


Abstract
Reactive oxygen species decrease dopamine transporter (DAT) functionin vitro. Because of this, and the finding that METH administration causes oxygen radical formation in vivo, the effects of METH administration on DAT activity in rat striatum were investigated. A single METH injection caused a dose-dependent (0–15 mg/kg) decrease in [3H]dopamine uptake into striatal synaptosomes prepared 1 h after METH administration; an effect attributable to a decreased V max of [3H]dopamine uptake. Similarly, multiple high-dose administrations of METH (10 mg/kg/dose; four doses at 2-h intervals) decreased DAT function. The decreases in DAT activity after either single or multiple METH administrations were reversed 24 h after treatment. [3H]5HT transport into striatal synaptosomes was also affected by METH treatment. Taken together, these data suggest that METH decreases DAT activity, perhaps through a reactive oxygen species-mediated mechanism. These findings may have important implications regarding the role of oxidative events in the physiological regulation of monoaminergic systems.
Abuse of the amphetamine analog, METH, is a serious worldwide health problem. Hence, there is considerable effort to elucidate mechanisms whereby METH alters central nervous system function. Deleterious effects of high-dose METH administration on monoaminergic neurons in rodents and primates, including decreases in brain levels of dopamine and 5HT, and the activity of their respective synthetic enzymes, tyrosine hydroxylase and TPH, have been described (Hotchkiss and Gibb, 1980;Wagner et al., 1980; Bakhit et al., 1981;Woolverton et al., 1989). Although mechanisms resulting in these deficits remain unclear, METH administration promotes formation of ROS (Kondo et al., 1994; Giovanni et al., 1995; Fleckenstein et al., 1996a) which likely contribute to impairment of monoamine systems, such as the rapid and reversible decrease in activity of TPH observed after a single high dose of METH (Stone et al., 1989a, b).
Aminergic transporters are among the neuronal constituents susceptible to damage by ROS. After in vitro exposure to a variety of oxidative conditions, decreases in glutamatergic (Volterra et al., 1994), GABAergic (Braughler, 1985; Debler et al., 1986) and dopaminergic (Berman et al., 1996; Metzgeret al., 1996) transporter function have been investigated. The sites on these transporters that are susceptible to oxidative modification are unknown, although cysteinyl residues located on a putative extracellular loop of carriers such as the DAT are plausible candidates. We recently suggested a role for superoxide radicals in decreasing DAT function (Metzger et al., 1996). Because METH likely causes formation of this (Hirata et al., 1995, 1996) and other ROS, an oxidative effect of METH on DAT would be expected. The purpose of this study was to characterize the response of DAT to METH treatment. The results reveal that a single METH administration causes a rapid and reversible decrease in DAT function, reminiscent of its effects on TPH activity. Multiple high-dose administrations of METH also cause a reversible decrease in DAT activity. The significance of these findings to mechanisms responsible for METH neurotoxicity, as well as for the physiological regulation of dopaminergic systems is discussed.
Methods
Animals.
Male Sprague-Dawley rats (200–300 g; Simonsen Laboratories, Gilroy, CA) were maintained under conditions of controlled temperature and lighting, with food and water providedad libitum. Rats were killed by decapitation. All procedures were conducted in accordance with approved National Institutes of Health guidelines.
Drugs and chemicals.
(±)Methamphetamine hydrochloride and (-)cocaine hydrochloride were supplied generously by the National Institute on Drug Abuse (Rockville, MD). Analytical reference materials [+METH and deuterated METH (METH-d8)] for METH determination were obtained from Radian Corporation (Austin, TX). Citalopram hydrobromide and pargyline hydrochloride were kindly supplied by H. Lundbeck and Co., and Abbott Laboratories (North Chicago, IL), respectively. [7,8-3H]Dopamine (43 Ci/mmol) and [3H]5HT (30 Ci/mmol) were purchased from Amersham Life Sciences (Arlington Heights, IL) and New England Nuclear (Boston, MA), respectively. Drugs were administered as indicated in the legends of appropriate figures; doses were calculated as the respective free bases.
Synaptosomal [3H]dopamine and [3H]5HT uptake.
Uptake of [3H]monoamines was determined according to a modification of a method described by Boja et al. (1992). Fresh striatal tissue was homogenized in ice-cold 0.32 M sucrose and centrifuged (800 × g for 12 min; 4°C). The resulting supernatant (S1) was then centrifuged (22,000 × g for 10 min; 4°C), and the pellets (P2) resuspended in ice-cold 0.32 M sucrose. In experiments designated as “washout” experiments, resuspended P2 fractions were centrifuged (22,000 × gfor 10 min; 4°C). The resulting pellet (P3) was then resuspended in 0.32 M sucrose and once again centrifuged (22,000 × gfor 10 min; 4°C) to obtain a pellet (P4) which was subsequently resuspended and assayed. Assays were conducted in modified Kreb’s buffer (in mM: NaCl, 126; KCl, 4.8; CaCl2, 1.3; sodium phosphate, 16; MgSO4, 1.4; dextrose, 11; ascorbic acid, 1; pH 7.4). Each assay tube contained synaptosomal tissue (i.e,. resuspended P2 (or P4 in “washout” experiments) obtained from 1.5 mg (for [3H]dopamine uptake) or 7.5 mg (for [3H]5HT uptake) striatal tissue, original wet weight) and 1 μM pargyline. For kinetic experiments (fig. 2), striatal tissue was pooled from three control or METH-treated rats. In experiments designed to determine its IC50, METH (100 pM–10 μM) was likewise present in assay tubes. Nonspecific values were determined in the presence of 1 mM cocaine (for [3H]dopamine uptake) or 1 μM citalopram (for [3H]5-HT uptake). After preincubation of assay tubes for 10 min at 37°C, assays were initiated by addition of [3H]dopamine or [3H]5-HT (0.5 or 5 nM final concentrations, respectively, except as indicated in the legend to fig. 2). Samples were incubated at 37°C for 3 min and then filtered through Whatman GF/B filters soaked previously in 0.05% polyethylenimine. Filters were washed rapidly 3 times with 5 ml ice-cold 0.32 M sucrose using a Brandel filtering manifold. Radioactivity trapped in filters was counted using a liquid scintillation counter.

Effects of METH administration onKm and V max of [3H]dopamine uptake in striatal synaptosomes. Rats received METH (15 mg/kg s.c.) or saline vehicle (1 ml/kg s.c.) 1 h before decapitation. Synaptosomes were assayed for [3H]dopamine uptake, as described under “Methods,” with use of [3H]dopamine at final concentrations of 0.5 to 1000 nM. The Eadie-Hofstee plot depicts data from one of three experiments conducted with samples in each run in duplicate. The mean Km values from all experiments combined (n = 3) were 91 ± 7 and 78 ± 7 nM for synaptosomes prepared from control and METH-treated rats, respectively. The mean V max values from control and treated rats were 3282 ± 608 and 2175 ± 671 fmol/mg original wet weight tissue/3 min, respectively; these values differed significantly (P < .05).
METH determination.
Striatal synaptosomes were prepared as described above and maintained frozen at −70°C until assay. On the day of the assay, samples were allowed to equilibrate to room temperature. To each sample, 300 ng of deuterated METH (METH-d8) was added as internal standard. Samples were vortexed for 10 s. For tissue samples, 1 ml of 2 N NaOH was added to solubilize each sample, and the resulting preparations were then transferred to silanized glass tubes. Samples were incubated at 37°C for 1 h. After cooling samples to room temperature, 100 μl of concentrated ammonium hydroxide was added to each tube and then extracted into 4 ml of butyl chloride/chloroform (4:1, v/v) for 30 min. Samples were centrifuged for 45 min at 1200 × g and each organic phase (containing analytes of interest) was transferred to a clean tube. Trifluoroacetic acid anhydride (200 μl) was added to each organic phase and heated for 30 min at 70°C. The extracts were cooled to room temperature and then evaporated to dryness at 40°C. A 14-point standard curve ranging from 1 to 1000 ng/ml was prepared with human plasma and extracted as described above. Analytical accuracy and intra-assay precision were verified by concurrent analysis of quality control samples that were prepared in drug-free rat brain homogenate (5 and 500 ng/ml). Extracts were reconstituted in 100 μl of chloroform prior to analysis by gas chromatography/mass spectrometry.
Concentrations of METH were determined with a Finnigan 4500 MAT mass spectrometer operating in positive chemical ionization mode (methane/ammonia reagent gas) coupled to a DB5 MS-30 M-0.25 μ capillary column. Ions monitored were 263 m/z and 271m/z (METH and METH-d8, respectively). Accuracy was within 6–17% of spiked METH target values for brain homogenate quality control samples. The limit of quantitation in these experiments was 1 ng/ml.
Data analysis.
Statistical analyses between two groups were conducted using a 2-tailed Student’s t test. A paired 2-tailed t test was employed when comparing Vmax data. Analyses among three or more groups were conducted using analysis of variance followed by Fisher’s test. IC50 values were determined using EBDA [McPherson, 1986] computer software. Differences among groups were considered significant if the probability of error was less than 5%.
Results
Results presented in figure 1demonstrate that METH administration caused a dose-related decrease in [3H]dopamine uptake in striatal synaptosomes prepared from rats decapitated 1 h after METH administration. The highest dose of METH used, 15 mg/kg s.c., decreased [3H]dopamine uptake by 65%, and was used in all subsequent single-dose experiments. The decreased uptake was associated with a decrease in DAT V max(fig. 2). Km was virtually unaffected by METH administration.

Dose-response effects of METH administration on striatal [3H]dopamine uptake in rats. Rats received METH (0.5–15 mg/kg s.c.) or saline vehicle (1 ml/kg s.c.) 1 h before decapitation. Values represent means (fmol/mg original wet weight tissue) and vertical lines are 1 S.E.M. of determinations in six rats. *Values for METH-treated rats that are significantly different from saline-treated rats (P ≤ .05).
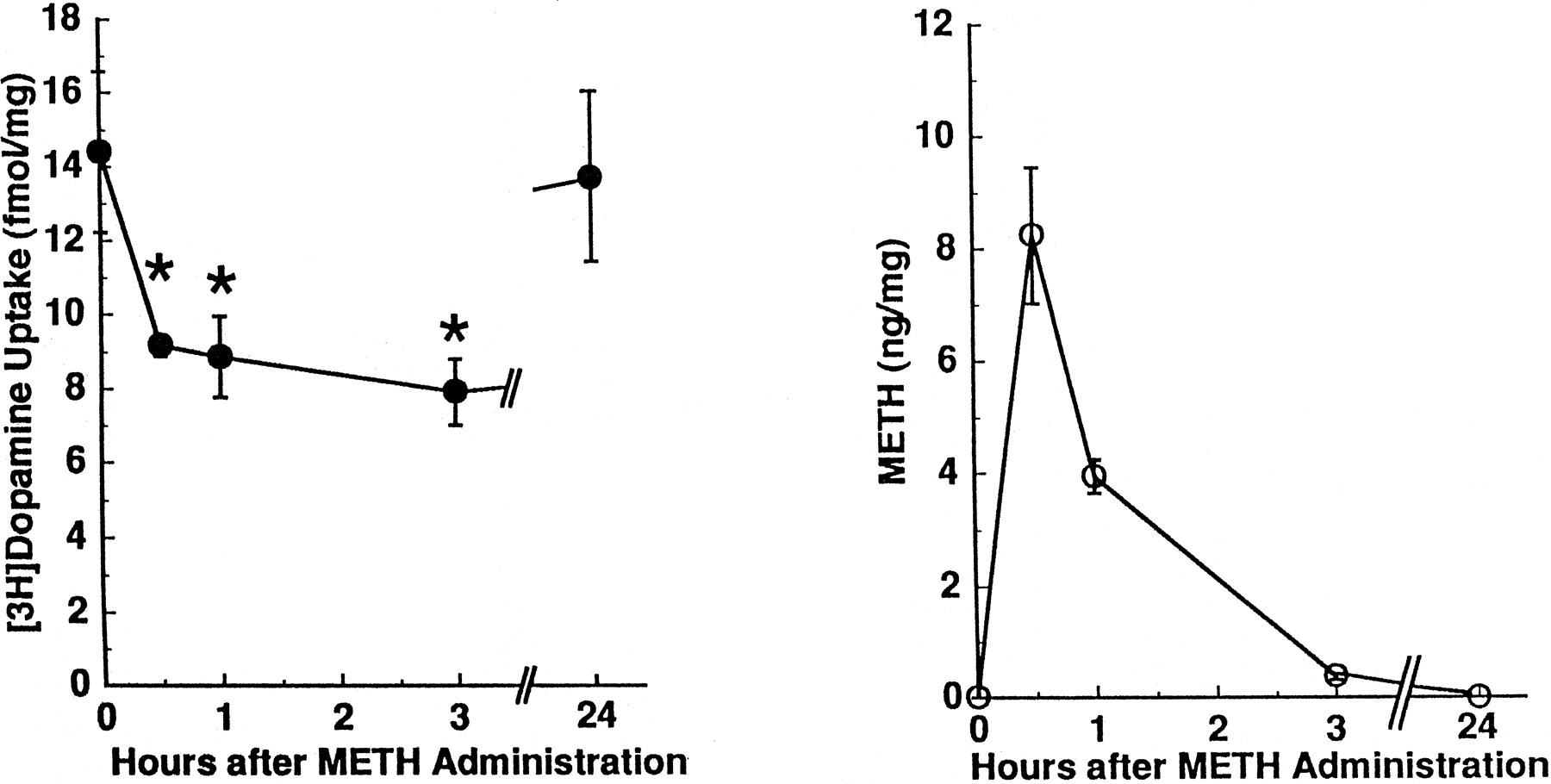
The METH-induced decrease in striatal [3H]dopamine uptake was reversible with time and persisted less than 24 h (fig.3, left panel). Because METH applicationex vivo can decrease directly striatal [3H]dopamine uptake (IC50= 291 ± 4 nM; table 1), the possibility that residual METH, introduced by the in vivosubcutaneous injection, directly decreased [3H]dopamine uptake was investigated. Results presented in figure 3 argue against this possibility: METH was virtually nondetectable in brain tissue (whole brain minus the striatum and cerebellum) 3 h after administration (fig. 3, right panel) even though [3H]dopamine uptake was decreased by 45% in these same animals.
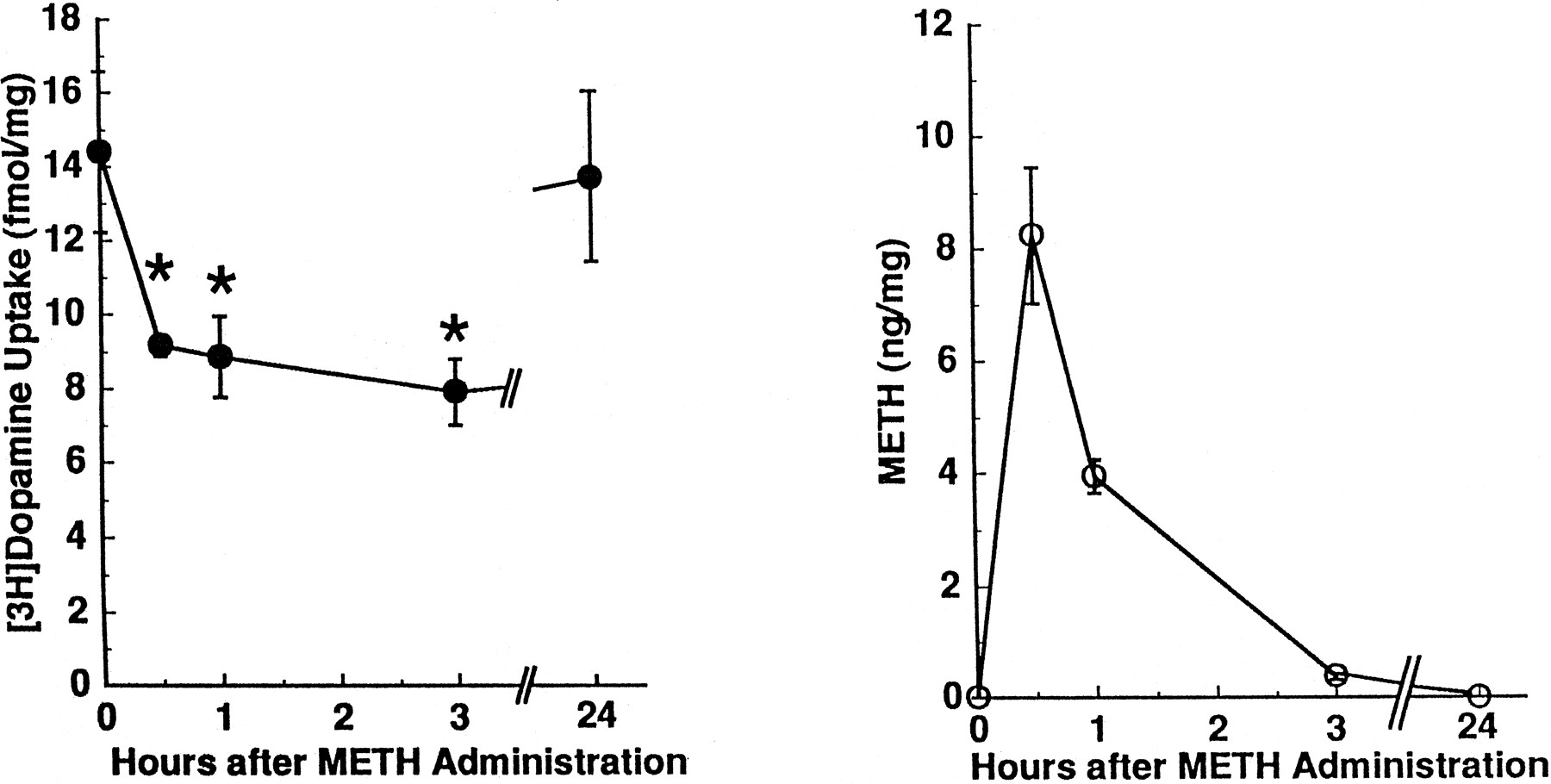
Time-response effects of METH administration on striatal [3H]dopamine uptake (left panel) and brain METH concentrations (right panel) in rats. Treated rats received METH (15 mg/kg s.c.) 0.5 to 24 h, and control rats received saline vehicle (1 ml/kg s.c.; zero time values) 0.5 h before decapitation. Values represent means (fmol or ng/mg original wet weight tissue) and vertical lines are 1 S.E.M. of determinations in six to eight rats. *Values for METH-treated rats that are significantly different from saline-treated rats (P ≤ .05).
Comparison of METH concentrations required to affect directly [3H]dopamine uptake with that present in synaptosomes prepared from METH-treated rats
To test more directly whether residual METH could cause decreased dopamine uptake (i.e., such as that depicted in figs. 1, 2, 3), a “washout” experiment (see “Methods”) was conducted. Despite successfully removing residual METH (table 1), successive washing of striatal synaptosomes did not diminish METH-induced decreases in [3H]dopamine accumulation (fig.4). Furthermore, the METH concentrations in unwashed and washed synaptosomes listed in table 1 were substantially less than the IC50 for [3H]dopamine uptake, and less than the concentration necessary to effect the METH-induced decrease in [3H]dopamine uptake depicted in figure 1.

Effects METH administration on [3H]dopamine uptake in zero and twice washed striatal synaptosomes. Rats received METH (15 mg/kg s.c.) or saline vehicle (1 ml/kg s.c.) 1 h before decapitation. “Washing” of synaptosomes was performed as described under “Methods.” Columns represent means and vertical lines are 1 S.E.M. of determinations in eight rats. *Values for METH-treated rats (fmol/mg original wet weight tissue) that are significantly different from saline-treated rats (P ≤ .05).
Results presented in figure 5demonstrate that, in addition to affecting [3H]dopamine uptake, multiple METH administrations (10 mg/kg/dose; four doses at 2-h intervals) decreased uptake of [3H]5HT uptake in striatal synaptosomal preparations. The time-course effect of multiple METH administrations (10 mg/kg/dose; four doses at 2-h intervals) on [3H]dopamine uptake is presented in figure6: a biphasic pattern of response was observed consisting of a reversal of the decrease in striatal [3H]dopamine uptake occurring 24 h after treatment, followed by a significant decrease 8 days later.

Comparative effects of METH administration on striatal [3H]dopamine and [3H]5HT uptake in rats. Treated rats received four injections of METH (2-h intervals; 10 mg/kg/injection s.c.) and were decapitated 1 h after the final METH administration. Control rats received four injections of saline vehicle (1 ml/kg/injection s.c.) and were decapitated 1 h after the final saline injection. Values represent means (fmol/mg original wet weight tissue) and vertical lines are 1 S.E.M. of determinations in five rats. *Value for METH-treated rats that is significantly different from corresponding saline-treated rats (P ≤ .05).
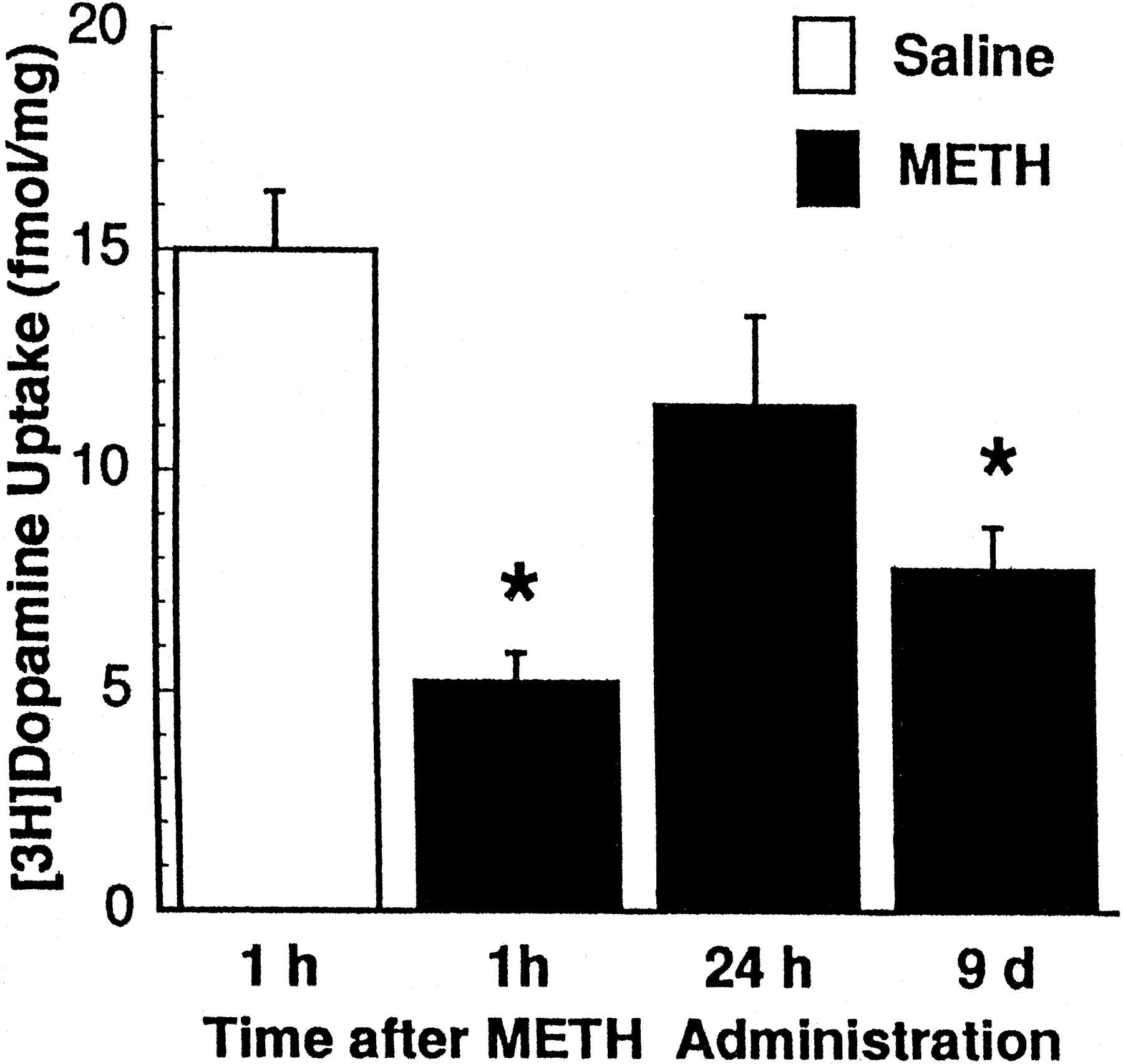
Time-response effects of multiple METH administrations on striatal [3H]dopamine uptake in rats. Treated rats received four injections of METH (2-h intervals; 10 mg/kg/injection s.c.) and were decapitated 1 h, 24 h and 9 days after the final METH administration. Control rats received four injections of saline vehicle (1 ml/kg/injection s.c.) and were decapitated 1 h after the final saline injection. Values represent means (fmol/mg original wet weight tissue) and vertical lines are 1 S.E.M. of determinations in eight rats. *Values for METH-treated rats that are significantly different from saline-treated rats (P ≤ .05).
Discussion
Administration of high doses of METH can cause long-term deficits in dopaminergic systems, including decreases in transporter numbers, dopamine concentrations and tyrosine hydroxylase activity (Hotchkiss and Gibb, 1980; Wagner et al., 1980; Ricaurte et al., 1982; Woolverton et al., 1989). These deficits can persist for months, and are likely associated with nerve-terminal degeneration. Results from the present study demonstrate that apart from this well characterized, long-term neurotoxic outcome, METH administration causes rapid and reversible changes in dopaminergic systems as well. As early as 30 min after a single administration, METH decreased striatal DAT function, as reflected by a decrease in [3H]dopamine uptake in synaptosomes prepared from METH-treated rats. This effect was apparently reversible, because DAT activity returned to control levels by 24 h after METH treatment (fig. 3). A similar finding of rapid and reversible reductions in [3H]dopamine uptake has been described after p-chloroamphetamine treatment bySanders-Bush et al. (1975).
The METH-induced reduction in [3H]dopamine uptake results from a decrease in transporterV max (fig. 2). These data are consistent with previous findings from this laboratory that exposure of DAT to the ROS-generating enzyme, xanthine oxidase, can decrease theV max of DAT as well (Fleckenstein et al., in press). The METH effect appears not to result from residual METH in the synaptosomal preparation (i.e., METH introduced by the in vivo drug treatment), because: 1) residual METH levels found in synaptosomes prepared from METH-treated rats were far less than those necessary to affect directly [3H]dopamine uptake in striatal preparations (table 1); and 2) successive washing of METH-treated synaptosomes did not diminish the decrease in [3H]dopamine uptake caused by METH administration (fig. 4) while lowering the METH below detectable levels (table 1). Furthermore, METH was virtually nondetectable in brain tissue 3 h after administration; a time point at which [3H]dopamine uptake was decreased by 45% (fig. 3).
The rapid decrease in DAT activity is not likely the result of neuronal cell death, because indicators of toxicity such as long-term deficits in tyrosine hydroxylase activity do not occur after a single dose of METH (15 mg/kg; Hotchkiss and Gibb, 1980). Moreover, the decrease in [3H]dopamine uptake does not appear to be associated with a loss of DAT protein, because the 24-h period demonstrated to restore METH-affected uptake to control levels is far less than the time likely required to synthesize replacement DAT (i.e., the t 1/2 of DAT turnover is approximately 6.3 days; Fleckenstein et al., 1996b). Rather, the METH-induced decrease in DAT activity may reflect regulation of DAT via a reversible modification of its structure: a comparable phenomenon has been proposed to result from DAT phosphorylation (Huff et al., 1996; Vaughan et al., 1996). Possible structural changes causing the present DAT effects may be the result of METH-induced ROS, because DAT are among the neuronal constitutents that are especially susceptible to oxidative damage and ROS are formed after METH administration. If protein oxidation were responsible for DAT impairment, the likely mechanism for its reactivation would be a functional restoration of the transporter by reducing events. For example, reducing agents such as glutathione and ascorbate found in high concentrations in the brain (Reiter, 1995) could help reverse oxidative inactivation of DAT.
Considerable similarity exists between the structures of the transporters for dopamine and 5HT, including cysteinyl residues on putative extracellular loops. Given the hypothesis that METH-induced ROS alter transporter function, it is not surprising that the activity of both DAT and the 5HT transporter are decreased after multiple administrations of METH (fig. 5). Also consistent with a role for METH-induced ROS in decreasing DAT activity is the relatively rapid onset of this effect after either single or multiple METH administrations. The rapid effect after multiple administrations does not likely result from residual METH (i.e., introduced by the injections) decreasing [3H]dopamine uptake, because greater than 99% of the residual drug is removed during preparation of the synaptosomes (see above). The decrease in DAT activity after multiple METH administrations was restored to control levels 24 h after the last METH injection, but was again decreased 8 days later. The recovery and subsequent loss of DAT function may reflect two distinct events: the first associated with a reversible DAT modification and the second with neurotoxicity and terminal degeneration. Consistent with the latter, long-term decreases in dopamine uptake sites after multiple METH administrations associated with neurotoxicity have been described (Wagner et al., 1980).
It is interesting to speculate about the implications and functional consequences of a METH-induced change in DAT function. Because DAT is the primary means whereby dopamine is cleared from the synaptic cleft, disruption of DAT could lead to increased extracellular dopamine and ultimately the production of highly damaging dopamine-related ROS. On the other hand, reduction in transporter activity may be neuroprotective, as evidenced by findings that monoamine transport inhibitors protect against damage associated with METH treatment (Schmidt and Gibb, 1985). Further investigation into the association among METH, ROS and DAT with regard to functional implications is required.
In conclusion, the present study demonstrates that either single or multiple METH administrations effect a rapid and reversible decrease in DAT activity. These data, taken together with previous findings that METH administration promotes ROS formation and that ROS can impair DAT function, support the hypothesis that the METH-induced effect on DAT is caused by ROS. The possibility that reactive species can modify other functional proteins is intriguing and is consistent with reports that such species can modify the function of other transporters (see above). The fact that the inactivation of DAT was reversible suggests the possibility that this is a significant mechanism for regulating DAT function under both drug-affected and normal physiological conditions.
Acknowledgments
The authors acknowledge gratefully the excellent technical assistance of Meisha Beyeler.
Footnotes
-
Send reprint requests to: Annette E. Fleckenstein, Ph.D., Department of Pharmacology and Toxicology, 112 Skaggs Hall, University of Utah, Salt Lake City, UT 84112.
-
↵1 This research was supported by grants DA 00869, DA 04222 and DA 05780 from the National Institute on Drug Abuse.
- Abbreviations:
- DAT
- dopamine transporter
- 5HT
- 5-hydroxytryptamine
- METH
- methamphetamine
- ROS
- reactive oxygen species
- TPH
- tryptophan hydroxylase
- Received December 23, 1996.
- Accepted April 7, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}