


Abstract
Use-dependent N-methyl-d-aspartate receptor (NMDAR) antagonists produce behaviors in human volunteers that resemble schizophrenia and exacerbate those behaviors in schizophrenic patients, suggesting that hypofunction of NMDAR-mediated neuronal circuitry may be involved in the etiology of clinical schizophrenia. Activation of the metabotropic glutamate receptor subtype 5 (mGluR5) enhances NMDAR-mediated currents in vitro. Thus, activation of mGluR5 could potentiate hypofunctional NMDARs in neuronal circuitry relevant to schizophrenia. To further elucidate the role of mGluR5, the present study examined the effects of mGluR5 antagonist administration, with and without coadministration of the use-dependent NMDAR antagonist phencyclidine (PCP), on locomotor activity and prepulse inhibition (PPI) of the acoustic startle response in rodents. We further examined PPI in mGluR5 knockout mice. Finally, we examined PPI after administration of the mGluR5 agonist 2-chloro-5-hydroxyphenylglycine (CHPG) alone and in combination with amphetamine. The data indicate that the mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine has no effect on locomotor activity or PPI by itself but does potentiate both PCP-induced locomotor activity and disruption of PPI. We further found that mGluR5 knockout mice display consistent deficits in PPI relative to their wild-type controls. Finally, the data indicate that CHPG has no effect on PPI by itself, but ameliorates amphetamine-induced disruption of PPI. Collectively, these data suggest that mGlu5 receptors play a modulatory role on rodent PPI and locomotor behaviors and are consistent with the hypothesis that mGlu5 agonist/potentiators may represent a novel approach for antipsychotic drug development.
Use-dependent N-methyl-d-aspartate receptor (NMDAR) antagonists, such as phencyclidine (PCP) and ketamine, induce psychotic states in normal human volunteers and exacerbate existing symptomatology in schizophrenic patients. Because NMDAR antagonist administration produces cognitive deficits and positive and negative symptoms reminiscent of those observed in schizophrenia, it more accurately reflects the schizophrenic state than administration of dopamine agonists (Javitt and Zukin, 1991). This observation has led to the hypothesis that NMDAR hypofunction may be critically involved in the etiology and/or underlying symptoms of schizophrenia (Olney et al., 1999). In support of this hypothesis, enhancement of NMDAR density has been reported in a variety of brain regions of schizophrenic patients (Ishimaru et al., 1992, 1994). Furthermore, enhancement of NMDAR function through administration of agonists at the allosteric glycine binding site results in a significant symptomatic improvement in schizophrenic patients (Javitt et al., 1994; Goff et al., 1995; Heresco-Levy et al., 1996, 1999, 2002; Tsai et al., 1998).
Metabotropic glutamate receptors (mGluR) are seven-transmembrane G protein-coupled receptors. There are currently eight known mGluR subtypes divided into three groups based on their homology, pharmacology, and second messenger coupling. Group I mGluRs (mGluR1 and mGluR5) couple to Gq and stimulate phosphoinositide hydrolysis (for review, see Conn and Pin, 1997). Clinical studies suggest that mGluR5 allele frequency is associated with schizophrenia among certain cohorts (Devon et al., 2001) and that a modest, yet significant, increase in mGluR5 message is found in cortical pyramidal cell layers of schizophrenic brains relative to controls (Ohnuma et al., 1998).
Preclinically, activation of group I mGluRs potentiates NMDAR-mediated function in a variety of brain regions and this activity is often mediated by mGluR5 (Awad et al., 2000; Mannaioni et al., 2001; Pisani et al., 2001; Benquet et al., 2002). Among mGluRs, this effect seems to be specific for group I mGluRs in that activation of group II or III mGluRs has no effect on NMDAR-mediated currents in vitro nor do group I agonists have any affect on α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor-mediated currents (Pisani et al., 1997). These clinical and preclinical findings have led investigators to hypothesize that activation of mGluR5 may normalize hypofunctional NMDAR-mediated activity and thereby represent a useful approach for the development of novel antipsychotic drug treatments (Chavez-Noriega et al., 2002; Marino and Conn, 2002).
Prepulse inhibition (PPI) of the acoustic startle response is an easily measured, quantifiable behavior that is disrupted in animals after the administration of dopamine agonists or NMDAR antagonists (for review, see Geyer et al., 2001). Because PPI is deficient in schizophrenic patients, this assay has been proposed to model the sensorimotor gating deficits observed in schizophrenic patients (Braff and Geyer, 1990). A recent report suggests that administration of the mGluR5 selective use-dependent antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP) can potentiate PCP-induced deficits in PPI in rats (Henry et al., 2002) at doses of MPEP that have no effect alone (Spooren et al., 2000; Henry et al., 2002). Furthermore, preliminary reports suggested that mGluR5 knockout mice may be deficient in PPI (Geyer et al., 2000). More recent reports, however, suggest that this may not be a reliable finding (O'Meara et al., 2002).
To more thoroughly describe the role of mGlu5 receptors in rodent behavior, the current study examined 1) the role of the mGluR5 antagonist MPEP in multiple schizophrenia-related animal models after acute PCP treatment; 2) mGluR5 knockout mice in a PPI assay; and 3) the effect of the selective mGluR5 agonist 2-chloro-5-hydroxyphenylglycine (CHPG) on PPI under normal conditions and after amphetamine-induced disruption of PPI.
Materials and Methods
Subjects. Male Sprague-Dawley rats (Harlan, San Diego, CA and Taconic Farms, Germantown, NY), 129S6/SvEvTac mice (Taconic Farms), mice homozygous for a Gprc1etm1Rod targeted mutation (mGluR5–/–), and wild-type controls (mGluR5+/+, stock 003121; The Jackson Laboratory, Bar Harbor, ME) were used in the present studies. Mice were housed on a reverse 12-h light/dark schedule (lights off at 6:00 AM) for prepulse inhibition studies; otherwise animals were housed on a normal light/dark schedule (lights on at 6:00 AM). Rats receiving i.c.v. administration of CHPG were commercially purchased (Taconic Farms) with a 22-gauge guide cannula implanted such that subsequent placement of the injection cannula allowed for infusion into the third ventricle. All animals were allowed access to food and water ad libitum before testing. Animals were housed and tested in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility in strict compliance with all applicable regulations.
Locomotor Activity. Activity was assessed as mean distance traveled (in centimeters) in standard 16 × 16 photocell testing chambers measuring 43.2 cm (length) × 43.2 cm (width) × 30.5 cm (height) (MED Associates, St. Albans, VT). Animals were habituated to individual activity chambers for at least 60 min before PCP administration. After administration of PCP or vehicle, activity was recorded for a 3-h time period. Data are expressed as the mean ± S.E.M. distance traveled recorded in 10-min intervals over the test period. The data were analyzed using repeated measures analysis of variance followed by post hoc testing using Tukey's honestly significant difference test, when appropriate. A difference was considered significant when p ≤ 0.05.
Prepulse Inhibition. SR-LAB (San Diego Instruments, San Diego, CA) acoustic startle chambers were used in the present studies. SR-LAB software controlled the delivery of all stimuli to the animals and recorded the response. Startle amplitude was measured as the mean value during a 65-ms period beginning at the onset of the startle-eliciting stimulus for mouse studies and during a 100-ms period for rat studies. Before the first session of any day the chambers were calibrated for both movement, using equipment provided by SR-LABS, and for sound levels, using a Tandy brand sound level meter. In each session, animals were randomly assigned to an experimental group, received their appropriate treatment, and were placed in the chambers. Animals were given a 5-min acclimation period during which a 65-db background noise was continuously present. This background noise remained present throughout the entire testing session. After the acclimation period, animals received a series of five 40-ms, 118- to 120-db bursts of white noise to partially habituate the animals to the startle-eliciting stimulus (Davis, 1988). After these five presentations, the test session, which consisted of 10 repetitions of trials, began. For mouse PCP/MPEP combination studies, six different trial types were presented during the session. These consisted of the following: a 10-ms prepulse at 75 or 80 db followed 100 ms later by the 118- to 120-db, 40-ms startle pulse (prepulse pulse conditions), the startle pulse alone (pulse alone), a period where no stimulus was presented (nostim), and the 10-ms, 75- or 80-db prepulse (i.e., 10 or 15 db above background) presented by itself (prepulse alone). Rat PCP/MPEP combination studies were performed in the same manner; however, a 70-db prepulse intensity (i.e., 5 db above background) was added to the test for this study. The mGluR5 knockout mouse studies and the rat i.c.v. CHPG study were also performed similarly; however, the trial types consisted of a 10-ms prepulse at 70, 75, 80, or 85 db (i.e., 5, 10, 15, or 20 db above background) followed 100 ms later by the 118- to 120-db, 40-ms startle pulse, a pulse alone condition, and a nostim condition. Initial and pilot studies had determined that these prepulse intensities were insufficient to induce a significant startle response independent of the startle stimulus. The stimuli were presented in random order with interstimulus intervals averaging 15 s. Levels of prepulse inhibition were determined by the formula (100 – ((prepulse pulse/pulse alone) × 100)) and expressed as percentage of prepulse inhibition ± S.E.M. Data were analyzed using repeated measures analyses of variance with the prepulse condition as the within group factor followed by analyses of simple main effects and, when appropriate, post hoc analysis using the Dunnett or Tukey test.
Drugs. PCP hydrochloride and d-amphetamine sulfate were obtained from Sigma-Aldrich (St. Louis, MO), dissolved in sterile saline, and injected at a volume of 1 ml/kg s.c. MPEP and CHPG were obtained from Tocris Cookson, Inc. (Ellisville, MO). MPEP was dissolved in sterile water or a 10%:90% Tween 80/water vehicle and injected at a volume of 1 ml/kg i.p. CHPG was dissolved in 0.5 N NaOH (pH adjusted to 7.0 with 1 N HCl) and infused into the third ventricle at a rate of 2 μl/min. For PPI experiments, MPEP was delivered 30 or 70 min pretest for mouse and rat studies, respectively. PCP was delivered 20 or 10 min pretest for mouse and rat, respectively. CHPG and amphetamine were delivered 15 and 20 min pretest, respectively. For locomotor studies, MPEP was delivered 60 min before PCP administration. Where appropriate, drug doses were determined as the base.
Results
Locomotor Activity
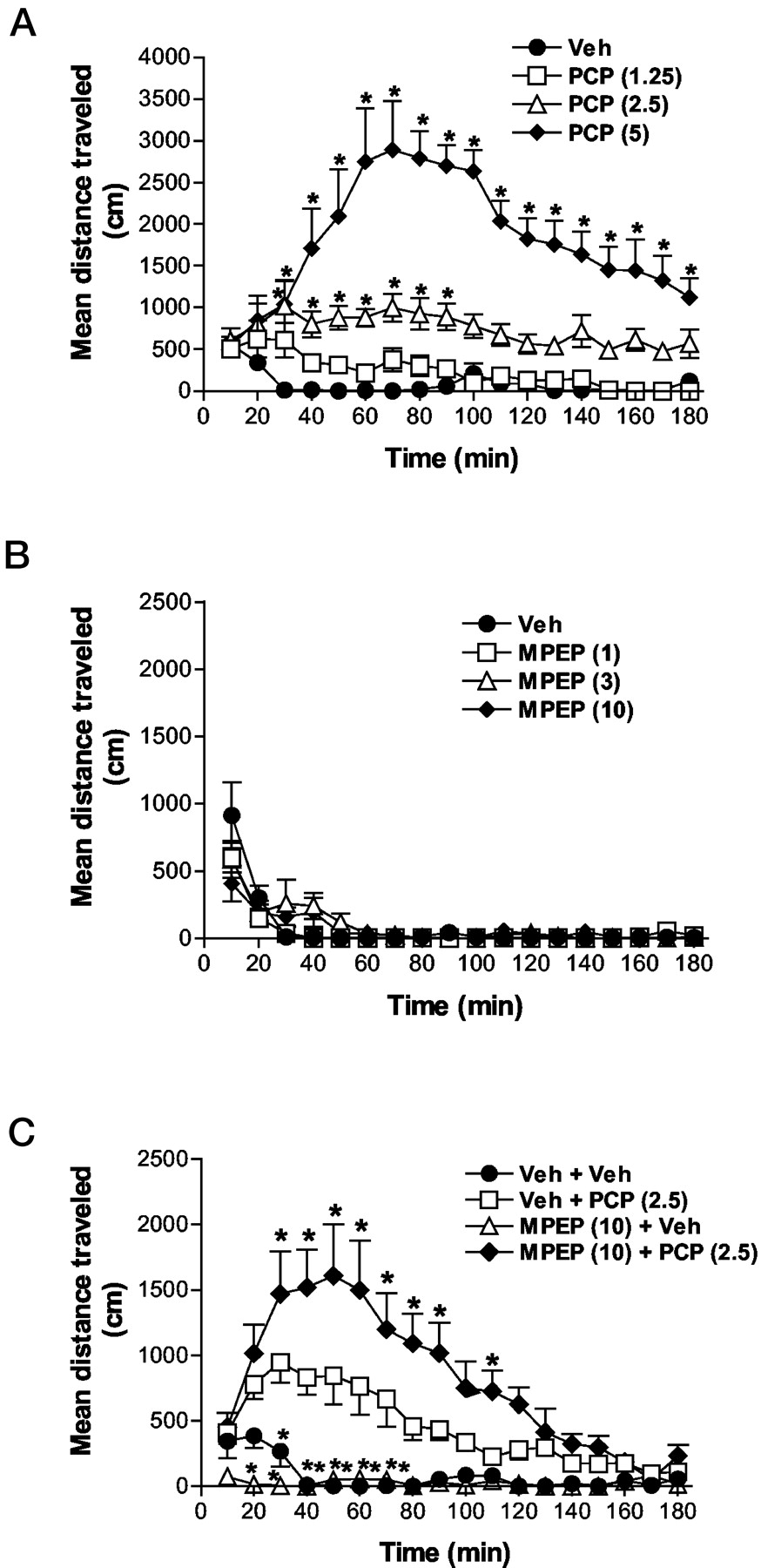
Systemic administration of PCP (1.25–5 mg/kg s.c.) produced a dose-dependent increase in locomotor activity in habituated rats in a time- and dose-dependent manner [F(3,28) = 35.12, p < 0.001; Fig. 1A]. In contrast, MPEP (1–10 mg/kg i.p.) had no effect on ambulatory activity in rats at any dose tested (Fig. 1B). However, when 10 mg/kg i.p. MPEP was administered 60 min before 2.5 mg/kg s.c. PCP a significant potentiation of PCP-induced locomotor activity was observed (Fig. 1C). Repeated analyses of variance confirmed a significant effect of treatment [F(3,36) = 16.53, p < 0.001], time [F(17,612) = 17.92, p < 0.001], and time by treatment interaction [F(51,612) = 7.03, p < 0.001]. Post hoc testing further confirmed significantly less activity in MPEP- and vehicle-treated animals relative to PCP-treated animals. In contrast, MPEP/PCP combination treatment enhanced activity relative to PCP alone treatment across a wide time range (Fig. 1C).

Effect of PCP (0–5 mg/kg s.c.; n = 8/group; A), MPEP (0–10 mg/kg i.p.; n = 6/group; B), or MPEP (10 mg/kg i.p. or vehicle) + PCP (2.5 mg/kg s.c. or vehicle) treatment (n = 10/group; C) in male Sprague-Dawley rats. All animals were habituated to the testing chambers a minimum of 60 min before PCP or vehicle administration. MPEP or vehicle was injected 60 min before PCP administration, which occurred at time 0. Asterisks represent a significant difference from vehicle (A) or vehicle + PCP treatment (C), *, p < 0.05. Error bars represent the S.E.M. Veh, vehicle treatment.
Prepulse Inhibition
MPEP/PCP Administration. The effect of PCP and MPEP alone, or in combination, on PPI was tested in 129S6 mice at two prepulse intensities (5 and 10 db above background). Figure 2A depicts the effect of vehicle, PCP alone (1–10 mg/kg s.c.), MPEP alone (3–30 mg/kg i.p.), and 10 mg/kg s.c. PCP combined with 30 mg/kg i.p. MPEP. Under these conditions, PCP, at doses up to 10 mg/kg s.c., did not significantly disrupt PPI at either prepulse level. MPEP, up to 30 mg/kg i.p., also had no effect on PPI by itself. When 30 mg/kg MPEP was combined with 10 mg/kg s.c. PCP, however, a significant disruption of PPI was observed during both prepulse conditions (Fig. 2A). These findings were confirmed by statistical analyses. Thus, a repeated measures analysis of variance using prepulse level as the within factor and treatment condition as the between factor revealed a significant main effect of treatment [F(7,68) = 6.94, p < 0.001], but no treatment by prepulse level interaction, suggesting that MPEP enhanced the disruptive PPI effects of PCP regardless of prepulse level. This was further confirmed by the finding of a significant simple main effect of treatment for each prepulse level [F(7,68) = 6.51, p < 0.001 and F(7,68) = 6.04, p < 0.001 for the 5- and 10-db prepulse levels, respectively]. Post hoc analysis using the Tukey procedure revealed a significant deficit in PPI at both the 5 and 10 prepulse intensity levels relative to vehicle + vehicle treatment when a combination of MPEP (30 mg/kg i.p.) and PCP (10 mg/kg s.c.) was administered (p < 0.01). No significant effect on PPI was found for either drug administered alone (p = 0.5 for MPEP, p > 0.1 for PCP). Likewise, no significant differences were observed during pulse alone presentations (Fig. 2B).

A, effect of vehicle (Veh), PCP (1–10 mg/kg s.c.), MPEP (3–30 mg/kg i.p.), and a combination of MPEP (30 mg/kg i.p.) + PCP (10 mg/kg s.c.) on PPI of the acoustic startle response in male 129S6 mice (n = 8/group) at two prepulse intensity levels (5 and 10 db above background). The 10-mg/kg dose indicated on the x-axis for the MPEP + PCP condition represents the dose of PCP administered with a 30 mg/kg i.p. dose of MPEP. MPEP and PCP were administered 30 and 20 min before placement of the mice into the testing apparatus, respectively. Asterisks represent a significant difference from the Veh + Veh condition, **, p < 0.01; ***, p < 0.001. B, effect of Veh, PCP, MPEP, and a combination of MPEP + PCP on startle amplitude during pulse alone trials in the same mice represented in A. No significant differences were observed in this measure of basal startle amplitude. Error bars represent S.E.M.
Similar to the findings using mice, we also found a significant MPEP potentiation of PCP effects on PPI in rats (Fig. 3). Thus, combining 10 mg/kg i.p. MPEP with 1 mg/kg s.c. PCP disrupted PPI, whereas neither treatment alone had any significant effect. Combination treatment resulted in a significant disruption of PPI at a prepulse level 10 or 15 db above background, whereas a similar, albeit nonsignificant, trend was noted at a prepulse level 5 db above background. These results were confirmed by the finding of a significant main effect of treatment at the 10- and 15-db prepulse levels [F(3,44) = 3.91, p <0.016 and F(3,44) = 6.07, p < 0.003, respectively]. At the 10-db level, post hoc testing using the Tukey procedure revealed a significantly enhanced disruption of PPI after MPEP + PCP treatment over PCP treatment alone; however, the disruption of PPI produced by PCP alone or combination treatment did not significantly differ from vehicle condition. At the 15-db level, post hoc testing revealed a significant reduction of PPI after combination treatment relative to both vehicle and PCP alone treatment. All other treatments failed to differ from vehicle under these conditions. As with the previous study, none of the treatments or combinations significantly altered response during pulse alone trials (Fig. 3B).
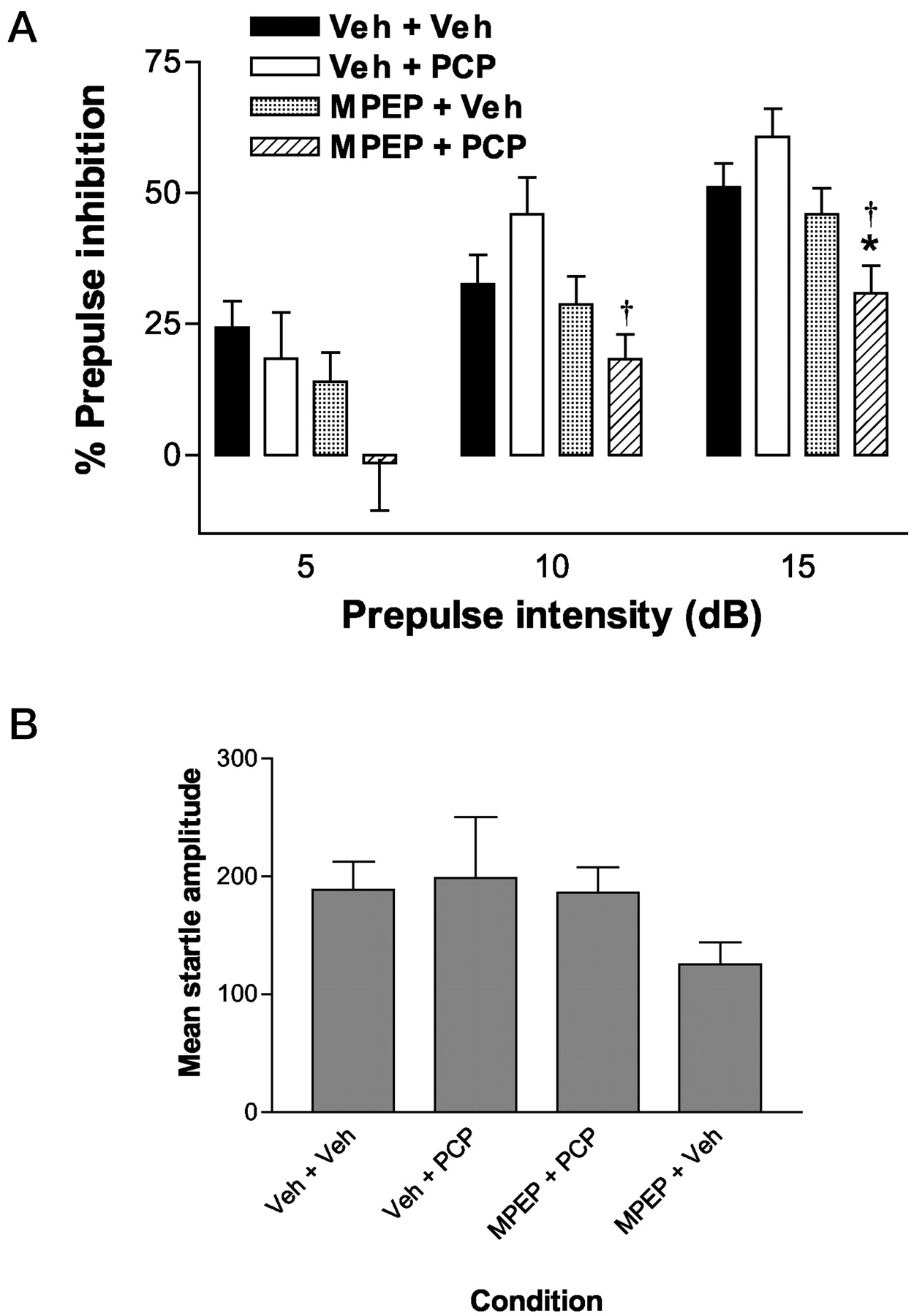
A, effect of vehicle (Veh), PCP (1 mg/kg s.c.), MPEP (10 mg/kg i.p.), and a combination of MPEP + PCP on PPI in male Sprague-Dawley rats (n = 12/group) at three prepulse intensity levels (5, 10, and 15 db above background). MPEP and PCP were administered 70 and 10 min before placement of the rats into the testing apparatus, respectively. Asterisks represent a significant difference from the Veh + Veh condition, *, p < 0.05. †, significant difference from Veh + PCP (1 mg/kg s.c.) condition, †, p < 0.05. B, effect of Veh, PCP, MPEP, and MPEP + PCP combination treatment on startle amplitude during pulse alone trials in the same rats represented in A. No significant differences were observed in this measure of basal startle amplitude. Error bars represent S.E.M.
mGluR5 Knockout Mice. Levels of PPI were tested three times in wild-type (mGluR5+/+) and knockout (mGluR5–/–) mice. Study 1 and 2 occurred within the same week, whereas study 3 occurred 10 days after the completion of study 2. The same mice were tested in all studies. The primary purpose of study 3 was to examine PPI effects under different light/dark conditions. The results of these tests are presented in Fig. 4A and demonstrate a significant and reproducible deficit of PPI in these mice. Repeated measures analysis of variance revealed a significant main effect of genotype [F(1,21) = 6.24, p < 0.03], a significant main effect of test number [F(2,42) = 6.18, p < 0.006], a significant effect of prepulse level [F(3,63) = 158, p < 0.001], but no significant prepulse level by genotype (p = 0.055) nor test number by genotype (p > 0.9) interactions. The lack of significant interactions suggest a stable deficiency of PPI in mGluR5–/– mice relative to wild-type controls that is neither dependent on prepulse intensity nor the activity phase of the animals during testing. Analysis of the simple main effect of genotype for each test revealed a significant disruption of PPI for prepulse levels 15 and 20 db above background for study 1 [F(1,23) = 5.07 and 4.74, p values < 0.04, respectively] and study 2 [F(1,23) = 7.99 and 7.92, p values = 0.01, respectively]. Similar analysis revealed a significant disruption of PPI in mGluR5–/– mice in study 3; however, this difference only reached the level of significance at the 20-db prepulse intensity [F(1,23) = 5.02, p < 0.04]. No significant differences were observed during pulse alone presentations (Fig. 5B).

A, percentage of PPI in mGluR5 knockout (mGluR5–/–) or wild-type (mGluR5+/+) mice in three separate studies using four prepulse intensities (5, 10, 15, and 20 db above background). Note that the same mice were examined in all studies (n = 12 and 13 for mGluR5+/+ and mGluR5–/–, respectively). Study 1 and 2 examined these mice during the active phase of their dark/light cycle, whereas study 3 examined these mice during their inactive phase. Asterisks represent a significant difference from wild-type controls, *, p < 0.05; **, p < 0.01. B, startle amplitude during pulse alone trials in the same mGluR5–/– and mGluR5+/+ mice represented in A. No significant differences were observed in this measure of basal startle amplitude. Error bars represent S.E.M.

A, effect of amphetamine (AMPH; 0–2 mg/kg s.c.; n = 8/group) on PPI in male Sprague-Dawley rats at four prepulse intensities (5, 10, 15, and 20 db above background). B, lack of effect of CHPG (500 nmol/8 μl i.c.v.; n = 5/group) on PPI in rats at four prepulse intensities. C, effect of AMPH (2 mg/kg s.c.) + vehicle (Veh; 8 μl i.c.v.) (n = 11) or AMPH + CHPG (500 nmol/8 μl i.c.v.) (n = 9) on PPI in rats at four prepulse intensities. D, effect of various treatments, or treatment combinations, on startle response during the pulse alone condition. Vehicle treatments were injected s.c. or i.c.v. AMPH/i.c.v., i.c.v. vehicle in AMPH (2 mg/kg s.c.)-treated rats. CHPG, CHPG (500 nmol/8 μl i.c.v.) delivered alone. Also illustrated are the effect of multiple doses of AMPH (0.5–2 mg/kg s.c.) and the effect of AMPH (2 mg/kg s.c.) + CHPG (500 nmol/8 μl i.c.v.) on basal startle amplitude. No significant differences were found for any treatment relative to its appropriate control. Error bars represent S.E.M. Asterisks represent a significant difference from vehicle (A) or AMPH + Veh (C) conditions, *, p < 0.05; **, p < 0.01; ***, p < 0.001.
mGluR5 Agonist Treatment. Because previously discussed data demonstrate that mGluR5 agonist treatment potentiates NMDAR function (Mannaioni et al., 2001; Pisani et al., 2001), it is reasonable to expect that mGluR5 agonist administration could potentiate the effect of a use-dependent NMDAR antagonist such as PCP. The corollary hypothesis that mGluR5 activation should reverse the effect of PCP-mediated behavior is also suggested by previous reports that glycine or glycine transport inhibitors reverse the behavioral effect of PCP, presumably through a coagonist property of glycine at the NMDA receptor (Javitt et al., 1997). In this way, both an enhancement and reversal of PCP-induced behavior after mGluR5 activation would be consistent with enhancement of NMDAR-mediated behaviors. To examine the role of mGluR5 activation on PPI that was disrupted by interaction with relevant neuronal circuitry distinct from a direct antagonism of NMDARs, we examined the effect of the mGluR5 agonist CHPG on amphetamine-induced disruption of PPI. The effect of multiple doses of amphetamine (0.5–2 mg/kg s.c.) on PPI in these rats is depicted in Fig. 5A. As expected, amphetamine dose dependently disrupted PPI across all prepulse levels examined as indicated by a significant main effect of dose [F(3,28) = 4.32, p < 0.02] and the absence of a significant prepulse level by dose interaction. The decrease in PPI was found independent of any significant change in basal startle response (Fig. 5D). Because a dose of 2 mg/kg s.c. was found to disrupt PPI at prepulse levels 10, 15, and 20 db above background, this dose was chosen for interaction studies with CHPG. Figure 5B depicts the effect of CHPG (500 nmol/8 μl i.c.v.) in normal rats. As shown, CHPG had no effect on PPI at any prepulse level. Furthermore, CHPG had no significant effect on startle amplitude during the pulse alone condition (Fig. 5D). In contrast to these results, CHPG (500 nmol/8 μl i.c.v.) delivered to amphetamine (2 mg/kg s.c.)-treated rats significantly ameliorated the disruptive effects of amphetamine at prepulse intensities 10, 15, and 20 db above the background level (Fig. 5C) independent of any significant change in basal startle response (Fig. 5D). These differences were confirmed by the finding of a significant main effect of treatment [F(1,18) = 8.78, p < 0.009]. Analyses of simple effects at each prepulse level revealed significant differences between treatment conditions at the 10-, 15-, and 20-db prepulse intensities (p values < 0.05).
Discussion
The present results demonstrate that administration of the selective mGluR5 antagonist MPEP, at doses having no effect on the measured behaviors, potentiates PCP actions in rodent PPI and locomotor behavior. We also found modest, yet significant and reproducible deficits in PPI in mGluR5–/– mice relative to their wild-type controls. Finally, i.c.v. administration of the selective mGluR5 agonist CHPG was shown to reverse amphetamine-induced deficits in PPI. Collectively these results suggest that potentiation of mGluR5 activity may play a modulatory role in rodent behaviors.
The finding that MPEP has little effect by itself in locomotor or PPI assays is consistent with a growing body of literature (Spooren et al., 2000; Tatarczyñska et al., 2001; Henry et al., 2002). In the present report, we found that, at doses up to 10 and 30 mg/kg i.p., MPEP had no activity on either locomotor activity or PPI. Nonetheless, when MPEP was combined with PCP, we found a significant enhancement of PCP response in each instance. These data are consistent with a recent report by Henry et al. (2002) where MPEP potentiated the effect of PCP on activity and PPI in rats at doses that had no effect alone. Despite the lack of effect of MPEP alone, mGluR5–/– mice did show consistent, albeit, modest deficits in PPI. Because the role of mGluR5 in these behavioral measures is likely modulatory (see discussion below), we suggest that complete removal of the contribution of this receptor system may be required before any emergent behavior can be reliably measured in normal animals. Thus, for any mGluR5 antagonist, receptor occupancy values in excess of the ∼90% expected in the present study (Anderson et al., 2002) may be needed to produce data consistent with mGluR5–/– mice. However, after pharmacological disruption of the relevant neuronal systems (e.g., using PCP or amphetamine), the role of mGluR5 may be more pronounced and subtle differences in behavior may then be revealed.
The potentiation of PCP activity in these animal behaviors seems to be selective for mGluR5 antagonist application in that the mGluR2/3 agonist LY314582 had no effect on PCP-induced responses (Henry et al., 2002). Although MPEP has been shown to antagonize NMDAR function in vitro (O'Leary et al., 2000), it is unlikely that MPEP, at these doses, augmented PCP effects via this mechanism. First, the MPEP dose range used in the present study does not exceed doses required for maximal in vivo mGlu5 receptor occupancy (Anderson et al., 2002). Second, we have found that doses of MPEP higher than those reported in the present study result in an inconsistent decrease of spontaneous locomotor activity in nonhabituated rats (data not shown). This effect is opposite of the predicted activity of NMDAR antagonist activity on this behavior. Finally, it has been reported that MPEP brain levels achieved at behaviorally active doses are well below the concentration required to inhibit NMDA currents in vitro (Khun et al., 2002). Related to this latter suggestion, the dose range of MPEP used in these studies is in line with those reported to produce mGluR5-mediated anxiolytic effects in rats (Tatarczyñska et al., 2001; Brodkin et al., 2002). Thus, available data suggest that the synergistic effect of MPEP on PCP-mediated behavior observed in the present study was due to its primary action as an mGluR5 antagonist.
As with MPEP, it is likely that the ability of the mGluR5 agonist CHPG to ameliorate amphetamine-induced disruption of PPI is due to selective agonist activity at mGlu5 receptors. Thus, previous publications demonstrate a selective mGluR5 agonist activity of CHPG at these doses using in vivo phosphoinositide hydrolysis measures (Johnson et al., 1999; Anderson et al., 2002).
One way in which mGluR5 may modulate neuronal circuitry responsible for the measured behaviors is through interaction with the NMDAR system. As discussed previously, mGluR5 activation potentiates NMDAR function in vitro. NMDAR activity leading to modest increases in Ca2+ concentrations can prevent mGluR5 desensitization via activation of a protein phosphatase and subsequent dephosphorylation of the receptor (Alagarsamy et al., 1999). Higher internal Ca2+ concentrations, however, may promote desensitization of mGluR5 via phosphorylation at a protein kinase C-sensitive site (Alagarsamy et al., 2002). These results reveal the existence of a synergistic feedback mechanism under conditions of low-to-modest internal Ca2+ levels. Higher Ca2+ levels are expected to limit mGluR5 function and consequently its ability to augment NMDAR function. These findings suggest that the role of mGluR5 on PPI and locomotor-related preclinical behaviors, which are also sensitive to NMDAR antagonists, might be less pronounced under conditions of normal NMDAR function coincident with higher basal intracellular Ca2+ levels relative to conditions of NMDAR hypofunction. Consistent with this hypothesis, we have presently demonstrated a modest effect in knockout mice, a lack of effect of MPEP alone, and a lack of effect of CHPG in normal rats (i.e., the role of mGluR5 was only unmasked after disruption of relevant neuronal circuitry).
In addition to biochemical interactions, an emerging body of evidence suggests that mGluR5 and NMDARs may also physically interact in a manner that mediates receptor trafficking to the cell surface. Thus, NMDA receptors are associated with the postsynaptic density protein shank, via interaction with guanylate kinase associate proteins (Kim et al., 1997; Naisbitt et al., 1999). Interestingly, shank and group I mGluRs are both homer-associated proteins, and homer proteins are known to dimerize (Tu et al., 1998; Xiao et al., 1998, 1999), thus providing a physical link between mGluR5 and NMDARs. Because homer proteins have recently been shown to regulate mGluR5 trafficking (Ango et al., 2002), this physical interaction between mGluR5 and NMDAR could potentially impact signaling, receptor trafficking, or both.
The functional consequence of mGluR5 deletion on NMDA function has been previously demonstrated. Thus, deficits in the NMDAR-mediated component of long-term potentiation in mGluR5–/– mice have been described (Lu et al., 1997; Jia et al., 1998). We hypothesized that a more widespread modulatory role of mGluR5 on NMDAR-mediated function would result in a modest deficit of PPI. Our finding of reproducible deficits in mGluR5–/– mice relative to their wild-type controls is consistent with this hypothesis. These data are also consistent with deficits previously reported in abstract form by Geyer et al. (2000). However, this finding is inconsistent with abstract reports by other groups (O'Meara et al., 2002). In previous studies, the Geyer laboratory has housed animals on a reverse dark/light cycle and tested animals during the dark phase (Henry et al., 2002). Thus, we were additionally interested in testing the contribution of this variable to the observation of significant deficits in these mice. Our mice were housed on a reverse dark/light cycle, initially tested during their dark phase (Fig. 4, studies 1 and 2), and subsequently tested during their light phase (Fig. 4, study 3). We found significant and reproducible deficits in these animals regardless of testing time within the dark/light cycle. Because previous reports have been restricted to abstract publications, details that could account for inconsistencies between these reports remain to be elucidated. One obvious possibility is sample size. In the present study, we used relatively large group sizes. In our present study, a group size of 12 to 13 resulted in effects modest in magnitude (p values ∼0.04). In comparison, we have replicated these results (data not shown) with a separate group of mice where the group size was increased to 25 to 26 animals per group. In these data, the difference between mGluR5 knockout and wild-type mice, although similar in magnitude, is more statistically robust (i.e., p values < 0.001).
Similar to the present studies using PCP, preliminary reports indicate that MPEP also potentiates methamphetamine-induced hyperlocomotion and disruption of PPI (Kinney et al., 2002). Furthermore, we have presently demonstrated that CHPG can ameliorate the disruptive effects of amphetamine on PPI. Collectively, these data suggest that the mGluR5 system can modulate PPI and stimulant-induced locomotor activity regardless of the stimulant used (i.e., PCP versus methamphetamine). Thus, mGluR5/NMDAR interactions may not wholly account for the contribution of mGluR5 in the behaviors measured herein. Group I mGlu receptors are known to modulate dopamine release in the prefrontal cortex. Thus, the nonselective group I agonist (+)-3-hydroxyphenylglycine has been shown to significantly reverse PCP-induced dopamine release in the rat prefrontal cortex (Maeda et al., 2003). Because amphetamine similarly enhances dopamine release in the prefrontal cortex (Moghaddam et al., 1990), it remains possible that mGlu5 receptors are having the effects reported herein via a dopamine-dependent mechanism independent from a primary effect on NMDARs. Although mGluR5 localization is typically regarded as postsynaptic, fiber labeling has also been described (Hay et al., 1999), consistent with known dopamine release sites on axonal varicosities (Wong et al., 1999). Furthermore, it has been demonstrated that enhancement of locomotor activity in response to infusion of nonselective group I mGluR agonists into the nucleus accumbens is dependent on intact dopaminergic neurotransmission (Attarian and Amalric, 1997; Meeker et al., 1998). Collectively, these data could suggest that mGlu5 receptors modulate amphetamine- and PCP-mediated activity via a direct interaction with dopaminergic systems or, alternatively, neuronal circuitry commonly affected by NMDA and dopaminergic mechanisms. The possibility that mGluR5 interacts with NMDA and dopamine systems independently must also be considered, given the lack of evidence to the contrary.
In summary, the results of the present manuscript demonstrate that PCP-induced behavior is augmented after coapplication of the mGluR5 antagonist MPEP. We further found that infusion of the mGluR5 agonist CHPG reverses the disruptive effects of amphetamine on PPI in rats and that mGluR5–/– mice demonstrate a consistent and reliable, albeit modest, deficit in PPI relative to their wild-type controls. Collectively, these data are consistent with a modulatory role for mGluR5 in preclinical behaviors sensitive to antipsychotic drug treatment and further support a potential therapeutic role for mGluR5 agonists/potentiators in the treatment of schizophrenia, psychosis, and related disorders.
Footnotes
-
DOI: 10.1124/jpet.103.048702.
-
ABBREVIATIONS: NMDAR, N-methyl-d-aspartate receptor; PCP, phencyclidine; mGluR, metabotropic glutamate receptor; PPI, prepulse inhibition; MPEP, 2-methyl-6-(phenylethynyl)pyridine; CHPG, (R,S)-2-chloro-5-hydroxyphenylglicine; NMDA, N-methyl-d-aspartate; LY314582, racemic (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid.
- Received January 2, 2003.
- Accepted March 20, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}