adrenoceptors in AV12-664 cells (Wainscott et al., 1998). Thus, the bell-shaped curve for cAMP stimulation that was produced by (–)-norepinephrine in the present work (Fig. 1A) suggested the possibility that the inhibitory phase of the curve could be due to stimulation of α2-adrenoceptors, which are known to inhibit adenylyl cyclase through coupling to Gi (Bylund et al., 1994). Consistent with this hypothesis, the potent α2-adrenoceptor antagonist RX821002 (Audinot et al., 2002) blocked the inhibitory phase of the norepinephrine curve, resulting in a monophasic stimulatory curve due to β-adrenoceptor stimulation (Fig. 1B).
The α2- and β-adrenoceptor antagonists had no effect on the response to β-PEA in human TAAR1-expressing rGαsAV12-664 cells. In addition, β-PEA had no activity in the control cell line rGαsAV12-664 not transfected with TAAR1. These data demonstrate that RX821002 and alprenolol effectively block the adrenergic responses in this cell line and that the response to β-PEA is clearly through the cloned human TAAR1. In addition, they demonstrate that RX821002 and alprenolol have little affinity for the cloned human TAAR1. Therefore, all subsequent experiments for the human TAAR1 receptor were performed in the presence of RX821002 and alprenolol.
There was a very different result in AV12-664 cells expressing the cloned rat TAAR1. In addition to blocking the α2-adrenoceptor response to (–)-norepinephrine, RX821002 shifted the β-PEA concentration-response curve 3-fold, as seen in Fig. 2B compared with Fig. 2A. Even more dramatic was the effect of the β-adrenoceptor antagonist alprenolol on the β-PEA concentration-response curve. Alprenolol shifted the β-PEA curve 30-fold to the right. By using the equation Kb = [antagonist]/([A′]/[A] – 1), the Kb values for RX821002 and alprenolol at the cloned rat TAAR1 would be 1310 ± 240 and 450 ± 156 nM, respectively. To determine the potency of compounds at the rat TAAR1, because RX821002 and alprenolol could not be used, the compounds were either 1) shown to not be affected by alprenolol and RX821002 in the human TAAR1-expressing rGαsAV12-664 cell line and therefore to have no affinity for the endogenous adrenoceptors or 2) shown not to be active in untransformed AV12-664 cells. These compounds were then presumed to be acting via the TAAR1 receptor.
Once the α2- and β-adrenoceptors were blocked with RX821002 and alprenolol, respectively, the human TAAR1 receptor displayed a pharmacology that was distinct from that of the other monoaminergic receptors (Fig. 3; Table 1). The highest potency of the tested endogenous monoamines was for β-PEA, followed by p-tyramine. These studies show β-PEA to be ∼4-fold more potent than p-tyramine. This result is in contrast to Borowsky et al. (2001), in which the two compounds were equally potent at the human TAAR1 transiently expressed in COS-7 cells. Lindemann et al. (2005) found β-PEA (EC50 = 300 nM) to be more potent than p-tyramine (EC50 = 1070 nM), although the receptor used in those studies was the human TAAR1 modified with rat sequences, including the G-loop. Navarro et al. (2006) have reported that β-PEA (EC50 = 160 nM) is more potent than p-tyramine (EC50 = 570 nM) against unmodified human TAAR1 expressed in Chinese hamster ovary (CHO-K1) cells.
The starting point for exploring structure-activity relationships at TAAR1 was β-PEA, the most potent of the endogenous amines tested. Because the phenylethylamine backbone is a common motif in compounds that interact with various adrenergic and serotonergic receptors, there was a wealth of historical information about how modifications of β-PEA have produced alterations in interactions with the classic monoamines. Thus, one logical starting point was to look at the effects of different substitution patterns on the phenyl ring, e.g., changing size, electronegativity, and the ability to act as hydrogen donors or hydrogen acceptors of substituents as they were systematically walked around the ring. Another logical region to explore was the ethylamine side chain and how substitutions on either the α-or β-carbons or the amino group affected potency and efficacy. Several series of substituted phenylethylamines were investigated for activity at the human TAAR1 (Table 2). A surprising finding was the potency of phenylethylamines with substituents at the phenyl C2 position relative to their respective C4-substituted congeners. In each case, except for the hydroxyl substituent, the C2-substituted compound had 8- to 27-fold higher potency than the C4-substituted compound. The C3-substituted compound in each homologous series was typically 2- to 5-fold less potent than the 2-substituted compound, except for the hydroxyl substituent. The most potent of the 2-substituted phenylethylamines was 2-chloro-β-PEA, followed by 2-fluoro-β-PEA, 2-bromo-β-PEA, 2-methoxy-β-PEA, 2-methyl-β-PEA, and then 2-hydroxy-β-PEA.
The effect of β-carbon substitution on the phenylethylamine side chain was also investigated (Table 3). A β-methyl substituent was well tolerated compared with β-PEA. In fact, S-(–)-β-methyl-β-PEA was as potent as β-PEA at human TAAR1. β-Hydroxyl substitution was, however, not tolerated compared with β-PEA. In both cases of β-substitution, enantiomeric selectivity was demonstrated.
In contrast to a methyl substitution on the β-carbon, an α-methyl substitution reduced potency by ∼10-fold for d-amphetamine and 16-fold for l-amphetamine relative to β-PEA (Table 4). N-Methyl substitution was fairly well tolerated; however, N,N-dimethyl substitution was not.
A number of nonselective monoaminergic compounds were tested for agonist and/or antagonist activity at the human TAAR1. This set of compounds included molecules with known multiple receptor cross-reactivities, including serotonergic, dopaminergic, adrenergic, and histaminergic interactions. None of the compounds had an EC50 or IC50 < 10 μM at the human TAAR1 receptor (Table 5). These results illustrate the unique structural requirements for activity at TAAR1 and suggest the possibility of development of TAAR1 receptor-selective agonists and antagonists.
A comparison of the rat and human forms of TAAR1 demonstrated some species differences in pharmacology (Table 6). A number of the compounds showed very similar potencies between the rat and human forms of the receptor, e.g., β-PEA, 2-chloro-β-PEA, and 2,6-dichloro-β-PEA (suggesting that ortho substitution on the phenyl ring is advantageous in the rat as it is at the human form). However, compounds having a simple substitution at position C4 tended to show striking differences between the two species. For example, p-tyramine and 4-chloro-β-PEA both have lower potency at the human receptor than β-PEA, whereas both compounds have greater potency than β-PEA at the rat receptor. Bunzow et al. (2001) also found p-tyramine to be more potent than β-PEA at N-terminal-tagged rat TAAR1 expressed in HEK-293 cells as did Lindemann et al. (2005), for rat TAAR 1 expressed in HEK-293 cells. (±)-Octopamine, which has hydroxyls at both the C4 position and on the β-carbon of the side chain, has very low potency at the human receptor but is ∼10-fold more potent at the rat receptor. The most extreme example of this ring C4 substituent effect was the differential potency of 3-iodothyronamine. This compound was previously reported to be very potent at the rat TAAR1, with an EC50 of 14 nM (Scanlan et al., 2004). Although the present study confirms the potency of 3-iodothyronamine at the rat TAAR1, it demonstrates that this compound has 67-fold lower potency at the human TAAR1 receptor.
In summary, the present work shows that the human TAAR1 has a unique pharmacology compared with the so-called classic monoamine receptors for norepinephrine, dopamine, histamine, or serotonin. In particular, it is interesting that no high-affinity or even moderate-affinity antagonists for the human TAAR1 were identified from a group of notoriously nonselective monoaminergic antagonists. After submission of this article, a description of the TAARs present in mouse olfactory epithelium was published (Liberles and Buck, 2006), showing that quite different volatile amines activated different members of the TAAR family via cAMP stimulation. In those studies, only human TAAR1 and mouse TAAR4 were activated by β-PEA, illustrating the unique pharmacologic requirements for activation among the TAARs. Taken together, these data suggest the possibility of development of agonists and antagonists that are selective for trace amine receptors and that could be used to elucidate the physiologic roles of these receptors.
Acknowledgments
The authors thank Jennifer Oler for assistance and expertise in automating the cAMP assay using the Tecan Freedom EVO 200 platform.
Footnotes
-
This work was funded by Eli Lilly and Company.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.112532.
-
ABBREVIATIONS: TAAR1, trace amine-associated receptor 1; β-PEA, β-phenylethylamine; PCR, polymerase chain reaction; h, human; HA, hemagglutinin; r, rat; RX821002, 2-methoxyidazoxan, 2-[2-(2-methoxy-1,4-benzodioxanyl)]-imidazoline hydrochloride; Emax, maximal response of the compound relative to the maximal response produced by β-PEA.
- Received August 21, 2006.
- Accepted October 11, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}