Article Text

Statistics from Altmetric.com
Introduction
Inflammatory bowel disease (IBD) encompasses two major entities: ulcerative colitis (UC) and Crohn's disease (CD).1 Both are chronic, progressive, disabling conditions that require lifelong medical treatment in most cases. IBD has a major impact on the patient's health-related quality of life,2 and the treatment-related costs place a significant burden on healthcare systems.3
Historically, the medical management of IBD has been based on the use of several small-molecule drugs (SMDs), including corticosteroids, immunomodulators (such as azathioprine, 6-mercaptopurine and methotrexate) and aminosalicylates.4 The introduction of biologic anti-tumour necrosis factor-α (TNF-α) agents in the first few years of this century has revolutionised the clinical management of IBD. In parallel, treatment goals have shifted from symptomatic control towards more objective endpoints (such as mucosal healing and deep remission) associated with better long-term outcomes.5 ,6 Over the past 20 years, drug research in the field of IBD has focused on the development of new, large-molecule biologics; hence, several anti-TNF-α monoclonal antibodies (including infliximab,7 ,8 adalimumab,9 ,10 certolizumab pegol,11 ,12 and golimumab)13 and, most recently, antibodies with other targets (such as vedolizumab14 ,15 and ustekinumab)16 have become available in clinical practice.
However, monoclonal antibodies have limitations in terms of efficacy, safety and cost. First, the available biologics are only moderately efficacious17 since up to 30% of patients show a lack of improvement after induction therapy with anti-TNF drugs (ie, primary non-response).18 Furthermore, a significant proportion of patients (between 13% and 25% per year)19–21 may develop a loss of response to anti-TNF agents over time (ie, secondary non-response).20 ,21 This loss of response may be due to pharmacodynamic, pharmacokinetic and/or immunogenic factors.22 Combination therapy is the best way to prevent the formation of antibodies,23 which are known to decrease efficacy and increase the risk of infusion reactions.24 However, the increasing use of combination therapy raises other concerns, such as a potential increase in serious treatment-emergent adverse events (eg, infections and malignant conditions).25 We shall probably face the same problems with vedolizumab and ustekinumab, although gut specificity of the former might be associated with a better safety profile.
At present, there are no curative treatments for IBD; hence, management typically involves lifelong treatments. It is not yet clear when to de-escalate treatment with immunomodulators or anti-TNF agents as this is often associated with disease relapse.26 Currently, the high treatment-related costs in IBD are primarily due to the anti-TNF agents,27 ,28 although less expensive biosimilars are now appearing.
Lastly, these large biological drugs require parenteral (ie, intravenous or subcutaneous) administration, which can be burdensome in patients requiring maintenance therapy for long periods of time.29
For the above-mentioned reasons, there is now a need for new, cost-effective, orally administered drugs with greater efficacy and tolerability. Hence, there is a growing interest in new, specific, SMDs in the field of IBD and in other immune-mediated inflammatory diseases, such as rheumatoid arthritis, psoriasis and multiple sclerosis.30 The present article reviews novel SMDs for the treatment of both UC and CD. It focuses on molecules in late-stage clinical development and gives insights into the agents' possible roles in the future.
Small molecules versus monoclonal antibodies
The treatment of inflammatory conditions like IBD is based on anti-inflammatory drugs. The latter can be classified as biologics or SMDs, depending on their chemical structure.30 The most obvious difference between an SMD and a biologic is molecular weight. By definition, SMDs have a molecular weight <1 kDa (and usually <500 Da).31 They can have any kind of chemical structure, although most are organic compounds composed of carbon, nitrogen and oxygen.32 This low molecular weight enables SMDs to diffuse more easily through cell membranes.33 In contrast, biologics are large, macromolecular structures with a molecular weight >1 kDa,34 although many monoclonal antibodies weight as much as 150 kDa (eg, ∼144.1 kDa for infliximab).35 Most biologics are complex proteins, composed of polypeptide chains with secondary and tertiary structures.36 This enormous difference in size notably influences the administration route, target location, pharmacokinetic features, antigenicity and drug–drug interactions (table 1).
One of the main advantages of SMDs over biologics is the potential of oral administration. When taken orally, low-molecular-weight drugs resist gastric degradation and can rapidly enter the systemic circulation. As mentioned above, patients with IBD may be inconvenienced or distressed by the long-term need for injections or infusions. Fear of needles is common in the general population and is associated with reluctance to seek medical care.38 Furthermore, patients being treated with parenteral medications for chronic conditions may consider injections to be a part of the disease burden rather than the best available treatment,39 and this may lead to poor adherence. Oral SMDs can boost patient satisfaction and increase treatment adherence, persistence and thus efficacy.40
SMDs usually have a short serum half-life (due to metabolism and binding to plasma proteins),32 whereas biologics exhibit a much longer half-life (with slow degradation by proteases). This is why most SMDs must be taken daily or twice daily. However, long-term adherence to treatment with oral drugs is often poor in clinical practice and the best way to improve this is still being debated.41 ,42 Nevertheless, adherence to intravenously or subcutaneously administered anti-TNF therapeutics in IBD is not optimal either.43
The short half-life of SMDs may constitute an advantage over biologics—especially in situations where rapid drug elimination is desired (infection, surgery, pregnancy, etc).
As mentioned above, the formation of antibodies against antigenic biologics is a major problem and may be the underlying cause of a lack or loss of response. Given that antigenic responses are usually directed against large molecules or fragments thereof, SMDs are not antigenic and thus avoid these reactions. The SMDs' lack of immunogenicity provides sustained efficacy over time and may increase drug persistence. In rheumatoid arthritis, the SMD tofacitinib showed very good drug persistence rates and low switch rates (to a biologic), regardless of whether it was given as a monotherapy or combined with immunomodulators.44 In contrast, anti-TNF agents showed lower persistence for monotherapy than for combination therapy.45 However, SMDs may induce idiosyncratic drug hypersensitivity and drug allergies.32
Another drawback of SMDs versus biologics is the fairly high probability of drug–drug interactions; these are mainly pharmacokinetic interactions produced by competition for clearance pathways.37 Drug–drug interactions are less frequent with biologics. Furthermore, SMDs may induce toxicity through ‘off-target’ effects, in which the agent binds to a target for which it was not intended.
Lastly, and although the research and development costs for SMDs are variable, the manufacturing process (based on chemical synthesis) is much simpler; in contrast, biologics have to be produced in a living cell46 via complex cellular processes (including transcription, translation, post-translational modification and protein folding).47 Less expensive production of SMDs may help to reduce drug costs and increase cost-effectiveness.
Tofacitinib and other JAK inhibitors
The Janus kinase (JAK) family comprises four intracellular tyrosine kinases: JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2).48 Many cytokines exert their biological functions by activating the JAK-STAT pathway: binding of a cytokine to its receptor leads to activation of intracellular JAKs, which then phosphorylate and activate STAT proteins. In turn, the STATs dimerise, translocate to the nucleus and regulate the expression of various target genes.49 The JAK-STAT pathway has an important role in innate immunity, adaptive immunity and haematopoiesis, and is implicated in the pathogenesis of several conditions (including IBD).50 JAKs are activated in pairs, and different combinations of JAKs are associated with different cytokine receptors. Thus, JAK inhibition can potentially block several cytokine and inflammatory pathways simultaneously; this contrasts with currently available biologics that inhibit a single, specific target.51
Tofacitinib (Xeljanz, Pfizer) is an oral SMD (molecular weight ∼312.3 Da) JAK inhibitor that inhibits JAK1, JAK3 and (albeit to a lesser extent) JAK2.52 This inhibition blocks signalling for a large subset of inflammatory cytokines: interleukin (IL)-2, IL-4, IL-6, IL-7, IL-9, IL-15, IL-21 and interferon (IFN)-γ.53 Tofacitinib citrate has been approved by the US Food and Drug Administration (FDA) for the treatment of moderate-to-severe rheumatoid arthritis in patients with an inadequate response to (or poor tolerance of) methotrexate.54–56 The drug is currently being investigated for the treatment of psoriasis57 ,58 and IBD (both UC and CD).
The efficacy of tofacitinib in UC has been evaluated in a phase II multicentre, double-blind, placebo-controlled, randomised trial.59 A total of 194 patients with moderate-to-severe UC were randomised to four dose levels of tofacitinib (0.5, 3, 10 or 15 mg twice a day) or placebo for 8 weeks. The investigators reported a marked, statistically significance difference in the clinical response at week 8 (primary outcome) between the highest dose level (15 mg twice a day) and placebo (78% vs 42%, p<0.001). Secondary outcomes, such as clinical remission and endoscopic remission (defined as an endoscopy subscore of 0), also occurred more frequently in most of the tofacitinib groups. These results prompted a phase III programme (OCTAVE) investigating the efficacy and safety of induction and maintenance therapy with tofacitinib in UC (OCTAVE Induction 1, NCT01465763; OCTAVE Induction 2, NCT01458951).60 A total of 905 and 243 patients were randomised to tofacitinib 10 mg twice a day and placebo, respectively. The proportion of patients in clinical remission at week 8 (primary end point) was significantly higher in the tofacitinib group in both studies (18.5% vs 8.2%, p=0.007, and 16.6% vs 3.6%, p=0.0005). Furthermore, mucosal healing (defined as a Mayo Clinic endoscopy subscore ≤1) was also more frequent in the tofacitinib groups. Importantly, anti-TNF-naive and anti-TNF-experienced patients appeared to benefit from tofacitinib to the same extent. Two other phase III studies of maintenance therapy with tofacitinib are underway (OCTAVE Sustain, NCT01458574; OCTAVE Open-Label Extension, NCT01470612).
Tofacitinib has also been evaluated in patients with CD. In a randomised, double-blind, placebo-controlled, phase II study, 139 patients with moderate-to-severe CD were randomised to three dose levels of tofacitinib (1, 5 or 15 mg twice a day) or placebo for 4 weeks.61 The investigators did not find any intergroup differences in neither the clinical response rate nor the clinical remission rate. To establish whether or not these results were due to the short duration of treatment, a phase IIb trial (NCT01393626) was recently performed in patients with CD who were randomised to receive tofacitinib 5 mg twice a day (n=86), tofacitinib 10 mg twice a day (n=86) or placebo (n=91) for 8 weeks.62 Most of the enrolled patients (76–79%) were anti-TNF-experienced. Again, there was no significant intergroup difference in the clinical remission rate (primary end point). However, the proportion of patients with a clinical response was slightly and significantly higher in the tofacitinib arms; and a significant reduction in C reactive protein levels was also seen in the two tofacitinib groups, although there were no changes in faecal calprotectin levels. Patients from the induction phase could enter a 26-week maintenance therapy phase (phase IIb study).63 At week 26, the clinical remission and clinical response rates were numerically higher in the tofacitinib 10 mg twice a day group (vs placebo), but the difference was not statistically significant. The future of tofacitinib in CD is uncertain.
Tofacitinib's mechanism of action gives it immunosuppressant properties; hence, a major concern in treated patients is the possible occurrence of infectious events and malignancies. Overall, tofacitinib was well tolerated in IBD trials. In UC trials, the most commonly reported infection-related adverse events were influenza and nasopharyngitis, although herpes zoster and other serious infections were also reported (anal abscess, postoperative abscess, cellulitis, Clostridium difficile infection and pneumonia).59 Similar infectious adverse events were seen in CD trials, including nasopharyngitis, abscess formation, gastroenteritis and pneumonia.61 ,62 Furthermore, neutropenia (in the range of 1000–1500 cells/mL3) was observed in three tofacitinib-treated patients with UC.59 A dose-dependent increase in serum low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol levels in tofacitinib-treated patients was observed, although levels normalised after cessation of the drug; similar changes in lipid levels were observed in clinical trials of tofacitinib in rheumatoid arthritis.64 Interestingly, in active rheumatoid arthritis, levels of HDL and LDL are lower than in health. These changes in lipid profile seem to be driven by increases in cholesterol ester catabolism. Tofacitinib may reduce cholesterol ester catabolism, thereby increasing cholesterol towards levels of healthy individuals.65
Tofacitinib is the first and only JAK inhibitor to have been approved in >40 countries (including the USA) for the second-line treatment of moderate-to-severe rheumatoid arthritis. The drug has not yet been approved by the European Medicines Agency (EMA)—mainly due to safety concerns.66 Serious infection-related events (3.09 events per 100 patient-years, overall)67 and malignancies (0.85 events per 100 patient-years, overall)68 were higher than with placebo. The most common malignancies were lung cancer, breast cancer, lymphoma and gastric cancer (in Japan only for the latter). Infection-related events were the most common severe adverse events, and the most commonly observed were pneumonia, herpes zoster and urinary tract infections. Some severe and sometimes fatal infections and opportunistic infections (tuberculosis, candidiasis, pneumocystis pneumonia and disseminated herpes zoster, among others) were also recorded.69 Other major adverse events included intestinal perforation and elevated serum liver enzyme levels.
Overall, tofacitinib has a manageable safety profile in the routine clinical treatment of rheumatoid arthritis and leads to very few drug withdrawals. The drug has a favourable risk–benefit ratio in UC trials. Pfizer is expected to use all these clinical data in its application for regulatory approval in UC.
Other JAK inhibitors (see figure 1) are in phase II of clinical development: the selective JAK1 inhibitor filgotinib (GLPG0634) is being evaluated in CD (NCT02048618);70 the JAK1 inhibitor ABT-494 is also being evaluated in CD (NCT02365649) and peficitinib (ASP015K, a second-generation, non-selective JAK inhibitor) is being tested in UC (NCT01959282). JAK1 selectivity (ABT-494 and filgotinib) may be associated with an improved safety profile compared with non-selective JAK inhibitors.71 TD-1473 is a novel pan-JAK inhibitor designed to inhibit JAK in the GI tract upon oral dosing. It is expected to lack significant systemic exposure and thus minimise toxicity;72 in the light of promising preclinical data, a phase I trial has been designed (NCT02818686).
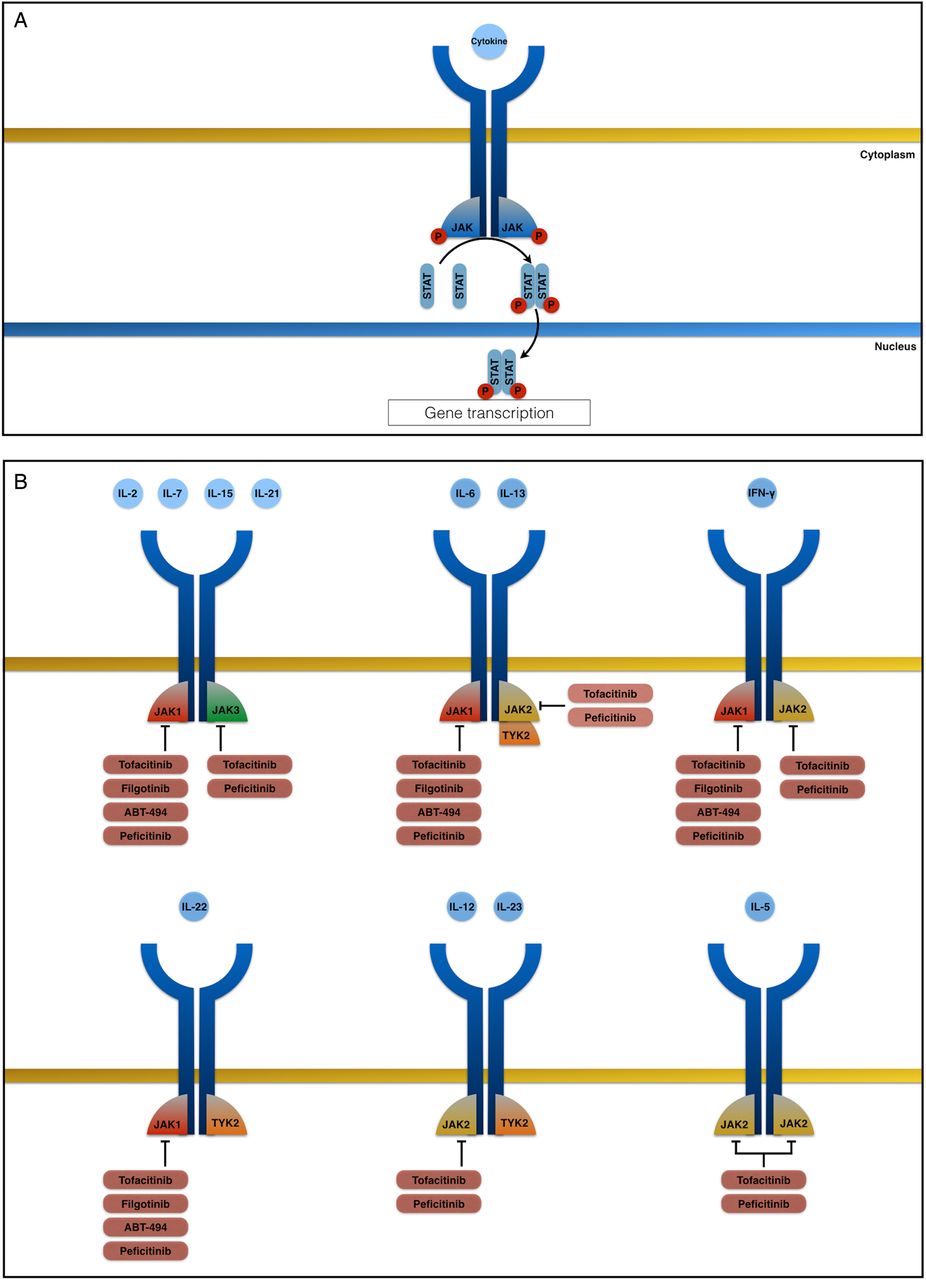
(A) The Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway is activated by many cytokines: binding of a cytokine to its receptor leads to activation of intracellular JAKs, which then phosphorylate and activate STAT proteins. In turn, the STATs dimerise, translocate to the nucleus and regulate the expression of various target genes. Each type of JAK binds preferentially to the intracellular domain of individual cytokine receptor chains. Hence, the inhibition of each type of JAK leads to inhibition of signalling by a specific subset of cytokines. (B) The common γ-chain receptor family (which includes interleukin (IL)-2, IL-7, IL-15 and IL-21) uses JAK1 and JAK3 to control signalling. The common glycoprotein-130 receptor family (which includes IL-6) mainly uses JAK1 but also uses JAK2 and tyrosine kinase 2 (TYK2); the IL-13 receptor is associated with JAK1 and either JAK2 or TYK2. The interferon (IFN)-γ receptor signals through JAK1 and JAK2. JAK1 and TYK2 pair up and control signalling of the IL-22 receptor, whereas the IL-12 and IL-23 receptors use JAK2 and TYK2. The common β-chain receptor family (which includes IL-3 and IL-5) only signals through JAK2.
Ozanimod and other S1P receptor modulators
Sphingosine-1-phosphate (S1P) is a sphingolipid metabolite73 that binds specifically to five widely expressed subtypes of a G-protein-coupled receptor (S1P1–5).74 These S1P receptors (S1PRs) regulate various cellular processes (such as adhesion, migration and endocytosis)75 ,76 and mediate a number of physiological events (including embryogenesis, angiogenesis, vascular tone and permeability, cardiac function and the trafficking of lymphocytes and other haematopoietic cells).77 Different S1PR expression patterns exist within the immune system: although most immune cells express S1P1, specific cell types express other S1PR subtypes (eg, S1P1 and S1P4 on lymphocytes).78 S1P has an important role in lymphocyte homing to lymphoid organs and lymphocyte migration into the circulation. S1P1 expression on effector lymphocytes regulates the cells' movement as a function of the S1P gradient. Lymphoid tissues have low S1P levels, whereas blood and lymph have high S1P levels; this determines the immune cells' egress into the circulation.77 S1P modulators bind to the receptor and induce its internalisation and degradation, which in turn prevent lymphocytes from leaving lymphoid tissues (see figure 2).75 This trapping leads to a reduction in the circulating effector T cell count and selective suppression of the immune system, although it does not downregulate overall immune function (in contrast to other immunosuppressants do).79 ,80

(A) Lymphocyte egression from the lymph nodes is mediated by the expression of sphingosine-1-phosphate receptor 1 (S1P1) on effector lymphocytes and the presence of a S1P concentration gradient; this determines the immune cells' egress into the circulation. (B) S1P receptor modulators bind to S1P1 and induce its internalisation and subsequent degradation, thus inhibiting the egress of lymphocytes from lymphoid tissues. Ozanimod binds to S1P1 and S1P5, whereas etrasimod is specific for S1P1. It is not known which subtype of receptor amiselimod (not shown in the figure) acts on.
Ozanimod (RCP-1063) is a novel, orally administered SMD (molecular weight ∼404.4 Da) that selectively modulates S1P1 and S1P5 receptors. The compound is currently being evaluated for the treatment of immune-mediated inflammatory diseases,81 such as multiple sclerosis82 and IBD. In a recent double-blind, placebo-controlled phase II clinical trial (the TOUCHSTONE study), Sandborn et al evaluated the efficacy and safety of induction and maintenance therapy with ozanimod in patients with moderate-to-severe UC.83 They randomised 197 patients to receive ozanimod 0.5 mg/day or 1 mg/day or placebo for up to 32 weeks. Ozanimod showed a slightly higher clinical remission rate at week 8—especially in the 1 mg group compared with placebo (16% vs 6%, p=0.048). At week 8, there were also differences in the proportion of patients with a clinical response and mucosal healing. Of the 197 patients who completed the induction phase, 103 patients entered the 32-week double-blind maintenance phase and 91 patients completed it. At week 32, there were significant differences in terms of clinical remission, clinical response and mucosal healing between both ozanimod levels and placebo. Interestingly, ozanimod treatment (especially at a dose level of 1 mg) was associated with a greater proportion of histological remission (defined as a Geboes score ≤2) at both week 8 and week 32.84 In terms of safety, ozanimod was well tolerated. However, the study was underpowered, and the follow-up period was too short for an adequate evaluation of safety issues. Elevation of serum liver enzymes was observed in four patients (3%) in the ozanimod group. Ozanimod had to be discontinued in a patient who developed sinus bradycardia and first-degree atrioventricular block.
Ozanimod is now being tested in patients with moderate-to-severe CD in a phase II trial (NCT02531113). A phase III trial of ozanimod's efficacy and safety in UC is also underway (NCT02435992). Two other compounds with S1PR modulation activity are currently being tested in phase II trials: etrasimod (APD-334) in UC (NCT02447302 and NCT02536404) and amiselimod (MT-1303) in CD (NCT02378688 and NCT02389790).
Although S1PR modulators appear to have a better safety profile than other classes of immunomodulators,85 some safety concerns persist. Fingolimod (Gylenia, Novartis) is a non-selective S1PR modulator (binding to S1P1, S1P3, S1P4 and S1P5) that has been approved by the FDA and the EMA for the treatment of relapsing-remitting multiple sclerosis.86 This non-selectivity may lead to a higher incidence of adverse events, including cardiovascular effects (bradycardia and atrioventricular blocks), pulmonary disorders, elevated liver enzymes and macular oedema. Serious infections (such as disseminated varicella zoster and herpes simplex infections) and cases of skin and breast cancer are rare but have been reported.87 In postmarketing surveillance studies, cases of cryptococcal meningitis and progressive multifocal leukoencephalopathy (PML) without prior natalizumab treatment88 have been reported in patients treated with fingolimod. As the risk of PML with natalizumab itself was also only characterised after its regulatory approval,89 PML risk assessment and management should probably be considered not only for fingolimod but also for all S1P modulators—including ozanimod and other compounds. Large, prospective postmarketing studies with long-term follow-up should definitively establish whether or not treatment with other S1PR modulators is associated with a potential risk of PML. Initial studies have suggested that S1P2 and S1P3 modulation with non-specific compounds (such as fingolimod) were associated with a decrease chronotropism (leading to bradycardia and possible atrioventricular block) and an increase in vascular tone (leading to vasoconstriction and increased blood pressure).90 The development of selective S1P1 modulators could theoretically bypass these cardiovascular side effects. Nevertheless, it has been shown that S1P1 is also expressed in atrial cardiomyocytes, leading to a dose-dependent heart rate reduction with selective S1P1 modulators.91 Although these effects appear to be transient, due to initial activation of S1P1 and before its internalisation and degradation, and could be minimised with a gradual dose titration regimen.92
Laquinimod
Laquinimod is an oral, synthetic, roquinimex-derived quinolone-3-carboxamide SMD (molecular weight ∼356.8 Da) with anti-inflammatory properties.93 In a recent phase III study, laquinimod displayed clear efficacy in relapsing-remitting multiple sclerosis94 and also has been evaluated for the treatment of CD95 and other chronic inflammatory diseases (such as lupus nephritis and arthritis).
Although laquinimod's exact mechanism of action and molecular target are not yet well defined, the drug has modulatory effects on antigen-presenting cells and directs T cells towards an anti-inflammatory Th2 profile (with a subsequent increase in IL-4, IL-10 and transforming growth factor-β1 (TGF-β) levels) and away from a pro-inflammatory Th1 and Th17 profile (with subsequent decreases in TNF-α, IFN-γ, IL-12 and IL-17 levels).96 ,97 Laquinimod also seems to have direct neuroprotective effects, although it is not clear whether it has protective effects on other tissues.98 ,99 In a murine model of colitis (the IL-10 knock-out mouse), laquinimod treatment was associated with a decrease in (i) the severity of spontaneous colitis and (ii) counts of T cells secreting with pro-inflammatory cytokines.100 Interestingly, an improvement in barrier function (with increased expression and distribution of tight junction proteins) and reduced expression of nuclear factor-κB pathway proteins were also observed.
In a phase IIa multicentre, randomised, placebo-controlled study, d'Haens et al95 evaluated the safety and efficacy of 8 weeks of induction therapy with laquinimod in patients with active, moderate-to-severe CD (defined as a Crohn's disease activity index (CDAI) of 220–450 and either a serum C reactive protein level >5 mg/L or endoscopic evidence of mucosal ulcerations). Overall, 117 patients were randomised to laquinimod 0.5, 1, 1.5 or 2 mg/day, and 63 patients were randomised to placebo. Surprisingly, the lowest dose level of laquinimod (0.5 mg/day) showed greater clinical benefits than the placebo in terms of the clinical remission rate (48.3% vs 15.9%, respectively) and the clinical response rate (55.2% vs 31.7%, respectively). Less benefit was seen with laquinimod 1 mg, and the results for the highest dose levels (1.5 and 2 mg) did not differ from placebo. A post hoc analysis showed that the percentage of patients with a reduction in faecal calprotectin levels was greater in all the laquinimod-treated groups than in the placebo group.
In terms of safety, laquinimod appears to be well tolerated. Exacerbation of CD was the most frequent serious adverse event, and other common adverse events included headache, abdominal pain and elevation of pancreatic amylase levels. Larger trials of laquinimod in other disease conditions (such as multiple sclerosis) have also reported abdominal pain, back pain, cough and mild elevation of serum liver enzymes as common adverse events.94
Laquinimod has not yet been tested in patients with UC. In CD, phase III trials of induction and maintenance therapy with lower doses of laquinimod (0.25 mg and 0.5 mg/day) are being planned.95
Mongersen
Low activity of TGF-β1, a cytokine produced by various cell types, is seen in patients with IBD.101 TGF-β1 has several functions, including immunosuppressive effects and the inhibition of T cells and antigen-presenting cells.102 TGF-β1 exerts its effects through a transmembrane receptor comprising two subunits (TGF-β1RI and TGF-β1RII). The binding of TGF-β1 to TGF-β1RII phosphorylates and activates the TGF-β1RI subunit, which in turn phosphorylates the intracellular proteins SMAD2 and SMAD3. These two proteins subsequently form a complex with SMAD4. This complex translocates into the nucleus and suppresses the expression of inflammatory genes.103 ,104 In contrast, the intracellular protein SMAD7 inhibits TGF-β1 signalling by binding to TGF-β1RI and thus blocking phosphorylation of SMAD2 and SMAD3.103 ,105 The low activity of TGF-β1 in patients with IBD appears to be related to overexpression of SMAD7.101
The orally administered pharmaceutical formulation mongersen (formerly known as GED-0301) contains a 21-base single-strand synthetic oligonucleotide that hybridises to SMAD7 mRNA and induces the latter's degradation via an antisense mechanism (figure 3). A pH-dependent coating protects the oligonucleotide from gastric degradation and delivers the active antisense oligonucleotide mainly to the terminal ileum and right colon.107 In pharmacological terms, mongersen is not a conventional SMD (table 1). Nevertheless, we have included the molecule in the present review by virtue of (i) its oral administration, (ii) the intracellular target and the novel pathway involved and (iii) the promising results observed to date.

(A) Binding of transforming growth factor-β1 (TGF-β1) to TGF-β1RII phosphorylates and activates the TGF-β1RI subunit, which leads to phosphorylation of the intracellular proteins SMAD2 and SMAD3. These SMAD proteins then form a complex with SMAD4. The complex translocates into the nucleus and suppresses the expression of inflammatory genes. SMAD7 inhibits TGF-β1 signalling by binding to TGF-β1RI and thus blocking SMAD2 and SMAD3 phosphorylation. (B) SMAD7 overexpression is observed in patients with IBD, which prevents the TGF-β1-mediated suppression of inflammatory genes and thus upregulates several cytokines (such as tumour necrosis factor-α (TNF-α), interleukin (IL)-2, IL-6, IL-8, IL-17 and interferon (IFN)-γ).106 Mongersen is an oligonucleotide that binds to SMAD7 mRNA and induces its degradation via an antisense mechanism. It thus inhibits SMAD7 production and restores TGF-β1 activity.
Mongersen's efficacy and safety in patients with active CD have been evaluated in a phase II, multicentre, double-blind, placebo-controlled, randomised trial.108 In total, 166 patients with moderate-to-severe CD (defined as a CDAI between 220 and 400 but no objective markers of disease activity) and involvement of the ileum and/or right colon were randomised to receive placebo or one of three dose levels of mongersen (10, 40 or 160 mg/day) for 2 weeks. The patients were then followed up for 84 days. The primary end point was the clinical remission (defined as a CDAI <150 at day 15 and maintenance of CDAI <150 until day 28), which was significantly higher in the two groups with higher dose regimens levels of mongersen (65% and 55% vs 10%, p<0.001). The clinical response (CDAI reduction of >100 points by day 28) was also significantly greater in all the mongersen groups than in the placebo group.108 Surprisingly, a post hoc analysis showed that neither disease duration nor elevated C reactive protein (≥3 mg/dL) had an impact on mongersen's efficacy. Furthermore, no association between the baseline CDAI and remission/response rate was seen in the 160 mg group. However, patients in the 40 mg group showed lower remission and response rates with a baseline CDAI ≥260, which accounts for the need for a higher dose of mongersen in individuals with a higher CDAI.109 The remission and response rates seen in this study were much higher than in pivotal induction studies with biologics.110 Two potential weakness of the study were (i) the lack of objectively confirmed mucosal ulceration as an inclusion criteria and (ii) a high percentage of participants with normal baseline C reactive protein levels. Hence, these results should be considered with a degree of caution. In view of the well-known dissociation between clinical symptoms and mucosal lesions in CD,111 data on mucosal healing were eagerly awaited. To address this important issue, a randomised, double-blind, multicentre phase Ib trial is underway (NCT02367183). The primary outcome is to evaluate the endoscopic effects after a 12-week treatment phase followed by a 54-week observation phase; 63 patients with endoscopic confirmation of disease activity were enrolled. A recent press release by Celgene112 stated that in this trial a significant proportion of patients treated with mongersen showed an endoscopic improvement (defined as a 25% improvement from baseline Simplified Endoscopic Activity Score for Crohn's Disease) at week 12, but the exact percentage is not yet in the public domain.
In terms of safety, mongersen was well tolerated, with similar frequencies of adverse events in the placebo group (64%) and the mongersen group (65%). Most of the adverse events (mainly nausea, vomiting, abdominal pain and influenza) were mild. The serious adverse events were usually related to CD activity. Nonetheless, the relatively short duration of drug exposure (2 weeks), the short follow-up period and the small sample size mean that larger, longer safety trials are needed. One important concern with mongersen is the known role of TGF-β1 in tissue remodelling with pro-fibrogenic effects,113–115 which might lead to fibrosis and narrowing of stenosis; this might limit the use of mongersen to induction treatments of acute exacerbations of CD.116
Following the very promising results reported for mongersen in phase II in CD, a phase III trial (as well as the phase Ib endoscopic trial) in CD (NCT02596893) and a phase II trial in UC (NCT02601300) are underway. These studies should provide a better understanding of the drug's safety profile and more robust information on objective markers of anti-inflammation efficacy (such as mucosal healing).117
Perspectives
With the advent of biologics, the care of patients with IBD has changed radically over the last two decades. However, treatment with today's biologics has some major drawbacks: moderate efficacy (with only one-third of patients in remission 1 year after treatment initiation), infection-related complications, a potential risk of malignancy, the need for parenteral administration and elevated treatment-related costs.
The appealing intrinsic characteristics of SMDs may address the problems associated with currently available biologics. Recently, a clear paradigm shift has taken place in the treatment of chronic hepatitis C infection, with a move from the biologic interferon to SMD-based treatments with outstanding results.118 Evidently, the pathogenesis of IBD is far more complex and diverse, although a better understating of the critical mechanisms driving inflammation will be essential for better targeting.
Several new SMDs and other orally administered formulations are currently being investigated as therapeutic options in both UC and CD. These agents have shown promising levels of efficacy and an acceptable safety profile, although larger trials are needed before definitive conclusions can be drawn. Recently, tofacitinib was found to be effective in phase III clinical trials, and Pfizer is seeking marketing authorisation in UC.
The SMDs presented in the present review are probably pioneers in a novel field of therapeutic research in IBD. Other molecules are in the pipeline (figure 4 and table 2); some have the same molecular targets as the drugs discussed above (JAK inhibitors and S1PR modulators) and others modulate completely different pathways (eg, phosphodiesterase type 4, ATPases, etc). Interestingly, nutritional small molecules like phosphatidylcholine, aim to a restoration of the intestinal mucosal barrier. After promising results in a phase IIa study,119 a retarded release phosphatidylcholine formulation (LT-02) is being tested in phase III trials (NCT02849951, NCT02280629 and NCT02142725). The extended release nicotinamide developed by Ferring also looks promising in IBD.
Small-molecule drugs under clinical development in IBD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Small-molecule drugs pipeline for IBD. CD, Crohn's disease; JAK, Janus kinase; MOA, mechanism of action.
Other appealing compounds are the orally administrated anti-TNF agents, like AVX-470, which has shown promising results in a phase I study.120 However, data regarding these large molecules, which are orally administrated biologics, are preliminary, and it remains to be confirmed whether they share the advantages of SMDs other than oral administration.
The use of SMDs raises several issues in clinical practice. First, long-term adherence to other oral drugs used in IBD (such as azathioprine and aminosalicylates) is poor.42 Second, the SMDs' positioning will remain a matter of debate until head-to-head trials against monoclonal antibodies or between SMDs are performed.
It should be noted that not all patients are likely to respond to a particular drug (whether SMDs or biologics). Hence, a better understanding of the mechanisms involved in these differences and the discovery of corresponding biomarkers will be critical. It is probable that a given drug will not achieve all treatment goals in some patients. Therefore, combinations of SMDs and SMD-biologic might inhibit several pathways at the same time and this augments efficacy—although this would probably increase the treatment cost and the frequency of adverse events.
In summary, this is an exciting time in the field IBD; many novel agents are being developed and some will probably become available in clinical practice in the foreseeable future. Our patients with IBD will benefit from a broader therapeutic armamentarium. Tofacitinib may soon be available in the clinic for the treatment of UC. Given that controlled trials are not sufficiently powered for the assessment of a drug's safety profile (especially for rare events such as serious infections and malignancy), the risk–benefit ratio of SMDs in IBD should be evaluated in large, prospective studies such as I-CARE (NCT02377258).
References
Footnotes
Twitter Follow Pablo Olivera at @poliverasendra
Contributors PO and LP-B wrote the article. PO drafted all figures. SD critically reviewed the article for intellectual content.
Competing interests PO declares no conflict of interest. SD has served as a speaker, a consultant and an advisory board member for Abbvie, Ferring, Hospira, Johnson & Johnson, Merck, Millennium Takeda, Mundipharma, Pfizer Inc, Tigenix, UCB Pharma, and Vifor. LP-B: consulting fees from Merck, Abbvie, Janssen, Genentech, Mitsubishi, Ferring, Norgine, Tillots, Vifor, Therakos, Pharmacosmos, Pilège, BMS, UCB-pharma, Hospira, Celltrion, Takeda, Biogaran, Boerhinger-Ingelheim, Lilly, Pfizer, HAC-Pharma, Index Pharmaceuticals, Amgen, Sandoz. Lecture fees from Merck, Abbvie, Takeda, Janssen, Ferring, Norgine, Tillots, Vifor, Therakos, Mitsubishi, HAC-pharma.
Provenance and peer review Commissioned; externally peer reviewed.