


Abstract
Dinapsoline is a new potent, full agonist at D1 dopamine receptors with limited selectivity relative to D2receptors. The efficacy of this compound was assessed in rats with unilateral 6-hydroxydopamine lesions of the medial forebrain bundle, a standard rat model of Parkinson's disease. Dinapsoline produced robust contralateral rotation after either subcutaneous or oral administration. This rotational behavior was attenuated markedly by the D1 receptor antagonist SCH-23390, but not by the D2 receptor antagonist raclopride. During a chronic 14-day treatment period in which rats received dinapsoline either once or twice a day, dinapsoline did not produce tolerance (in fact, some sensitization of the rotational response was observed in one experiment). Because dinapsoline shows less D1:D2 selectivity in vitro than other D1 agonists, the contribution of D2 activity to tolerance was assessed. Chronic daily cotreatment with dinapsoline and raclopride did not enable the development of tolerance to chronic dinapsoline treatment. In contrast, when dinapsoline was administered by osmotic minipump, rapid tolerance was observed. To explore further the contribution of D1 and D2 receptors to tolerance, experiments were performed with the selective D1agonist A-77636. Daily dosing with A-77636 rapidly produced complete tolerance, as previously observed, whereas coadministration of the D2 agonist quinpirole plus A-77636 failed to either delay or prevent tolerance. Taken together, these results indicate that the development of tolerance to D1 receptor agonists is influenced by the pattern of drug exposure but not by the D1:D2 selectivity of the agonist.
Parkinson's disease (PD) is a neurological disorder involving progressive degeneration of dopaminergic neurons that arise from the substantia nigra and innervate the caudate and putamen (Hughes et al., 1993;Marsden, 1994). The principal approach in pharmacotherapy of PD has been dopamine (DA) replacement therapy using l-DOPA (l-dihydroxyphenylalanine), a drug that can provide significant palliative effects for several years. The principal limitations of the long-term use of l-DOPA, however, include the development of unpredictable “on-off” phenomena, as well as dyskinesias and psychiatric symptoms such as hallucinations, sleep disturbances, and psychoses. To avoid these adverse events, direct acting agonists targeted for various DA receptors have been tried. D2-preferring agonists, such as bromocriptine, ropinirole, and pramipexole, are useful monotherapy only in the early stages of the disease, losing efficacy as the illness progresses (Brooks et al., 1998). Notably, however, their parenteral administration can have marked beneficial effects for some patients (O'Sullivan and Lees, 1999).
Initial efforts to develop D1 agonists for PD met with limited success as SKF-38393 and CY 208–243 showed excellent effects in rodent models but were less effective in primates and patients (Nomoto et al., 1985; Braun et al., 1987; Temlett et al., 1988; 1989). These compounds were identified subsequently as partial agonists at D1 receptors, and as a result, the need for full intrinsic activity at the D1receptor was hypothesized (Lovenberg et al., 1989; Brewster et al., 1990; Watts et al., 1993). This hypothesis is supported by recent studies with the D1 receptor full agonists dihydrexidine (DHX), A-77636, and ABT-431, which had robust effects in primate PD models and efficacy in patients (Taylor et al., 1991;Kebabian et al., 1992; Shiosaki et al., 1996; Blanchet et al., 1998;Rascol et al., 1999). These compounds have not evolved as clinical drugs due to pharmacokinetic limitations (DHX and ABT-431), toxicity (A-68930), or rapid development of behavioral tolerance (A-77636). Therefore, minimum requirements for a novel D1agonist for PD therapy include full efficacy at D1 receptors, robust activity after oral dosing, and no evidence for the development of tolerance.
The present experiments assessed whether the new full-efficacy D1 agonist dinapsoline (DNS; Ghosh et al., 1996) fits the desired behavioral profile. The experiments all utilized the rat unilateral 6-hydroxydopamine (6-OHDA) rotation model of PD in which 6-OHDA is infused unilaterally into the medial forebrain bundle, the substantia nigra, or the striatum. This results in the destruction of DA terminals and neurons and a loss of striatal DA, and a profound functional dopaminergic supersensitivity develops on the lesioned side. When challenged with direct-acting DA receptor agonists, unilateral 6-OHDA rats turn contralaterally (away from the side of the lesion) because of the increased sensitivity of the postsynaptic DA receptors on the lesioned side (Ungerstedt 1968, 1971; Heikkila et al., 1989). Although the unilateral 6-OHDA-lesioned rat model is often considered the principal rodent model for PD (Schwarting and Huston, 1996), it has substantial limitations in comparison to the MPTP-lesioned primate. Specifically, whereas positive results in the rat unilateral 6-OHDA lesion model demonstrate dopamine agonism, they do not always predict efficacy in MPTP-lesioned primates or patients (Nomoto et al., 1985;Braun et al., 1987; Temlett et al., 1988, 1989). Thus, the unilateral 6-OHDA model, although not predictive of responses in PD, provides a relevant system that is amenable to mechanistic studies.
One of the primary foci of the present experiments was to determine whether dinapsoline would cause tolerance as has been reported for another D1 full agonist, A-77636. In addition to tolerance, some drugs, including both indirect (e.g., amphetamine) and direct dopamine agonists (e.g., apomorphine and bromocriptine), may cause behavioral sensitization under some administration paradigms (Robinson, 1984; Gancher et al., 1995; Henry et al., 1998). In the present studies, however, the focus was not only on the consequences of dinapsoline administration, but also why one direct-acting, full D1, full agonist (i.e., A-77636) produced tolerance (Asin and Wirtshafter, 1993), whereas others (e.g., A-68930 or ABT-431) did not (Shiosaki et al., 1996). One way in which these D1 full agonists differ is their degree of D2 affinity. ABT-431 has moderate D1:D2 selectivity (20-fold:Shiosaki et al., 1996), whereas A-77636 is 150-fold selective for the D1 versus D2 receptor (Kebabian et al., 1992). These findings lead to the hypothesis that D2 receptor coactivation may decrease the propensity for tolerance. If this hypothesis is true, DNS, a D1 receptor agonist with high affinity for both D1 and D2 receptors (Ghosh et al., 1996) should provide robust rotational behavior in unilateral 6-OHDA rats, yet not induce behavioral tolerance.
Materials and Methods
Subjects.
Adult male Sprague-Dawley Rats (Hilltop Laboratories, Chatsworth, CA), weighing between 280 and 320 g, were used as subjects. Animals were housed individually with food and water available ad libitum, except as noted below. The light:dark schedule was 12 h:12 h, and testing was performed during the light cycle. All methods were approved by the Bristol-Myers Squibb Animal Care and Use Committee and adhere to the guidelines in the Guide for the Care and Use of Experimental Animals published by the National Institutes of Health (Pub. 85-23, 1985).
Surgery.
Rats were pretreated with 25 mg/kg desipramine (s.c.) approximately 30 min before surgery. Rats were anesthetized by inhalation of isoflurane (1.5 to 4.0%) and placed in a stereotaxic apparatus. An infusion cannula was placed in the medial forebrain bundle 3.8 mm posterior to bregma, 1.5 mm lateral to midline, and 7.2 mm ventral to the surface of the brain according to the atlas of Paxinos and Watson (1986). Ten micrograms of 6-OHDA (Sigma Chemical Co., St. Louis, MO) in a volume of 4 μl was infused at a rate of 0.5 μl/min in a vehicle of 0.01% ascorbate. After a 14-day recovery period, rats were prescreened for rotation in response tod-amphetamine (5 mg/kg) and to apomorphine (0.3 mg/kg) 1 week later. Animals that responded to both d-amphetamine (>800 rotations in 3 h) and apomorphine (>100 rotations in 1 h) were retained for further study.
Testing of compounds began on day 28 postsurgery in each case. A new group of 6-OHDA-lesioned rats were used for each new study. In some studies, rats were implanted with a subcutaneous 14-day osmotic minipump (model 2 ML2, Alza, Palo Alto, CA) with a flow rate 5.0 μl/h. The rats were re-anesthetized with 1.5 to 4% isoflurane, a small incision was made on the back of the neck, and the minipump was placed subcutaneously in the cavity. The incision was closed with sterile wound clips. Before implantation, minipumps were incubated in sterile saline (37°C) to ensure outflow at the time of implantation. The minipumps were used to administer DNS, or vehicle [50% dimethyl sulfoxide (DMSO), 12.5% ascorbic acid].
Striatal Dopamine Content.
In a subset of animals, striatal DA content was measured to confirm the extent of the 6-OHDA lesion. At the completion of the study, animals were anesthetized deeply by CO2 inhalation and rapidly decapitated using a guillotine. Brains were removed quickly, and kept on ice while right and left striata were isolated, removed, and weighed in individual nonfilter microcentrifuge tubes containing 0.5 ml of a homogenizing buffer (0.22 N perchloric acid, 0.5% EDTA, 0.15% sodium metabisulfite). The samples were homogenized by sonication for 10 s and then centrifuged at 14,000g for 20 min. The supernatant was transferred to microcentrifuge tubes with a filter (0.2 μm) and centrifuged at 14,000g for 2 min. The samples were frozen at −80°C to await HPLC analysis.
HPLC Analysis.
Thawed samples were analyzed for DA content using established high performance liquid chromatography (HPLC)-electrochemical detection methods. Briefly, 50-μl samples were injected into the sample loop of an HPLC system using an acetate buffer mobile phase (17% methanol) pumped at 0.4 ml/min. Peaks were separated with a C-18 reverse phase column (3-mm diameter, MD-180, ESA, Chelmsford MA) and detected with a dual coulometric cell (5014B, ESA) and detector (Coulochem II, ESA). Dopamine was analyzed by sequential reduction (−100 mV) and oxidation (350 mV) and was quantified at the latter electrode. Dopamine concentration in each sample was calculated in reference to established standard curves and was represented as picomoles per milligram of striatal tissue. Depletion was calculated as the percentage of DA content on the lesioned side relative to the nonlesioned side.
Apparatus, Procedure, and Statistics.
Rats were tested for rotation in automated rotation chambers (Rotoscan, Accuscan, Columbus, OH). The apparatus consisted of a cylindrical Plexiglas chamber 30 cm in diameter in which the animal is fitted to a harness attached to a flexible rod connected to a rotating microswitch. Animals were allowed to habituate to chambers for 30 min before drug treatment in each case. Data were collected for 1 to 12 h after injection, using 15-min time bins. Treatments were compared using one-way and repeated measures of analysis of variance (ANOVA), as appropriate; post hoc analysis was performed with Dunnett's test.
Acute DNS Administration.
Beginning 1 week after the screening dose with apomorphine, subjects (n = 12) were tested once per week with DNS (0.02, 0.2, or 2 mg/kg) or vehicle (s.c.) using a counterbalanced design and rotation behavior was monitored for 10 h. After the final day of testing, rats were euthanized and brains were removed for subsequent assessment of DA depletion. In the oral dosing experiments, a separate group of subjects (n = 8) received DNS (0.02, 0.2, or 2 mg/kg) or vehicle once per week using a counterbalanced design. Rats were fasted for 16 h before dosing with oral gavage, and rotation behavior was monitored for 10 h.
In the experiments that included acute antagonist administration, subjects (n = 8/group) were pretreated with either the D1 antagonist SCH-23390 (0.5 mg/kg s.c.; D1 antagonist), the D2antagonist raclopride (2 mg/kg s.c.), or vehicle. After 30 min, they were injected with DNS (0.2 or 2 mg/kg s.c.), and rotation was monitored for 3 h. The shortened assessment period was chosen, because the D1 antagonist SCH-23390 is known to have a relatively short duration of action (approximately 3 h) in our assay.
Chronic DNS Administration.
Subjects (n = 5/group) were dosed daily for 14 days at 8:00 AM every day with either A-77636 (1 mg/kg s.c.) or DNS (2 mg/kg s.c.). In a separate group DNS (2 mg/kg s.c.) or vehicle was administered twice daily at 8:00 AM and 6:00 PM everyday. Rotation behavior was monitored in all animals every day for 3 h after the morning injection. In this case, the 3-h assessment period was used to minimize the time that the animals did not have access to food or water.
Coadministration of DNS with Raclopride.
Subjects (n = 8/group) were dosed with either raclopride (2 mg/kg s.c.) or vehicle, followed 30 min later by DNS (2 mg/kg s.c.) once daily for 6 days. Rotation was monitored for 3 h after DNS administration. On day 7, all animals were challenged with DNS (0.2 mg/kg s.c.) followed by rotation monitoring for 3 h.
Coadministration of A-77636 with Quinpirole.
Subjects (n = 8/group) were dosed with A-77636 (0.3 mg/kg s.c.) plus either the D2 agonist quinpirole (0.1 mg/kg s.c.), or vehicle for the 2 days. Rotation was monitored for 3 h immediately following quinpirole or vehicle administration. To assess tolerance on day 3, all animals were treated with A-77636 (0.3 mg/kg s.c.) alone followed by rotation monitoring for 3 h. To confirm that the tolerance was specific to D1 receptor desensitization, on day 4, all animals treated with quinpirole alone (0.1 mg/kg s.c.), and rotation was monitored for 3 h.
Minipump Studies.
Rats (n = 8/group) were subcutaneously implanted with minipumps calibrated to deliver DNS (0.006, 0.06, 0.6, or 6 mg/kg/day) or vehicle. Behavioral testing for rotation was started at 16 h postimplantation and was monitored for 1 h twice daily. On day 14 after minipump implantation, rats were challenged with DNS (0.2 mg/kg s.c.) and rotation was monitored for 3 h.
Drugs.
Dinapsoline was synthesized in-house using a synthetic route based on the one reported by Ghosh et al. (1996). SCH-23390, raclopride, A-77636, and quinpirole were obtained from Research Biochemicals International (Natick, MA). The vehicle used for DNS was 0.1% ascorbate (Sigma Chemical Co.), and in all other cases sterile water was used as vehicle. In the experiments employing osmotic minipumps, the vehicle was 50% DMSO and 12.5% EDTA in sterile water.
Results
Acute Effects.
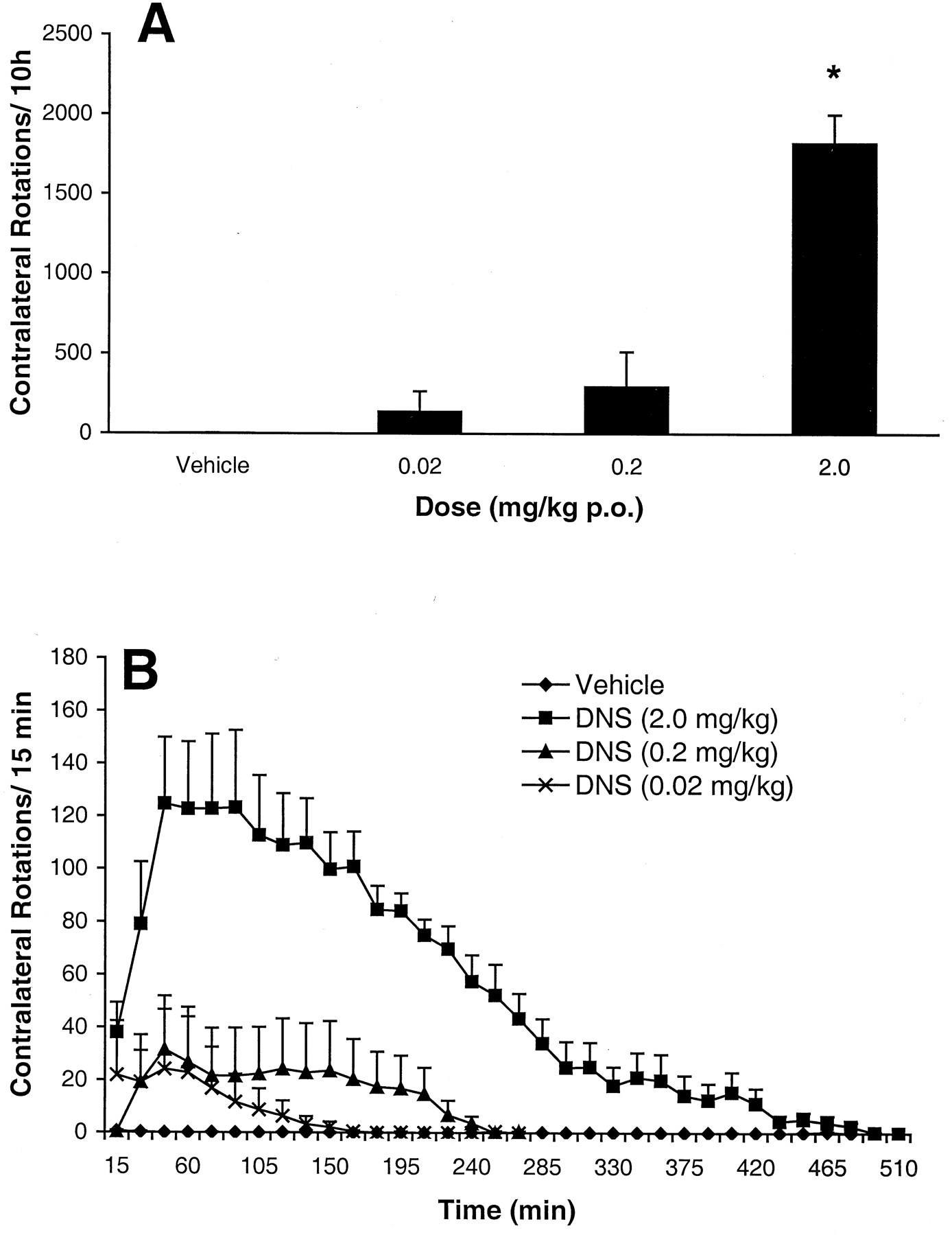
When dosed subcutaneously (Fig.1A), DNS produced robust, dose-dependent rotational behavior (F 3,40 = 77.3,p < 0.001). Statistically significant increases in rotation relative to vehicle were obtained at 2.0 and 0.2 mg/kg (p < 0.05, Dunnett's test), but not at 0.02 mg/kg. Figure 1B shows the time course of rotation for each dose. When dosed at 2 mg/kg, DNS produced rotation that lasted approximately 10 h, whereas the effects at 0.2 mg/kg lasted about 5 h. In contrast, the maximal rate of rotation produced by these two doses was comparable, around 150 to 200 rotations per 15-min time bin. Post-mortem analysis of the DA content from the striatum of these animals demonstrated a depletion of 98.1 ± 0.2% (mean ± S.E.M.), with a range of 97.3 to 99.8%. A subset of rats was sampled from subsequent experiments, and in all cases depletions were greater than 95%.

A, cumulative rotation (mean ± S.E.M.;n = 12/group) over 10 h produced by subcutaneous dosing of dinapsoline. B, mean (±S.E.M.) rotations for each 15-min time bin produced by dinapsoline during the 10-h test period. *Different from vehicle; p < 0.05, ANOVA, Dunnett's test.
Dinapsoline also produced robust rotation (Fig.2A) when administered orally (F 3,21 = 42.3, p < 0.001), but the response was not dose-dependent. Only the increase in rotation caused by the 2 mg/kg dose was significantly different from baseline (p < 0.05, Dunnett's test). When dosed orally at 2 mg/kg, rotation continued to be observed for 7 h. Administration of a higher dose of DNS (10 mg/kg p.o.) produced severe stereotypy, including self-injurious behavior, as well as the apparent induction of seizures, necessitating termination of the experiment.

A, cumulative rotation (mean ± S.E.M.,n = 8/group) over 10 h produced by oral administration of dinapsoline. B, dinapsoline-induced rotation (mean ± S.E.M., n = 8/group) for each 15-min time bin during the 8-h test period. *Different from vehicle;p < 0.05, ANOVA, Dunnett's test.
The rotational response to DNS (Fig. 3A) was blocked completely by the D1 receptor antagonist SCH-23390 (0.5 mg/kg s.c.). SCH-23390 blocked the rotation produced by DNS administered at 0.2 mg/kg s.c. (F 1,14 = 63.8, p < 0.001) and 2.0 mg/kg (F 1,14 = 95.4,p < 0.001). In this experiment rotational behavior was quantified for only 3 h to match the known duration of action of SCH-23390.

A, effect of SCH-23390 (0.5 mg/kg s.c.) on cumulative rotation (mean ± S.E.M., n = 8/group) produced by dinapsoline (0.2 or 2.0 mg/kg s.c.) in a 3-h period. B, effect of raclopride (2 mg/kg s.c.) on cumulative rotations (mean ± S.E.M., n = 8/group) produced by dinapsoline in a 3-h period. *Different from vehicle plus DNS; p< 0.05, ANOVA.
The rotational response to DNS was not altered by pretreatment with the D2 antagonist raclopride (2 mg/kg s.c.; Fig. 3B). Raclopride (2 mg/kg s.c.) did not reduce the rotational response to DNS at 0.2 mg/kg s.c. (F 1,14 = 2.5,p > 0.05) or 2 mg/kg s.c. (F 1,14 = 0.03, p > 0.05). In contrast, the D2 agonist quinpirole (0.25 mg/kg s.c.) produced robust rotation that was blocked completely by raclopride (2 mg/kg s.c.: data not shown). These results indicate that the rotational response in the unilaterally 6-OHDA-lesioned rats can be attributed to activation of D1 receptors.
Behavioral Tolerance.
When A-77636 was dosed daily at 1 mg/kg s.c. for 14 days, dramatic behavioral tolerance was observed (Fig.4). When dosed in naı̈ve animals, A-77636 (1 mg/kg s.c.) produced robust rotation, but as early as the second day of dosing, A-77636 produced significantly less rotation than on the first day (F 1,13 = 8.5,p = 0.012). By the fourth day of dosing, the amount of rotation was no greater than that seen with vehicle (F 1,14 = 3.2, p > 0.05), indicating that complete tolerance had occurred.

Cumulative rotation (mean ± S.E.M.,n = 5/group) over 3 h produced by daily dosing with A-77636 or dinapsoline for 14 days. A-77636 was dosed once daily, and dinapsoline was dosed once or twice daily.
In contrast, no evidence for behavioral tolerance was observed for DNS when dosed once or twice daily at 2 mg/kg s.c. (Fig. 4). As described above, the duration of response to DNS at this dose was about 10 h, whereas A-77636 produced rotation for approximately 18 h when dosed at 1 mg/kg s.c. (Kebabian et al., 1992). Our own experiments confirmed this; A-77636 (1 mg/kg s.c.) produced rotation for greater than 14 h at rates comparable to those seen with DNS, 150 to 200 rotations per 15-min time bin (data not shown). To account for this difference in duration, a group of animals was dosed twice daily with DNS. Rather than a decrease in response, dinapsoline produced a significant increase in responding over time whether it was dosed once daily (F 13,52 = 42.0,p < 0.001) or twice daily (F 13,52 = 3.0, p = 0.006). This observation indicates that DNS may produce behavioral sensitization, rather than tolerance, under intermittent dosing regimens. The once per day dosing regimen produced a stronger sensitizing effect than did the twice per day regimen (F 13,104 = 3.1, p = 0.009).
Contribution of D2 Receptor Activity to Tolerance.
To assess the basis for the difference in tolerance-producing properties between A-77636 and DNS, we examined the possibility that D2 receptor activity confers some resistance to tolerance. First, we compared the effect of chronic coadministration of raclopride (2 mg/kg s.c.) with DNS (2 mg/kg s.c.) for 6 days (Fig.5A). There was no significant difference in rotational response to DNS with or without raclopride on days 1 through 6 (F 5,45 = 0.2,p > 0.05). On day 7, DNS alone was given to both groups (Fig. 5A), to confirm the lack of behavioral tolerance; again no difference was observed (F 1,9 = 0.1,p = 0.72). These results indicate that D2 agonist activity is probably not responsible for the lack of tolerance observed with DNS.

A, cumulative rotation (mean ± S.E.M.,n = 8/group) over 3 h produced by daily dosing with dinapsoline (2 mg/kg s.c.) with and without coadministration of raclopride (2 mg/kg s.c.). Day 7 represents a challenge dose in which neither group received raclopride pretreatment. B, cumulative rotation (mean ± S.E.M., n = 8/group) over 3 h produced by a single daily injection of A-77636 (0.3 mg/kg) with and without coadministration of quinpirole (0.1 mg/kg). On days 1 and 2, rats received either A-77636 plus vehicle or plus quinpirole. As challenge doses, all animals received only A-77636 on day 3 and only quinpirole on day 4.
To explore this further, the D2 agonist quinpirole was coadministered subcutaneously in combination with the more selective D1 agonist A-77636. As shown in Fig. 5B, A-77636 alone (black bars) caused a maximal rotational response on day 1, yet significant tolerance by day 2. On day 1, the response in rats treated with both A-77636 and quinpirole (white bars) was somewhat less than that in rats treated with A-77636 alone (F 1,13 = 5.9, p = 0.03). Conversely, by day 2 (F 1,12 = 12.4, p = 0.004), the rats treated with A-77636 plus quinpirole had a greater response than those treated with A-77636 plus vehicle (probably due solely to the actions of quinpirole). The challenge dose of A-77636 (0.3 mg/kg s.c.) on day 3 demonstrated equal tolerance in both groups (F 1,13 = 0.1,p > 0.05), indicating that cotreatment with quinpirole was not “protective”. Similarly, on day 4, quinpirole produced equal rotation in both groups, indicating that tolerance was specifically related to D1 receptor function.
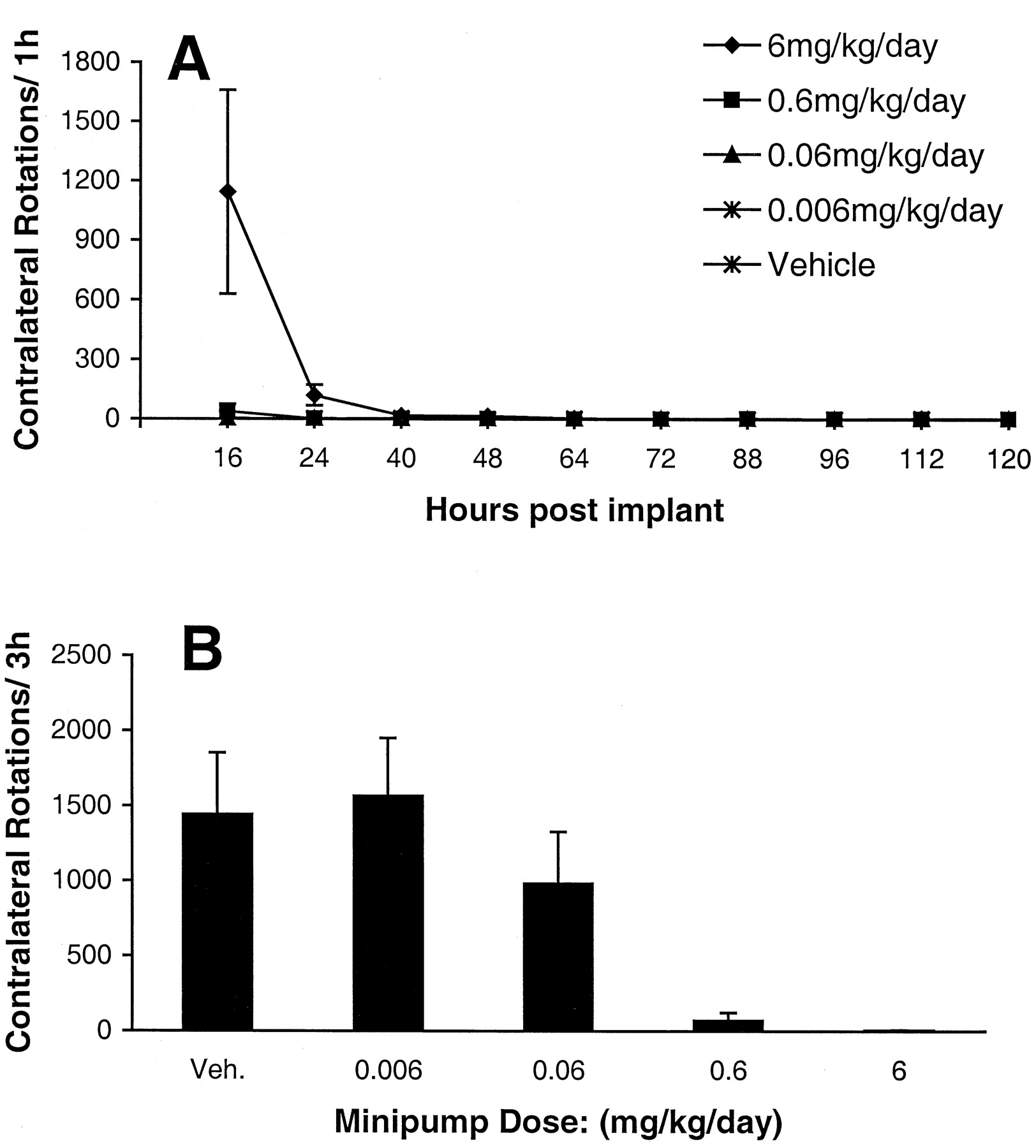
Further experiments tested whether the cause of the difference in response to chronic treatment with A-77636 and DNS was related to the pattern of exposure to the drug. DNS was administered via osmotic minipump, and behavioral testing was performed for 1 h twice daily beginning 16 h after implantation (Fig.6A). DNS was administered at four different doses; the highest dose produced a brief behavioral response that was tolerated by 24 h, whereas the lower doses produced no evidence of rotation. To confirm that the loss of response represents tolerance rather than a technical problem, a test dose of DNS (0.2 mg/kg s.c.) was given on day 14 after minipump implantation (Fig. 6B). This DNS challenge produced no rotation in the groups that received either 6 or 0.6 mg/kg/day of DNS by minipump, confirming that the loss of effect represented tolerance

A, rotations (mean ± S.E.M.,n = 8/group) per 1-h time bin at various time points following implantation of osmotic minipumps administering dinapsoline subcutaneously. B, effect of a challenge dose of dinapsoline (0.2 mg/kg s.c.) on rotation (mean + S.E.M.) in rats that received various doses of dinapsoline by osmotic minipump for 14 days.
Discussion
Rats with unilateral 6-OHDA lesions in the medial forebrain bundle exhibited robust contralateral turning behavior when challenged with DNS. This turning behavior in response to DNS demonstrated a strong dose-response relationship with doses below 0.2 mg/kg s.c. inducing no significant rotation behavior (Fig. 1A). The duration of rotation was proportional to the dose of DNS administered; higher doses of DNS (2 mg/kg s.c.) produced rotation that lasted approximately 10 h (Fig.1B). Dinapsoline also produced robust rotation after oral administration (Fig. 2A) when administered at a dose of 2 mg/kg. This oral efficacy of DNS offers significant advantage over other full D1 agonists like DHX and ABT-431 that have demonstrated anti-parkinsonian effects in humans only when given parenterally (Blanchet et al., 1998; Rascol et al., 1999).
The rotational behavior induced by DNS appears to be attributable to D1 receptor stimulation in vivo. The D1-selective antagonist SCH-23390 (Fig. 3A) completely blocked DNS-induced rotation, whereas the D2-selective antagonist raclopride did not affect rotation (Fig. 3B). In contrast, this dose of raclopride fully blocked rotation produced by the D2 agonist quinpirole (data not shown). This functional profile in vivo contrasts with the in vitro data that suggest that, although DNS is a high affinity full D1 agonist (K 0.5= 5.9 nM), it also has substantial affinity for D2 receptors (K 0.5 = 31 nM; Ghosh et al., 1996). One possible explanation is that DNS is not a pure D2 agonist but may have some partial agonist or antagonist properties at selected D2-like receptors, a concept sometimes termed “functional selectivity” (Mailman et al., 1998; Lawler et al., 1999) or “agonist trafficking” (Kenakin, 1995). Future experiments are needed to assess the possibility of such differential D2functional responses in more detail.
Chronic DA agonist treatment has the potential to produce tolerance by producing postsynaptic subsensitivity. Repeated daily dosing with A-77636 produced complete tolerance by day 4 (Fig. 4), consistent with a previous study (Asin and Wirtshafter, 1993). In contrast, DNS administered either once or twice daily for 14 days consistently caused turning behavior in rats without producing tolerance. In fact, both chronic administration paradigms for DNS caused behavioral sensitization over the 14-day period, although a second set of rats demonstrated no evidence of sensitization when dosed once daily (Fig.5). Because these studies relied upon automated measuring devices, it cannot be determined retrospectively whether competing behaviors (e.g., various stereotypies) influenced these results. Moreover, the ability to demonstrate sensitization is often critically dependent on the regimen used, making the impact of this aspect of the current results further unclear. It is interesting that other DA agonists that may produce sensitization in unilateral 6-OHDA-lesioned rats such as apomorphine, l-DOPA, and bromocriptine (Gancher at al., 1995; Henry et al., 1998) are not reported to produce sensitization in the clinic. Indeed, the relationship of sensitization/tolerance in the rat unilateral 6-OHDA model as compared with Parkinson's patients may be tenuous, at best, based on differences in species, as well as type and extent of lesion.
The difference in the tolerance profiles for DNS and A-77636 may be influenced by several factors, including the pattern of exposure of the D1 receptors, the relative activation of D1 versus D2 receptors, and intrinsic pharmacodynamic differences of the drugs at D1 receptors. Indeed, in at least one cell line transfected with the primate D1A receptor, DNS causes less desensitization than does A-77636 (Lewis et al., 1998), although there is no proven relationship between the molecular desensitization of the D1 receptor in vitro and tolerance in vivo. Thus, it is important to determine how the behavioral effects of repeated DA agonist treatment depend on drug doses and treatment regimens, as well as pharmacokinetic and pharmacodynamic properties of the administered compound.
A dopaminergic agonist can either induce up- or down-regulation with the duration of the drug-free period between successive administrations being the important factor determining the direction of the effect (Robinson, 1984; Castro et al., 1985; Henry et al., 1998). A-77636 has a long plasma half-life (>6 h) and a long duration of action (≈18 h) resulting in persistent D1 receptor stimulation (Asin and Wirtshafter, 1993) that may contribute to the receptor desensitization. In contrast, DNS has a moderate duration of action (about 10 h) that allows for a limited duration of D1 receptor activation. Dinapsoline, administered subcutaneously by minipump to simulate conditions of extended receptor activation, produced complete tolerance to contralateral rotation behavior in about 30 h. Thus, intermittent administration of DNS did not produce tolerance, whereas constant infusion produced rapid tolerance. These observations are similar to other preclinical studies in mice and rats where evidence of tolerance was observed upon constant, but not intermittent administration of apomorphine (Winkler and Weiss, 1986; Grandas et al., 1992; Gancher et al., 1995). In addition, clinical studies using constant infusions of the dopaminergic agents l-DOPA (Mouradian et al., 1990; Nutt et al., 1993;Schuh and Bennett, 1993) and apomorphine (Gancher et al., 1992) have demonstrated a reduction in dopaminergic sensitivity.
The propensity of D1 agonists to produce tolerance seems to relate to duration of receptor activation, but the relationship is complex. Twice daily dosing with DNS produced levels of rotation comparable to that from once daily dosing with A-77636, yet A-77636, but not DNS, showed tolerance. Thus, some component of repeated dosing, in addition to duration of action, is involved in the response. It would be useful in future studies to have parallel data on the plasma (and even brain) concentrations of both agents over time.
Dinapsoline is a full D1 agonist with moderate activity at the D2 receptor and thus has an appropriate profile to explore the role of D2receptors in conferring resistance to tolerance under chronic administration paradigms. In the present study, chronic intermittent administration of DNS did not produce tolerance in a 14-day study. If agonist activity at the D2 receptor is indeed responsible for the resistance to tolerance, concurrent administration of a D2 antagonist should have produced tolerance by eliminating this “protective” mechanism. This was not observed, however, as concurrent administration of raclopride had no significant effect on DNS-induced contralateral rotation in the chronic dosing paradigm. This idea was explored in parallel by looking at the role of D2 receptor activation in the tolerance development induced by A-77636. In this case, chronic D2 receptor stimulation by concurrent administration of a D2 agonist would be predicted to inhibit induction of tolerance. Again, this was not observed, as chronic concurrent administration of quinpirole had no significant effect in either inhibiting or delaying the rapid tolerance produced by A-77636. Thus, in the unilateral 6-OHDA model, activation of the D2 receptor does not appear to impact on the development of tolerance to chronic administration of full D1 agonists.
It is interesting to note that coadministration of quinpirole with A-77636 produced less rotation than A-77636 alone, whereas this combination would be expected to produce additive effects. It is possible that this combination dose resulted in stereotypy that interfered with the rotational behavior. Indeed, the dose of A-77636 (0.3 mg/kg) used in combination with quinpirole was lower than that (1 mg/kg) used in the other experiments, because higher doses of A-77636 administered in combination with quinpirole produced behavioral toxicity that was sometimes lethal.
In summary, DNS is a full D1 DA receptor agonist that produces robust rotational activity in the unilateral 6-OHDA rat model. This compound has advantages over previous D1 agonists in that it is orally active while showing a low probability for producing tolerance. Other animal studies have indicated that D1-selective agonists may be less likely to produce dyskinesias than either l-DOPA or D2-selective agonists (Falardeau et al., 1988;Blanchet et al., 1993). Thus, DNS may offer advantages over D2 agonists in providing comparable efficacy with a lower tendency to induce dyskinesia. Final assessments of the efficacy of DNS and its dyskinesia liability will be performed in MPTP-lesioned primates, a model that is considered more predictive of effects in patients.
Footnotes
- Received July 11, 2000.
- Accepted October 26, 2000.
-
Send reprint requests to: Dr. Matthew Taber, Bristol-Myers Squibb, Dept. 405, 5 Research Parkway, Wallingford, CT 06492. E-mail:taberm{at}bms.com
-
This research was funded by Bristol-Myers Squibb Company. R.B.M. and D.E.N. were supported in part by National Institutes of Health Grants MH40537 and MH42705, respectively.
Abbreviations
- PD
- Parkinson's disease
- 6-OHDA
- 6-hydroxydopamine
- A-77636
- (1R,3S)-3-(1′-adamantyl)-1-aminomethyl-3,4-dihydro-5,6-dihydroxy-1H-2-benzopyran
- A-68930
- 5,6-dihydroxy-3-phenyl-1-aminomethyl-isochroman
- ABT-431
- 9,10-acetoxy-2-propyl-4,5,5a,6,7,11b-hexahydro-3-thia-5-azacyclopent-1-ena[c]phenanthrene hydrochloride
- CY 208–243
- (−)-4,6,6a,7,8,12b-hexahydro-7-methylindolo[4,5-ab] phenanthridine
- DA
- dopamine
- DMSO
- dimethyl sulfoxide
- l-DOPA
- l-dihydroxyphenylalanine (levodopa)
- DHX
- dihydrexidine
- DNS
- dinapsoline
- MPTP
- N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride
- SKF-38393
- (±)-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazapine
- SCH-23390
- R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}