


Abstract
Acetaminophen (AAP) is metabolized by a variety of pathways such as sulfation, glucuronidation, and fatty acid amide hydrolase–mediated conversion to the active analgesic metabolite AM404. CYP2E1-mediated metabolism to the hepatotoxic reactive metabolite NAPQI (N-acetyl-p-benzoquinone imine) is a minor metabolic pathway that has not been linked to AAP therapeutic benefits yet clearly leads to AAP liver toxicity. N-acetylcysteine (NAC) (an antioxidant) and fomepizole (a CYP2E1 inhibitor) are clinically used for the treatment of AAP toxicity. Mice treated with AAP in combination with fomepizole (plus or minus NAC) were assessed for liver toxicity by histology and serum chemistry. The anticancer activity of AAP with NAC and fomepizole rescue was assessed in vitro and in vivo. Fomepizole with or without NAC completely prevented AAP-induced liver toxicity. In vivo, high-dose AAP with NAC/fomepizole rescue had profound antitumor activity against commonly used 4T1 breast tumor and lewis lung carcinoma lung tumor models, and no liver toxicity was detected. The antitumor efficacy was reduced in immune-compromised NOD-scid IL2Rgammanull mice, suggesting an immune-mediated mechanism of action. In conclusion, using fomepizole-based rescue, we were able to treat mice with 100-fold higher than standard dosing of AAP (650 mg/kg) without any detected liver toxicity and substantial antitumor activity.
SIGNIFICANCE STATEMENT High-dose acetaminophen can be given concurrently with CYP2E1 inhibition to allow for safe dose escalation to levels needed for anticancer activity without detected evidence of toxicity.
Introduction
Acetaminophen (AAP) is metabolized by a variety of pathways, including glucuronidation, sulfation, and fatty acid amide hydrolase–mediated conversion to the active analgesic metabolite N-acylphenolamine (AM404) (Ohashi and Kohno, 2020). Additionally, CYP2E1-mediated conversion of AAP into a transient reactive intermediate, N-acetyl-p-benzoquinone imine (NAPQI), is a minor metabolic pathway, comprising 10% of AAP metabolism. Within the liver, NAPQI is rapidly detoxified by glutathione (GSH), and in overdose, hepatic GSH becomes depleted, leading to free radical–mediated liver toxicity (Heard, 2008).
The CYP2E1 metabolic pathway is clearly responsible for the toxicity of AAP; CYP2E1 knockout mice are essentially immune from high-dose AAP–induced liver damage (Lee et al., 1996). However, considering NAPQI is a transient reactive intermediate byproduct of a minor metabolic pathway that is rapidly detoxified by GSH in the liver, it is unlikely that CYP2E1 metabolism is required for AAP therapeutic efficacy. In fact, we have demonstrated in multiple models that glutathione depletion selectively occurs in the liver and not the tumor (Wu et al., 2013; Pingali et al., 2021); the selective glutathione depletion within the liver is likely secondary to increased CYP2E1 expression in hepatocytes relative to other organs (Bieche et al., 2007). Despite observing no glutathione depletion in the tumor, we have consistently observed anticancer efficacy of high-dose AAP, suggesting a CYP2E1-independent mechanism of action (Neuwelt et al., 2009, 2014).
High-dose AAP has demonstrated promising results for the treatment of cancer, both preclinically and clinically. In a phase I clinical trial of high-dose AAP using N-acetylcysteine (NAC) rescue, the response rate was 20% among assessable patients without achieving dose-limiting toxicity (Kobrinsky et al., 1996). Drugs with at least a 20% response rate in phase I (fewer than 10% of drugs tested) meet primary endpoints in subsequent phase II trials 51% of the time (Bugano et al., 2017). Despite demonstrating promise in phase I, AAP has not been studied in subsequent phase trials. The lack of further clinical evaluation is in part attributable to a lack of mechanistic understanding.
NAC is the Food and Drug Administration (FDA)-approved antidote for AAP toxicity. However, there are no large human randomized studies demonstrating the efficacy of NAC; the efficacy is mostly based on case series-level data. A Cochrane report acknowledges the limited evidence for NAC monotherapy (Brok et al., 2006). There are multiple clinical case reports of combination rescue strategies involving fomepizole and NAC with or without dialysis for the management of massive AAP overdose (Chiu et al., 2021; Pourbagher-Shahri et al., 2022). Fomepizole is generally safe and widely available considering it is FDA-approved for methanol and ethylene glycol toxicity (Rasamison et al., 2020). Fomepizole is a potent CYP2E1 inhibitor (Hazai et al., 2002), thus preventing AAP-induced NAPQI formation and resultant hepatotoxicity (Akakpo et al., 2018). Additionally, fomepizole inhibits c-Jun N-terminal kinase, thus decreasing free radical–mediated stress signaling in the mitochondria (Akakpo et al., 2019). As a result, fomepizole is increasingly being used as an adjunctive therapy for massive AAP poisoning in the clinic (Filip et al., 2022).
In the present manuscript, we evaluate the novel approach of using fomepizole concurrently with high-dose AAP for enhanced therapeutic benefits while preventing hepatotoxicity.
Materials and Methods
Cell Lines
Lewis lung carcinoma (LLC)-Luc cells were obtained from American Type Culture Collection. 4T1 and MDA-MB-231 cells were obtained from collaborator Joseph Landry [Department of Genetics, Virginia Commonwealth University (VCU)]. Cell lines were routinely validated using morphology and were checked for mycoplasma using a commercial kit (American Type Culture Collection). Authenticity of lines was confirmed using short-tandem repeat analysis.
Mouse Studies
Eight- to 12-week-old BALB/c mice and C57bl/6 mice were obtained from Jackson laboratories. NOD-scid IL2Rgammanull (NSG) mice were obtained from VCU Cancer Mouse Modeling Core facility. All studies were conducted with the approval of our institutional animal care and use committee (Protocol 02444). Alanine transaminase (ALT) and blood urea nitrogen (BUN) were measured in the clinical laboratory at the Richmond Veterans Affairs Medical Center. Liver H&E slides were prepared at the VCU Tissue and Data Acquisition Analysis Core facility and analyzed by pathologist Won Lee. Fomepizole was obtained from Selleck and diluted in PBS as appropriate. AAP was obtained from Sigma and diluted in 10% glucose in warmed water prior to injection. NAC was obtained from Sigma and dissolved in 10% glucose in water. For 2′,7′-Dichlorofluorescein diacetate (DCFH-DA) assays, 4T1 tumor-bearing mice were treated as indicated. Frozen sections were obtained of tumors, and slides containing 5-µm-thick tissue slices were incubated for 30 minutes with 5 µM DCFH-DA dye prior to imaging with a Zeiss LSM 700 confocal microscope (488 laser). The optical magnification was 40× (oil objective). At the time of animal sacrifice, serum was taken and used to measure lactate dehydrogenase (LDH) levels using Cyquant assay (Fisher), n = 3 replicates per condition. Portions of the tumor were homogenized into a protein lysate. Glutathione levels (reduced and total) were measured using the Glutathione Colorimetric Assay Kit (Fisher) and normalized for total protein levels, n = 3 replicates per condition. The experiment was repeated in two different tumor models, LLC and 4T1. Data was graphed using Graphpad Prism software.
Orthotopic Lung Tumor Models
Intratracheal Model.
LLC-luc cells (100,000) were injected intratracheally in 20% matrigel via intratracheal intubation. Tumor growth was monitored using IVIS Spectrum In Vivo Imaging System (PerkinElmer) after injection of D-luciferin (Gold Bio) intraperitoneally. IVIS quantification was performed using internal PerkinElmer system software for evaluation of the region of interest.
Surgical Model.
One hundred thousand LLC-luc cells were implanted orthotopically into the left lobe of male C57bl/6 mice using previously published protocols (Kwak et al., 2018). Treatment was twice per week starting about 5 days after inoculation (e.g., when visible signal appeared on IVIS imaging). Mice were sacrificed on about day 21. Tumor volume (V) was calculated after animal sacrifice using the equation V = 1/2 (length × width2). The number of replicates (mice) is shown in respective figure legends.
Orthotopic Breast Tumor Models
4T1 tumors were inoculated via direct injection in the breast tissue (mammary fat pad) of female BALB/c mice. Once tumors became palpable, treatment was initiated. Treatment was intraperitoneal twice per week. Tumor size was measured at least twice per week using digital calipers, and tumor size was calculated using equation V = 1/2 (length x width2). The vehicle for both AAP and rescue cocktails (NAC, fomepizole) was 10% glucose in warmed water. When indicated, propylene glycol 10% was also included in the vehicle.
Acetaminophen Levels
ELISA was used to evaluate acetaminophen levels using a kit from Immunalysis (ref 227-0096). N = 2 replicates per condition.
Viability and Cytotoxicity Assays
Cells were treated as indicated. After 48 hours, viability was assessed using a Cell Counting Kit-8 assay per manufacturer’s protocol (Dojindo). The experiment was performed in triplicate. The experiment was performed one time each in three separate cell lines (LLC, 4T1, and MDA-MB-231). Additionally, the conditioned medium was assessed for LDH levels using the Cyquant assay (Thermo Fisher). The experiment was performed in triplicate one time each in two separate cell lines (LLC and 4T1). Data were processed and graphed using Graphpad Prism software.
Quantitative Polymerase Chain Reaction
RNA quantification was performed using our previously published methodologies (Pingali et al., 2021). The primers used were CYP2E1 mouse forward TTCAGCGGTTCATCACCCT and reverse GAGGTATCCTCTGAAAATGGTGTC. GAPDH mouse forward CATGGCCTTCCGTGTTCCTA and reverse TGTCATCATACTTGGCAGGTTTCT. Experiment was performed in triplicate.
JC-1 Assay of Mitochondrial Membrane Potential
Mouse hepatocytes were obtained from collaborator Huiping Zhou using published protocols (Zhou et al., 2006). Hepatocytes were treated as indicated and incubated with JC-1 dye (Biotium) per the manufacturer’s protocol. Microscopy was performed at the VCU Microscopy Core Facility using a Zeiss LSM 700 confocal microscope. The optical magnification was 40× (oil objective). Live-cell imaging was performed using the 488 laser to image green and the 555 laser to image red.
Study Approval
All studies were conducted with the approval of our institutional animal care and use committee (Protocol 02444).
Protein Adduct Measurement
Snap-frozen liver and tumor tissues were homogenized in 10 mM sodium acetate (pH 6.5) using a blade-type homogenizer. Supernatants were collected after centrifugation of the homogenate at 16,000g for 5 minutes. To remove low-molecular-weight compounds that might interfere with the measurement, the supernatants were filtered through Bio-Spin 6 columns (Bio-Rad, Hercules, CA), which were prewashed with 10 mM sodium acetate. The filtrates were subjected to overnight protease digestion to separate the acetaminophen-cysteine adducts from proteins, which were then precipitated using ice-cold 40% trichloroacetic acid (Sigma Aldrich, St. Louis, Missouri). The supernatant was collected and filtered through microcentrifuge tubes. Acetaminophen-cysteine was then measured using high-performance liquid chromatography with electrochemical detection as described (Akakpo et al., 2018).
Statistical Analysis
All data are presented as mean ± S.D. The Student t test was used for the comparison of measurable variants between two groups. A two-way ANOVA with a Bonferroni post-test was used for statistical comparisons between groups in tumor growth. P ≤ 0.05 was considered statistically significant. The Grubbs outlier test was used to exclude extreme outliers (only Fig. 5; mouse 4, day 16 and mouse 5, day 24) for bioluminescence intensity statistics. Note that this exclusion does not affect biologic interpretation of data—outliers were in the same direction as overall trend (GraphPad Prism 6.0; Graph Pad Software). Data processing and graphing was performed using GraphPad Prism Software.
Results
Efficacy of Clinically Used AAP Antidotes for Preventing Hepatotoxicity.
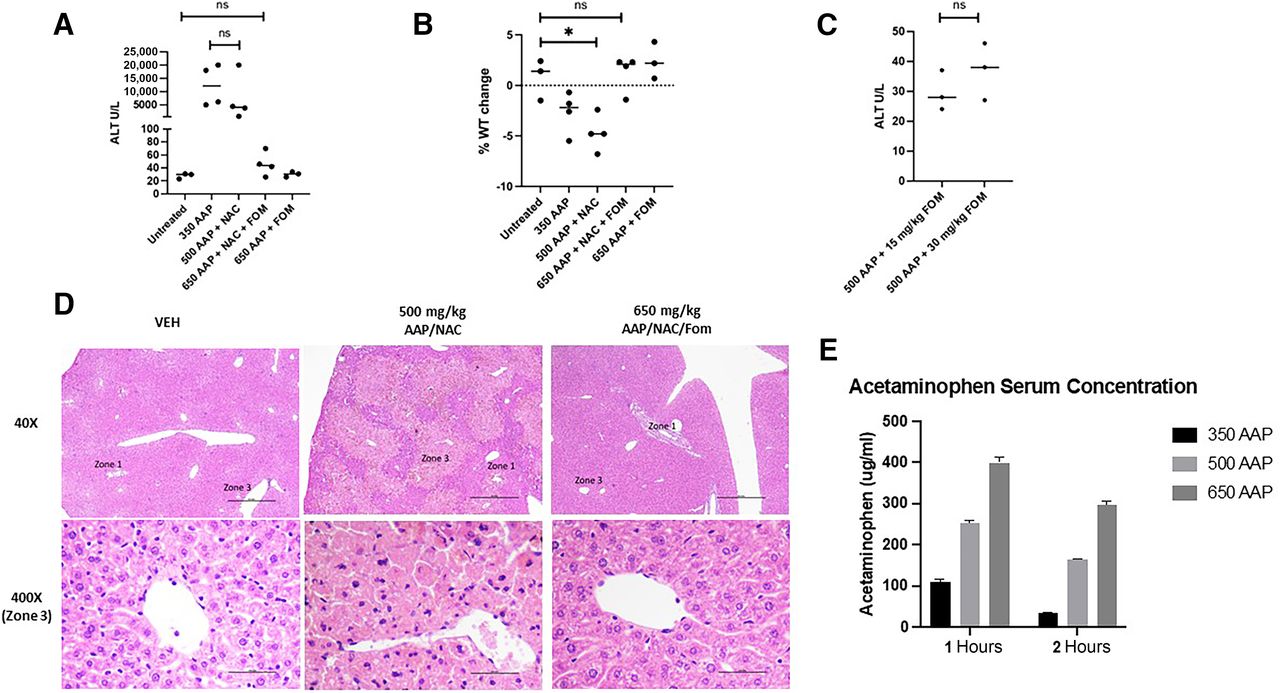
In the clinic, fomepizole and NAC are used to treat AAP poisoning, though only NAC is FDA approved for this purpose (Heard, 2008; Filip et al., 2022). We thus sought to evaluate the efficacy of fomepizole and NAC in preventing AAP toxicity in our mouse models. BALB/c mice were treated with AAP alone and in combination with fomepizole plus or minus NAC. The following day, the mice were weighed and sacrificed. Serum was collected for analysis, and liver histology was assessed. Fomepizole prevented AAP hepatotoxicity as assessed by serum ALT levels and liver morphology (per review by pathologist W.L.), but NAC did not. Additionally, the combination of NAC plus fomepizole was also an effective cocktail for preventing AAP liver injury and weight loss (Fig. 1, A–D). In fact, there was no significant difference between ALT values in untreated mice (average ALT, 28 ± 4) versus those treated with 650 mg/kg AAP concurrently with fomepizole/NAC rescue (average ALT, 46 ± 18; P = 0.16). ELISA was used to measure AAP levels at the doses used: at 1 hour post treatment of 650 mg/kg AAP, levels were 399 µg/mL and decreased to 298 µg/mL at 2 hours post-treatment (Fig. 1E). These serum levels are comparable to what was achieved in a clinical trial of high-dose oral AAP with delayed NAC rescue (mean serum concentration, 245 ug/mL; range, 95–473 ug/mL) (Kobrinsky et al., 1996).

Fomepizole prevents AAP hepatotoxicity. (A–C) BALB/c mice were treated as indicated and sacrificed the following day when they were weighed and serum analyzed. AAP concentrations in mg/kg. Used NAC 100 mg/kg and fomepizole 30 mg/kg unless otherwise indicated. (D) Histology of liver analyzed 24 hours treatment as indicated. Scale bars, 0.5 mm (low magnification) and 0.05 mm (high magnification). Fomepizole and NAC concentrations were the same as above. (E) Serum AAP concentrations determined by ELISA at indicated time after treatment. AAP treatment concentrations in mg/kg. *P < 0.05. Error bars represent S.D. FOM, fomepizole; ns, not significant; VEH, vehicle.
NAC/Fomepizole Rescue Does Not Compromise Anticancer Activity of High-Dose AAP in Vitro.
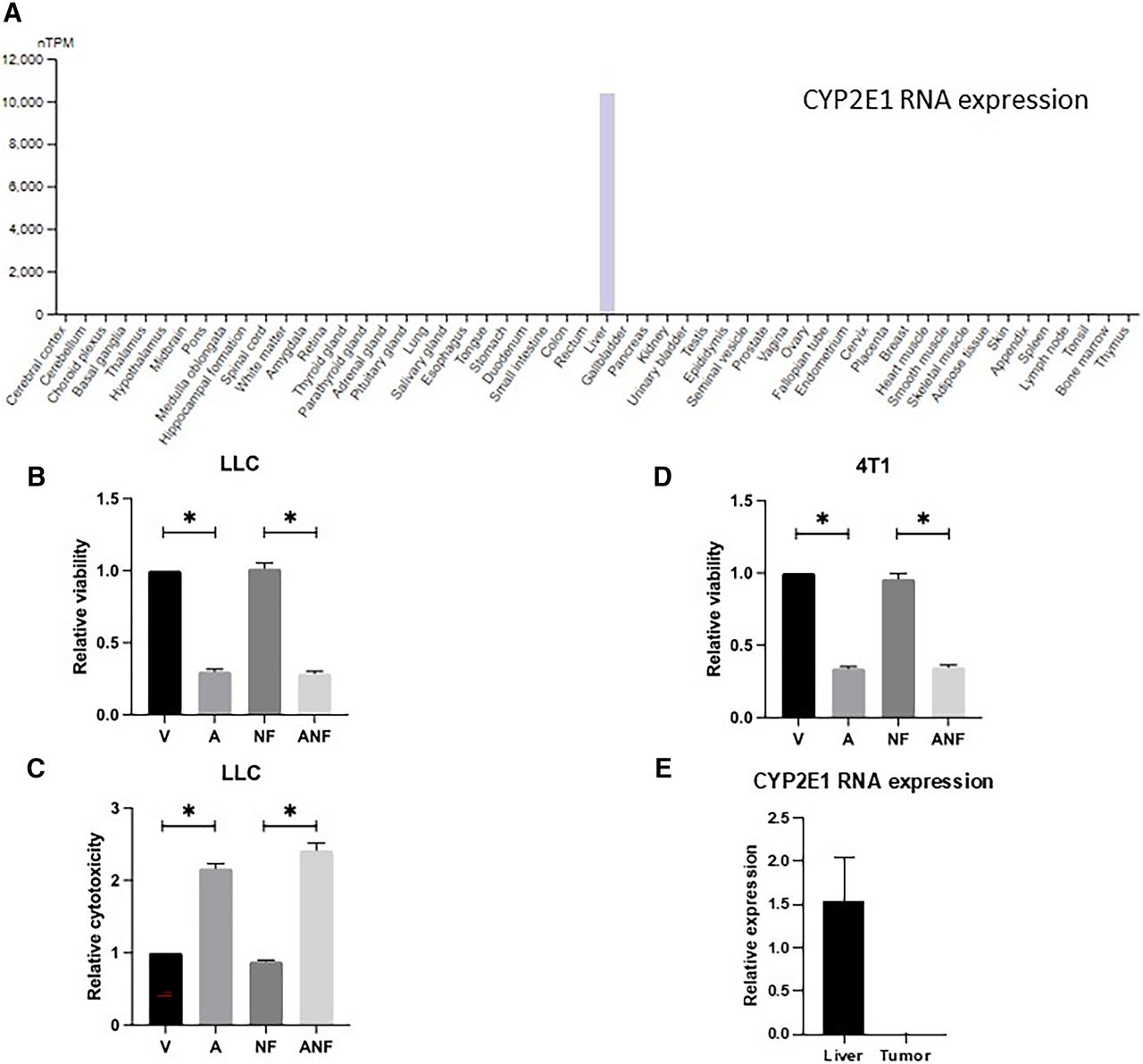
The Protein Atlas was used to evaluate expression of CYP2E1 in various organs. It was demonstrated that CYP2E1 is expressed in the liver and no other organ (Fig. 2A) [note: CYP2E1 levels have been determined in mouse and human kidneys in other studies (Akakpo et al., 2020)]. Because CYP2E1 mediates conversion of AAP into a toxic reactive metabolite that can be neutralized by antioxidants such as NAC, we evaluated whether a rescue regimen comprised of the CYP2E1 inhibitor fomepizole and NAC reverses antitumor activity of high-dose AAP. We demonstrate in 4T1 and MDA-MB-231 breast cancer cells in addition to LLC-luc lung cancer cells that NAC/fomepizole rescue does not reverse AAP cytotoxicity in vitro (Fig. 2, B–D; Supplemental Fig. 1). In 4T1 tumor bearing mice, CYP2E1 was expressed at high levels in the liver, but not the tumor (Fig. 2E), potentially explaining the differential effects of NAC/fomepizole rescue on the tumor and liver.

NAC and fomepizole do not reverse AAP cytotoxicity against tumor cells in vitro. (A) Protein Atlas was used to assess CYP2E1 RNA levels in various organs. LLC cells were cultured for 48 hours in AAP (1 mM), NAC (0.3 mg/mL), and/or fomepizole (300 µM) and then analyzed via Cell Counting Kit-8 (CCK-8) assay (B) or LDH release assay (C). (D) CCK-8 experiment was repeated in 4T1 cells. AAP concentration, 3 mM; fomepizole and NAC concentrations were the same as above. (E) CYP2E1 expression as assessed by RNA quantitative polymerase chain reaction in the liver and tumor of a 4T1 tumor-bearing mouse. Normalized for GAPDH levels. Error bars represent S.D. *P < 0.05. A, acetaminophen; ANF, acetaminophen, n-acetylcysteine and fomepizole; NF, n-acetylcysteine and fomepizole; V, vehicle.
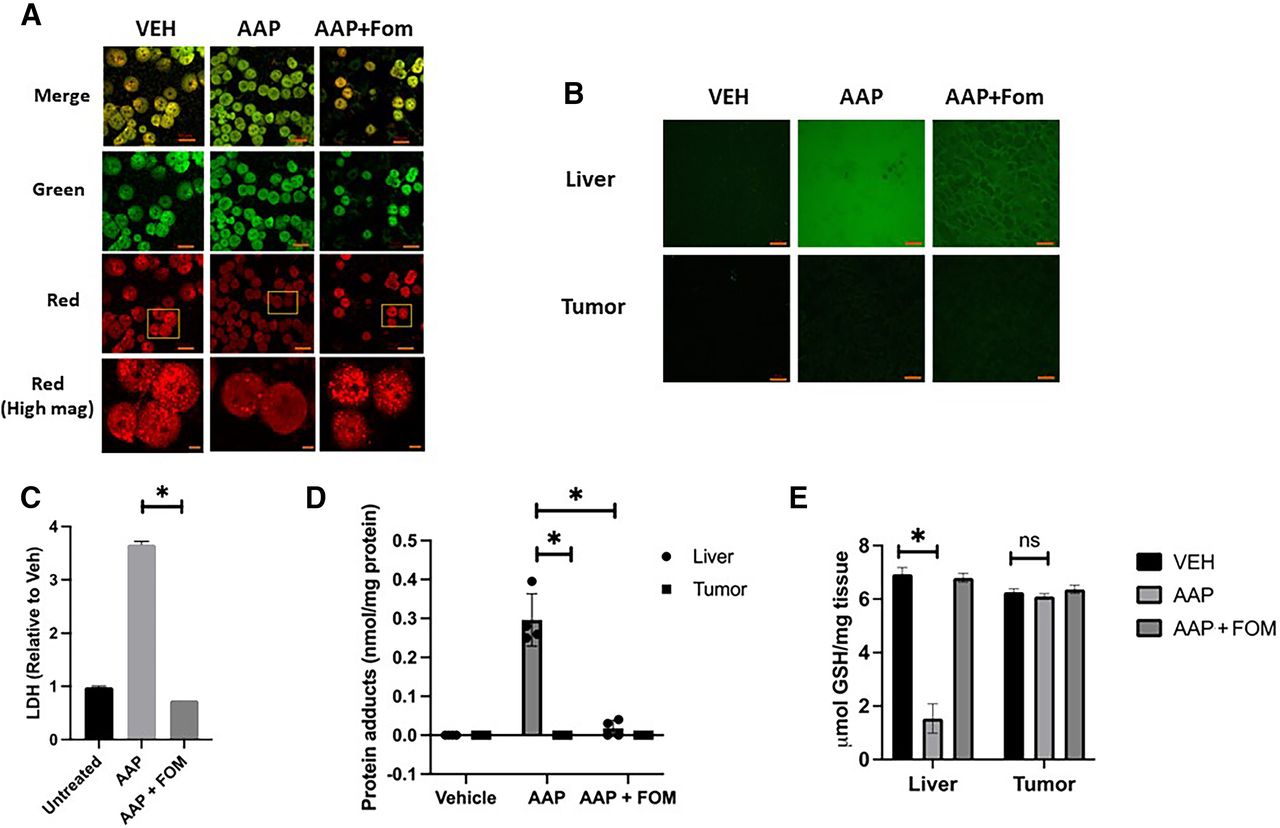
We have previously shown that AAP selectively depletes glutathione in the liver but not the tumor in multiple xenograft models (Wu et al., 2013; Pingali et al., 2021). Extensive prior research demonstrates that NAC helps prevent AAP-induced reactive metabolite-mediated injury and resultant glutathione depletion ultimately leading to mitochondrial injury in the liver (Saito et al., 2010). We next sought to determine if fomepizole has similar benefits. Hepatocytes isolated from a mouse liver were treated with AAP for 6 hours with or without fomepizole rescue and analyzed with JC-1 dye (a sensitive marker for mitochondrial membrane potential) using confocal microscopy. We demonstrate that AAP led to mitochondrial depolarization (decrease in red/green ratio) that was reversed by fomepizole (Fig. 3A). In vivo, 4T1 tumor-bearing mice were treated with AAP with or without fomepizole, and 6 hours later, the tumor and liver were assessed for free radical levels using DCFH dye (a sensitive indicator for reactive oxygen species in cells). AAP created high levels of free radicals in the liver but not the tumor, and fomepizole effectively rescued the free radical injury in the livers of AAP-treated mice (Fig. 3B). Plasma levels of LDH, which can be used as a marker of hepatotoxicity (Kotoh et al., 2011), were elevated in the AAP-treated mice and corrected with concurrent fomepizole rescue (Fig. 3C).

Fomepizole prevents free radical–mediated AAP hepatotoxicity. (A) Isolated hepatocytes from mice were treated with AAP (5 mM) and/or fomepizole (1 mM) for 6 hours and analyzed using live cell imaging via fluorescent microscopy. (B) 4T1 tumor-bearing mice were treated in vivo with 500 mg/kg AAP and/or fomepizole (30 mg/kg) and 6 hours later frozen sections made of tumor and liver. DCFH staining was used to image free radicals. Optical magnification was 40× (oil objective). (C) Serum from mice treated as in (B) was analyzed for LDH as a marker of hepatotoxicity. (D and E) C57BL/6 mice were treated with AAP (500 mg/kg) and/or fomepizole (30 mg/kg), and 6 hours later, liver and tumor (LLC) were analyzed for protein adduct formation (D) and reduced and oxidized glutathione content (E). For experiments shown in (D) and (E), n = 4 mice used per condition. Error bars represent S.D. *P < 0.05. FOM, fomepizole; ns, not significant; VEH, vehicle.
Protein adducts are formed by hepatic metabolism of acetaminophen to the reactive intermediate NAPQI, which covalently binds to hepatic proteins (James et al., 2009). AAP treatment led to a large accumulation of protein adducts in the liver but not the tumor. Fomepizole effectively prevented AAP-induced hepatic protein adduct formation (Fig. 3D). Similarly, high-dose AAP depleted the levels of GSH in the liver, consistent with the known conjugation of NAPQI. AAP-induced GSH depletion was reversed by fomepizole. On the other hand, no changes in GSH levels were observed in the tumor upon treatment with AAP (Fig. 3E; Supplemental Fig. 2). AAP concentrations were similar in the tumor and liver, suggesting that distinct patterns of drug distribution do not account for the observed changes in GSH depletion and protein adduct formation (Supplemental Fig. 3).
High-Dose AAP Has Antitumor Activity in Vivo without Liver Toxicity.
We next sought to evaluate the efficacy of high-dose AAP with NAC/fomepizole rescue in treating orthotopic 4T1 tumors (note that although NAC may not be necessary for rescue in our mouse models—see Fig. 1—we nevertheless used a NAC/fomepizole rescue regimen given the established role of NAC in treating clinical AAP toxicity). Our results demonstrated an approximately 50% decrease in tumor growth in 4T1 tumors treated with 650 mg/kg AAP and NAC/fomepizole rescue relative to NAC/fomepizole alone (Fig. 4A). The mice treated with high-dose AAP had no weight loss and, in fact, gained weight during the course of the study (Fig. 4A). NAC/fomepizole alone did not modulate tumor growth relative to untreated mice (Supplemental Fig. 4).

AAP has anticancer activity in vivo against 4T1 breast tumors. (A) BALB/c mice were injected with 4T1 breast cancer cells orthotopically. Treatment was twice per week once palpable tumors formed with vehicle (NAC, 100 mg/kg and fomepizole, 30 mg/kg) and/or AAP (650 mg/kg). Tumor size was monitored, and mice were weighed. n = 5 mice per condition. (B) Treatment was with vehicle (NAC 100 mg/kg and propylene glycol, 10%) or AAP 500 mg/kg. 4T1 tumor size was monitored, and mice were weighed. n = 5 mice per condition. At the time of sacrifice, serum was analyzed for ALT and BUN (C) and liver histology assessed (D). Scale bars, 0.5 mm (low magnification) and 0.05 mm (high magnification). Error bars represent S.D. *P < 0.05. FOM, fomepizole; ns, not significant.
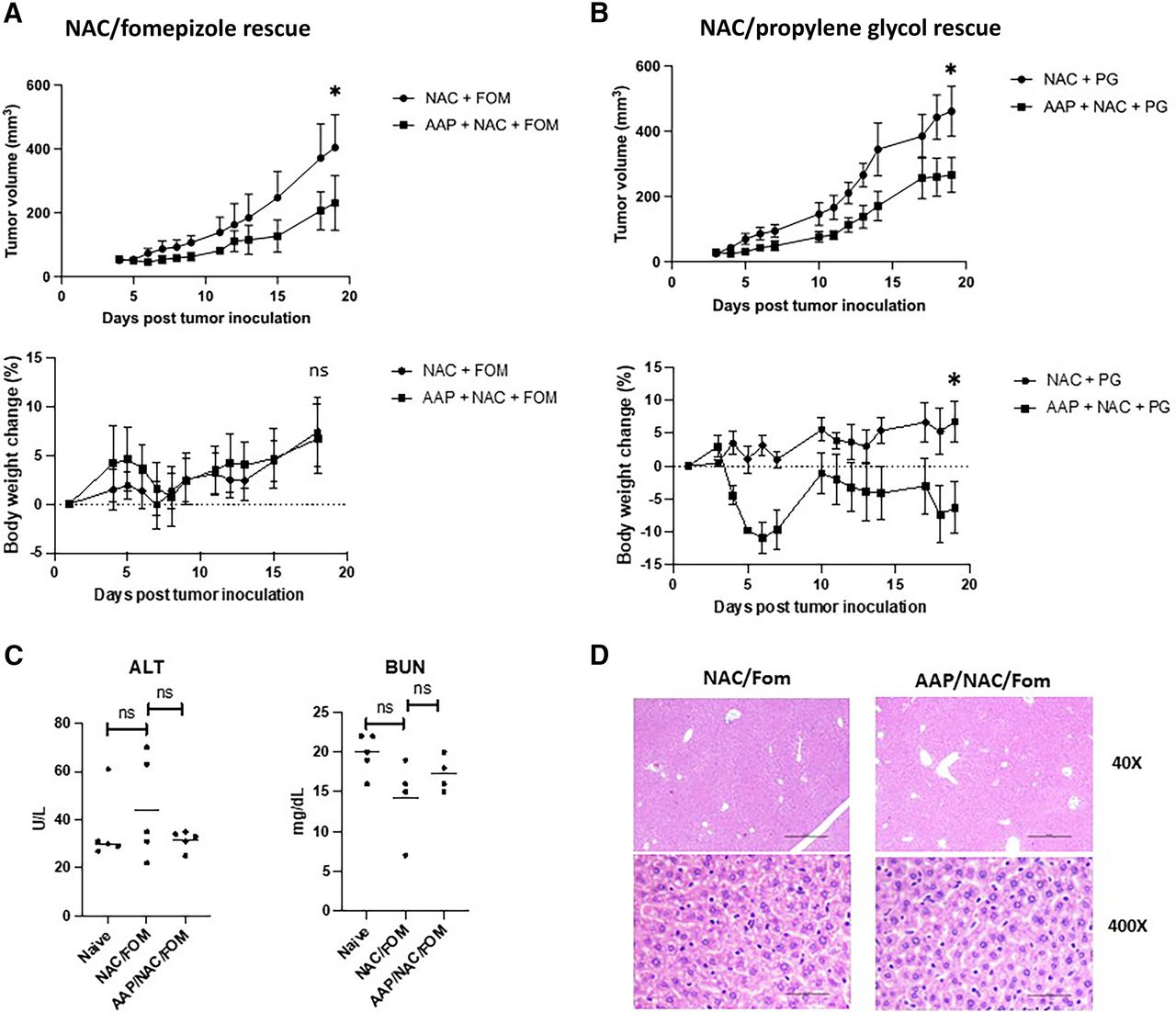
To determine if other CYP2E1 inhibitors besides fomepizole may be used to mitigate AAP toxicity while preserving antitumor efficacy, we performed a similar experiment using NAC/propylene glycol (PG) rescue. PG is known to be a CYP2E1 inhibitor that helps prevent AAP toxicity (Thomsen et al., 1995). The in vivo antitumor activity of high-dose AAP was virtually identical using PG/NAC rescue relative to fomepizole/NAC rescue (Fig. 4B). However, mice treated with high-dose AAP with PG/NAC rescue did lose weight (6% on average), whereas mice treated with PG/NAC alone gained weight (7% on average). Importantly, neither regimen was associated with liver toxicity as determined by serum ALT and liver histology (Fig. 4, C and D). A small increase in BUN was observed in the mice treated with AAP with PG/NAC rescue, but no increase in BUN was observed following NAC/fomepizole rescue (Fig. 4C; Supplemental Fig. 5).
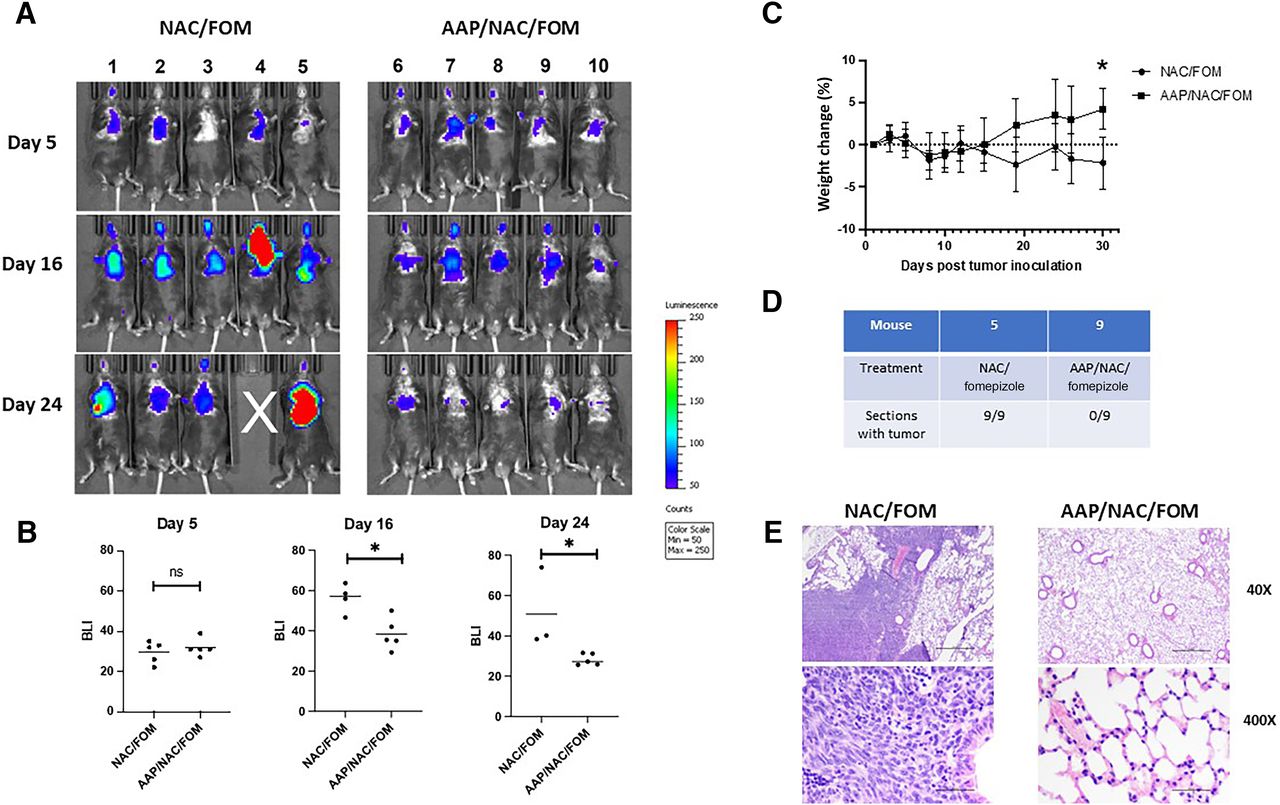
We next evaluated the antitumor efficacy of high-dose AAP using an orthotopic syngeneic model of lung cancer. LLC-luc cells were injected intratracheally into C57bl/6 mice and treated twice weekly with 650 mg/kg AAP with NAC/fomepizole versus rescue alone. It was demonstrated that AAP-treated mice had markedly reduced tumor burden relative to control mice (Fig. 5A). At the conclusion of the experiment, lung weights were lower in AAP-treated mice, likely reflecting lower tumor burden. Additionally, over the course of the experiment, mice treated with AAP gained weight, whereas control mice lost weight, likely a result of differences in tumor burden (Fig. 5, B and C; Supplemental Fig. 6). Confirming the IVIS results, histologic evaluation revealed tumors in 0 of 9 evaluated sections in mouse 9, treated with AAP/NAC/fomepizole, and 9 of 9 sections in mouse 5, treated with NAC/fomepizole alone (Fig. 5D). A representative section (Fig. 5E) reveals tumor-infiltrated lung in the vehicle-treated mouse and normal lung in the AAP-treated mouse.

AAP with fomepizole-based rescue has antitumor activity against an LLC intratracheal orthotopic model. (A) Mice were injected intratracheally with LLC cells, and 5 days later (upon getting an IVIS signal), treatment was begun with vehicle (fomepizole 30 mg/kg and NAC 100 mg/kg) or AAP (650 mg/kg) twice weekly, and IVIS signal followed over time. (B) Bioluminescence intensity was quantitated. (C) Body weight over time during experiment. (D) Serial sections of lungs from mice 5 and 9 were obtained every 150 µM. H&E stains were evaluated for tumor. (E) Representative H&E stain showing tumor in NAC/FOM-treated mice and no tumor in AAP/NAC/FOM-treated mice. Scale bars, 0.5 mm (low magnification) and 0.05 mm (high magnification). Error bars represent S.D. *P < 0.05. FOM, fomepizole; ns, not significant.
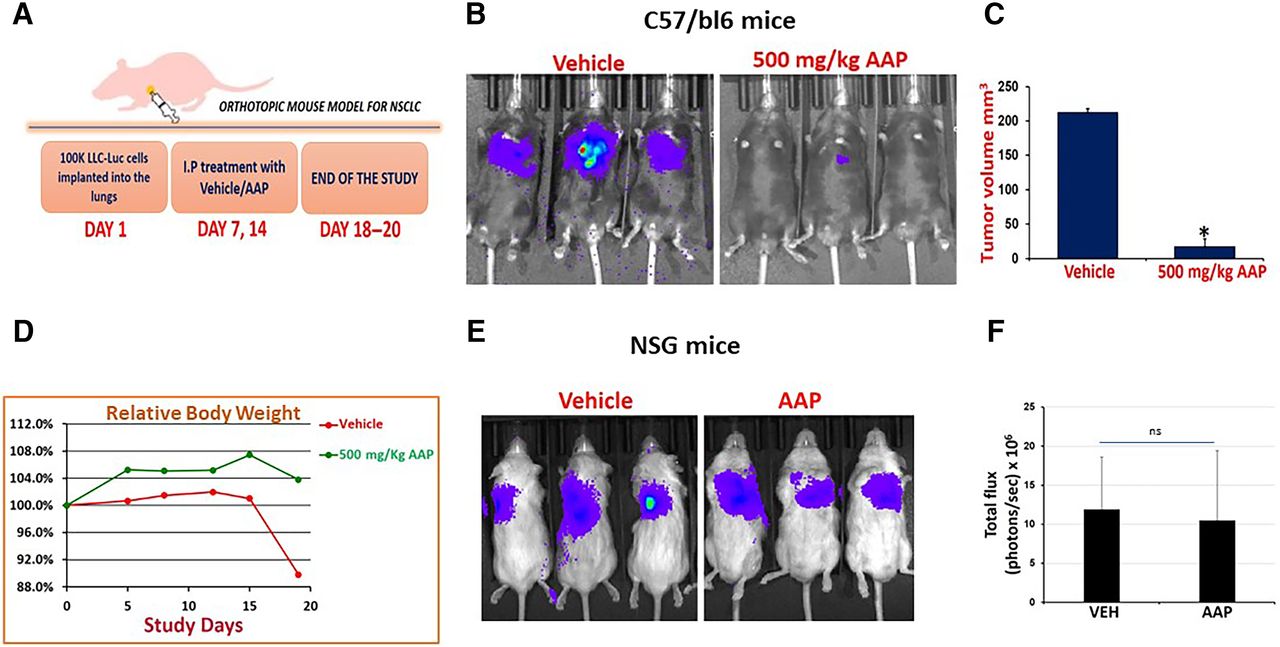
Using a surgical orthotopic technique, LLC-luc cells were implanted into the left lobe of C57bl/6 mice. Mice were treated with AAP and NAC/PG rescue versus NAC/PG alone. AAP again had profound antitumor activity in this model (Fig. 6, A–C; Supplemental Fig. 7). Only mice in the vehicle arm lost weight, likely a result of increased tumor burden in mice not receiving AAP (Fig. 6D). We then repeated the experiment using immune-compromised NSG mice. Interestingly, there was no antitumor activity of AAP in these mice (Fig. 6, E and F). These data altogether demonstrate that AAP exerts an immune-mediated mechanism of anticancer activity.

High-dose AAP has antitumor activity in a surgical orthotopic model with NAC/propylene glycol rescue and the effect appears at least in part immune mediated. (A) Schematic of surgical orthotopic model. Treatment was vehicle (100 mg/kg NAC and 10% propylene glycol) or AAP (500 mg/kg). (B) IVIS images at conclusion of study. (C) Tumor size at the time of animal sacrifice, n = 4 for vehicle (one mouse passed from disease burden), n = 6 for AAP. (D) Mouse body weight during study. (E) Experiment was repeated in NSG immune-compromised mice, and IVIS images are shown with quantification (F), n = 5/condition. Error bars represent S.D. P < 0.05. ns, not significant.
Discussion
CYP2E1-mediated metabolism has not been definitively linked to AAP therapeutic benefits such as analgesia and antitumor activity. In the present study, we demonstrate that concurrent administration with CYP2E1 inhibition with fomepizole allows for dose escalation to 100-fold higher than standard AAP dosing (650 mg/kg or about 50 g in average 75 kg human adult) without any detectable liver toxicity (Fig. 1) and without compromising antitumor activity (Fig. 2). Further, high-dose AAP with fomepizole/NAC rescue has profound antitumor activity against commonly used syngeneic orthotopic models of breast cancer (4T1) and lung cancer (LLC-luc) (Figs. 4–6). Mouse syngeneic tumor models (4T1 and LLC) were used for in vivo studies because AAP appears to lose activity in immune-compromised animals (NSG mice; Fig. 6) used to grow human tumors. Both 4T1 and LLC are known to be resistant to standard first-line therapies, including PD-1 (Kim et al., 2014; Li et al., 2017) and cisplatin (Merritt et al., 2003) (note: PD-1 is clinically used first line in both lung and breast cancer; cisplatin is first line in lung cancer but not breast cancer).
The mechanism of anticancer activity of high-dose AAP has not been definitively characterized. We have previously shown that AAP has anti-cancer stem cell activity via a STAT3-dependent mechanism. AAP directly binds to STAT3 and has a high degree of specificity for STAT3 relative to STAT1 (Pingali et al., 2021).
In addition to direct effects on the tumor cell (e.g., via STAT3 inhibition), AAP may also modulate JAK-STAT signaling in the tumor immune microenvironment. In the innate immune system, STAT1 signaling mediates conversion of macrophages to an activated antitumor “M1 phenotype,” whereas STAT3 and STAT6 are more closely associated with immune-suppressive “M2” tumor-associated macrophages (Xiao et al., 2020). Similarly, the various JAK-STAT signaling pathways modulate differentiation of T cells toward a cytotoxic versus regulatory phenotype (Villarino et al., 2015). Our data suggests that AAP has a substantially reduced efficacy in NSG mice with defective innate and adaptive immune systems, arguing that AAP may function, at least in part, via modulation of the antitumor immune response (Fig. 6). Although at standard doses AAP is not felt to have anti-inflammatory or immune modulatory properties, at high doses the effects of AAP on systemic inflammation have not been studied. The effect of AAP on the antitumor immune response (particularly tumor-associated macrophage polarization/activation) is of great interest and the subject of ongoing studies within our laboratory (Bryan, 2023).
Administering rescue agents to allow for dose escalation of anticancer therapeutics is an established approach. For instance, leucovorin is a folic acid derivative that allows for synthesis of nucleic acids even in the presence of high doses of methotrexate, thus mitigating methotrexate toxicities such as myelosuppression and gastrointestinal toxicity. Concurrent administration of methotrexate and leucovorin has been widely adopted, particularly in the setting of malignant lymphomas (Flombaum and Meyers, 1999). Concurrent administration of CYP2E1 inhibitors and AAP may have analogous benefits for the treatment of patients with advanced malignancies such as breast and lung cancer.
The relative lack of efficacy of NAC for preventing hepatotoxicity in our models is unexpected given the established role of NAC as an antidote to AAP toxicity (Heard, 2008; Akakpo et al., 2022). Prior preclinical studies of NAC yielded mixed results in preventing AAP liver toxicity (Saito et al., 2010; Khayyat et al., 2016; Wang et al., 2021). Khayyat et al. (2016) showed that NAC (106 mg/kg i.p.) administered 1.5 hours after AAP (400 mg/kg i.p.) decreased ALT from 940 (no NAC) to 860 U/L (plus NAC) relative to baseline ALT of 10 U/L. Wang et al. (2021) demonstrated no protection (e.g., reversal of ALT elevation) of 100 mg/kg NAC i.v. 30 minutes after AAP (350 mg/kg i.p.) in C57BL/6 mice. Additional in vitro evidence suggests that physiologically relevant concentrations of NAC have minimal protective effect against CYP2E1-mediated AAP toxicity (Dai and Cederbaum, 1995). Nevertheless, there is a large body of literature supporting the use of NAC both preclinically (James et al., 2003; Owumi et al., 2015) and clinically (Heard, 2008). Although there is conflicting preclinical data (an effect that is likely model dependent), in the clinic, NAC continues to have a well established role in the treatment of AAP poisoning.
In the present work, we observe complete prevention of liver toxicity from AAP using concurrent fomepizole (Fig. 1), a finding consistent with prior observations (Akakpo et al., 2018, 2019, 2022).
In conclusion, concurrent CYP2E1 inhibition is a novel approach that allows for safe dose escalation of AAP to levels that have profound antitumor activity in well established preclinical lung and breast cancer models.
Acknowledgments
The authors appreciate the assistance of Martha Joslyn, Rujul Kaul, and Shellie Galabow for technical assistance.
Data Availability
All data available upon request. This article contains no datasets generated or analyzed during the current study.
Authorship Contributions
Participated in research design: Bryan, Faber, Landry, Jaeschke, Li, May, Patel, Neuwelt.
Conducted experiments: Bryan, Pingali, Akakpo, Neuwelt.
Contributed new reagents or analytic tools: Lee.
Performed data analysis: Bryan, Neuwelt.
Wrote or contributed to the writing of the manuscript: Neuwelt.
Footnotes
- Received June 1, 2023.
- Accepted October 13, 2023.
Services and products in support of the research project were generated by the Virginia Commonwealth University Flow Cytometry; Cancer Mouse Models; Microscopy Shared Resource, supported, in part, with funding from a National Institutes of Health National Cancer Institute Cancer Center support grant [Grant P30CA016059]; the Analytical Core at KUMC, supported, in part, by a National Institute of General Medicine–funded Liver Disease COBRE grant [Grant P30GM118247] (to H.J.). The work was supported by a Swim Across America grant; VA [Grant 5IK2BX004914-02] (to A.N.); and a VCU Internal Medicine Seed Grant.
Department of Veterans Affairs (A.N. and A.B.) has a patent pending for concurrent use of CYP2E1 inhibitors and acetaminophen.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AAP
- acetaminophen
- ALT
- alanine transaminase
- BUN
- blood urea nitrogen
- DCFH-DA
- 2′, 7′-Dichlorofluorescein diacetate
- FDA
- Food and Drug Administration
- GSH
- glutathione
- LDH
- lactate dehydrogenase
- LLC
- lewis lung carcinoma
- NAC
- N-acetylcysteine
- NAPQI
- N-acetyl-p-benzoquinone imine
- NSG
- NOD-scid IL2Rgammanull
- PG
- propylene glycol
- VCU
- Virginia Commonwealth University
- U.S. Government work not protected by U.S. copyright




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}