


Abstract
The prodrug concept has been used to improve undesirable properties of drugs since the late 19th century, although it was only at the end of the 1950s that the actual term prodrug was introduced for the first time. Prodrugs are inactive, bioreversible derivatives of active drug molecules that must undergo an enzymatic and/or chemical transformation in vivo to release the active parent drug, which can then elicit its desired pharmacological effect in the body. In most cases, prodrugs are simple chemical derivatives that are only one or two chemical or enzymatic steps away from the active parent drug. However, some prodrugs lack an obvious carrier or promoiety but instead result from a molecular modification of the prodrug itself, which generates a new active compound. Numerous prodrugs designed to overcome formulation, delivery, and toxicity barriers to drug utilization have reached the market. In fact, approximately 20% of all small molecular drugs approved during the period 2000 to 2008 were prodrugs. Although the development of a prodrug can be very challenging, the prodrug approach represents a feasible way to improve the erratic properties of investigational drugs or drugs already on the market. This review introduces in depth the rationale behind the use of the prodrug approach from past to present, and also considers the possible problems that can arise from inadequate activation of prodrugs.
I. Introduction
In simplified terms, prodrugs are masked forms of active drugs that are designed to be activated after an enzymatic or chemical reaction once they have been administered into the body (Fig. 1) (Ettmayer et al., 2004; Stella et al., 2007; Rautio et al., 2008; Rautio, 2010; Stella, 2010). Prodrugs are considered to be inactive or at least significantly less active than the released drugs; therefore, salts of active agents and drugs, whose metabolites contribute to the overall pharmacological response, are not included in this definition. The rationale behind the use of prodrugs is generally to optimize the so-called “drug-like” properties [i.e., absorption, distribution, metabolism, and excretion (ADME1) properties] because they can cause considerable problems in subsequent drug development, if unfavourable (Table 1). In addition, the prodrug strategy has been used to increase the selectivity of drugs for their intended target. This can not only improve the efficacy of the drug but also decrease systemic and/or unwanted tissue/organ-specific toxicity (T). Development of a prodrug with improved properties may also represent a life-cycle management opportunity.

A simplified illustration of the prodrug concept.
The rationale behind the use of the prodrug approach
Unfortunately, the general opinion among scientists still is that prodrugs are “an act of desperation”; they are considered only when problems are encountered with the original drug candidate. The design of an appropriate prodrug should never be viewed as a last resort. If anything, this alternative should be considered in the early stages of preclinical development, bearing in mind that prodrugs may alter the tissue distribution, efficacy, and even toxicity of the parent drug. Therefore, modifying the ADMET properties of the parent drug requires a comprehensive understanding of the physicochemical and biological behavior of the drug candidate. Although prodrug design is very challenging, it can still be more feasible and faster than searching for an entirely new therapeutically active agent with suitable ADMET properties. Another fallacy is that prodrugs are simply of academic interest and do not have industrial applications in attempts to overcome bioavailability and toxicity problems. A very good indication of the success of the prodrug approach can be obtained by examining the prevalence of prodrugs on the market. It might come as a surprise to many people that currently approximately 10% of all globally marketed medicines can be classified as prodrugs, and in 2008 alone, 33% of all approved small-molecular-weight drugs were prodrugs (Rautio, 2010; Stella, 2010). Despite these impressive numbers, we have only begun to appreciate the full potential of the prodrug approach in modern drug development, and many novel prodrug innovations await discovery.
II. History and the Present of Prodrug Design
Adrien Albert first introduced the term “pro-drug” in 1958 (Albert, 1958). A few decades later, he apologized for having invented such an inaccurate term, because “predrug” would have been a more descriptive term. However, by that time, the original version was used too widely to be changed. Nonetheless, the prodrug concept has been invented long before Albert's publication. The first intentionally designed prodrug is most probably methenamine (or hexamine), which was introduced 1899 by Schering. Methenamine releases 6 Eq of the antibacterial formaldehyde along with 4 Eq of ammonium ions in acidic urine and serves as a good example of a site-selective prodrug (Testa, 2004; Stella et al., 2007). At the same time, Bayer introduced aspirin (acetylsalicylic acid) as a less irritating form of the anti-inflammatory agent sodium salicylate. However, it remains a matter of debate whether aspirin is a true prodrug or not. Although it was intended to work as a prodrug, aspirin inhibits irreversibly cyclooxygenase, the enzyme responsible for formation of the key biological mediators, prostaglandins and thromboxanes, by acetylating one hydroxyl group of serine residue (Ser 530) in the active site of the enzyme, while the parent drug, salicylic acid, is a weak reversible inhibitor of cyclooxygenase. In this respect, aspirin cannot be considered as a prodrug (Vane and Botting, 2003). However, aspirin is rapidly hydrolyzed in the intestinal wall and liver, as well as in the blood to salicylic acid, which means that it acts, in fact, like a prodrug (Testa and Mayer, 2003).
Many decades elapsed until Bayer introduced their next prodrug, the antibiotic prontosil, in 1935. However, prontosil was not intentionally developed as a prodrug, because only later in the same year was it found to release an active agent, para-aminophenylsulfonamide, by reductive enzymes. The launch of prontosil gave rise to the second generation of sulfonamide prodrugs, because the sulfanilamide moiety was easy to link to other molecules (Bentley, 2009). In a similar way, Roche discovered the prodrug activity of the antituberculosis drug isoniazid more than 40 years after its introduction in 1952. Today, it is known that the bioactivation of isoniazid is catalyzed by the mycobacterial catalase-peroxidase called KatG. The generated reactive species form adducts with NAD+ and NADP+ that inhibit the biosynthesis of mycolic acid required for the mycobacterial cell wall (Timmins and Deretic, 2006). Sometimes, unintentionally developed prodrugs can reveal a less appealing truth of the drug under the development. A good example is heroin (diacetylmorphine), which was marketed during the years 1898 to 1910 as a nonaddictive morphine substitute to suppress cough and cure both cocaine and morphine addictions. Bayer was embarrassed to appreciate later that heroin is, in fact, rapidly metabolized into morphine after oral administration (Inturrisi et al., 1983; Sawynok, 1986).
Since the 1960s there has been an explosive increase in the use of prodrugs in drug discovery and development. The beginning of 21st century, when property-based drug design became an essential part of the drug discovery and development process (van De Waterbeemd et al., 2001), has been a time of real breakthroughs in prodrugs. This can be seen in published journal articles, reviews, and patents, as well as in numbers of clinically studied and marketed medicines. Moreover, dozens of prodrugs are currently undergoing clinical trials. To emphasize the extent of the successful implementation of the prodrug approach, almost 15% of the 100 best-selling small-molecular-weight drugs in 2009 were prodrugs (Table 2) (Rautio, 2010). One interesting example of these blockbuster prodrugs is lisdexamfetamine dimesylate (Vyvanse), the l-lysine prodrug of the psychostimulant dextroamphetamine, which was designed to have less abuse potential than other amphetamines due to the slower release of the active parent drug if inhaled or injected. Another best-selling group of prodrugs are the proton pump inhibitors (PPIs) omeprazole and its analogs, which are site-selectively bioactivated to their active species in the acidic parietal cells of stomach (also discussed in section III.E).
The occurrence of prodrugs among the world's 100 top-selling pharmaceuticals in 2009
III. Rationale for Prodrug Design
The prodrug approach is a very versatile strategy to increase the utility of pharmacologically active compounds, because one can optimize any of the ADMET properties as well as prolong the commercial life cycle of potential drug candidates. In most cases, prodrugs contain an obvious carrier or promoiety, which will be removed by enzymatic or chemical reaction(s), whereas some prodrugs release their active drugs after molecular modification (e.g., after oxidation or reduction reaction). The latter are usually referred to as bioprecursor prodrugs. Conventional carrier-linked prodrugs often have a synthetic handle, a spacer or linker, between the active drug and the promoiety when the desired prodrug moiety cannot be attached directly to the parent molecule because of steric hindrance or functional properties. The spacers are usually cleaved spontaneously after the enzymatic or chemical decomposition of the prodrug bond between the promoiety and spacer (Papot et al., 2002). The prodrug candidate can also be prepared as a double prodrug, where the second promoiety is attached to the first promoiety linked to the parent drug molecule. These promoieties are usually different and are cleaved by a dissimilar mechanism. The term double prodrug should not be confused with the term bifunctional prodrug, which means that the parent drug molecule has been modified at two functional groups of the parent molecule. In some cases, two pharmacologically active drugs can be coupled together in a single molecule, called a codrug. In a codrug each drug acts as a promoiety for the other. In the following sections, the potential of the prodrug approach to improve unfavourable ADMET properties or to prolong the life cycle of drugs will be illustrated by providing several examples of clinically used and investigational prodrugs.
A. Improving Formulation and Administration
Dissolution of the drug molecule from the dosage form may be the rate-limiting step to absorption (Hörter and Dressman, 2001). In fact, achieving optimal solubility is one of the greatest challenges in the drug discovery process. It has been reported that more than 30% of drug discovery compounds have poor aqueous solubility (i.e., less than 10 μM) (Di and Kerns, 2007). Different formulation techniques, such as salt formation and solubilising excipients, have been used to overcome this barrier. Prodrugs are an alternative way to increase the aqueous solubility of the parent drug molecule by improving dissolution rate via attached ionizable or polar neutral groups, sich as phosphates, amino acids, or sugar moieties (Fleisher et al., 1996; Stella and Nti-Addae, 2007; Stella et al., 2007; Müller, 2009). These prodrugs can be used not only to enhance oral bioavailability but also to prepare parenteral or injectable drug delivery.
Phosphate esters are a widely used prodrug strategy for improving the aqueous solubility, not only oral but also and especially drugs intended for parenteral administration (Krise and Stella, 1996; Stella and Nti-Addae, 2007; Stella et al., 2007). The active parent drug molecule is rapidly released from phosphate prodrugs by endogenous phosphatases, such as alkaline phosphatase. This enzyme is particularly abundant on the brush border (or apical surface) of the enterocytes and in plasma. Because the phosphate group is cleaved before or during absorption after oral administration, the ionizable prodrug structure does not decrease the permeability of the parent drug molecule.
Prednisolone sodium phosphate (Pediapred; Fisons Corp., Bedford, MA) is a classic example of a phosphate prodrug (Fig. 2). It is a highly water-soluble (>30 times greater than prednisolone), oral liquid formulation used as an immunosuppressant for children in allergic and inflammatory conditions and used to mask the unpalatable taste of prednisolone tablets. The phosphate promoiety is linked directly to a free hydroxyl group on prednisolone, as is in the case with another well known oral phosphate prodrug, fosamprenavir (Lexiva, Telzir; Vertex Pharmaceuticals, Cambridge, MA) (see also section III.F). Amprenavir (Agenerase; Glaxo Group Ltd., Greenford, Middlesex, UK) is an HIV protease inhibitor that, because of its poor water solubility, was originally formulated in a capsule containing a high concentration of solubilizing excipients. To achieve the correct therapeutic concentration, patients had to take eight amprenavir capsules every day. By replacing amprenavir with its 10-fold more water-soluble prodrug fosamprenavir, one can achieve a simplified and patient-compliant dosage regimen (i.e., two tablets twice a day) that has the same therapeutic efficacy (Chapman et al., 2004; Furfine et al., 2004; Wire et al., 2006). Both of these orally administered phosphate prodrugs are rapidly hydrolyzed by gut epithelial alkaline phosphatases to their respective parent drugs during the absorption process, with only minimal concentrations of prodrugs reaching the circulation.

Bioactivation of phosphate prodrugs of prednisolone and phenytoin.
In another example, fosphenytoin sodium salt (Cerebyx; Warner-Lambert, Morris Plains, NJ), the phosphate ester is attached to an acidic amine of the antiepileptic agent phenytoin via an oxymethylene spacer (Fig. 2). Fosphenytoin is used to reduce drug precipitation and consequent local irritation by phenytoin at the injection site. It has an aqueous solubility more than 7000 times greater than that of phenytoin and is rapidly converted (t1/2 < 15 min in human) to phenytoin in blood (Unadkat and Rowland, 1985; Boucher, 1996; Browne et al., 1996). The enzymatic hydrolysis initially releases an unstable intermediate, which is spontaneously converted to phenytoin (Fig. 2).
Amino acid esters or amides are also commonly used ionizable groups, which can be introduced into the hydroxyl, thiol, amine, or carboxylic acid functionalities of the parent drug molecule to compensate for poor aqueous solubility. A variety of esterases, amidases, and/or peptidases in plasma or in other tissues can bioconvert these prodrugs to their active counterparts. Quite often, however, amino acid ester and amide prodrugs suffer from poor aqueous stability or incomplete in vivo bioconversion, respectively, and therefore phosphate prodrugs are the preferred prodrug structures if one wishes to improve the aqueous solubility of the parent drug (Stella et al., 2007). Paradoxically, amino acid esters and amides can also be used to enhance absorption and consequently oral drug delivery of parent drugs, because the brush-border membrane of intestinal epithelium possesses a considerable number of transporters for amino acids and peptides (Majumdar et al., 2004; Dobson and Kell, 2008). Examples of these kinds of prodrugs will be given in the next chapter. Sugar moieties, such as glucose, galactose, or glucuronic acid, have also been used as ways to improve the aqueous solubility of poorly water-soluble drugs. These prodrugs are bioconverted to their parent drug molecules by β-glucosidase, β-galactosidase, or β-glucuronidase and usually have spacers between the parent drug and the promoiety (Leenders et al., 1999).
B. Enhancing Permeability and Absorption
The transport of a drug to its site of action usually requires passage through several lipid membranes; therefore, membrane permeability has a considerable influence on drug efficacy (Chan and Stewart, 1996). In oral drug delivery in particular, which is the preferred route for the majority of drugs, the most common absorption routes are unfacilitated and largely nonspecific, passive transport mechanisms. The lipophilicity of poorly permeable drugs can be increased by modifying the hydrocarbon moieties. However, good activity sometimes requires a structure, which is far from ideal one for good membrane permeability. In such situation, the prodrug strategy can be an extremely valuable option. Improvements of lipophilicity have been the most widely studied and therefore now also the most successful field of prodrug research. It has been achieved by masking polar ionized or nonionized functional groups to enhance either oral or topical absorption (e.g., for drugs to be given by transdermal and ocular administration) (Beaumont et al., 2003).
A hydrophilic hydroxyl, thiol, carboxyl, phosphate, or an amine group on the parent drug can be converted to more lipophilic alkyl or aryl esters, and these prodrugs are readily bioconverted to their active species by ubiquitous esterases, which are present throughout the body (Taylor, 1996; Liederer and Borchardt, 2006). The attractiveness of this prodrug approach is that the alkyl chain length can be modified to obtain precisely the desired lipophilicity. Currently, a considerable number of ester prodrugs have advanced into clinical use.
Oseltamivir (Tamiflu; Hoffmann-La Roche Inc., Nutley, NJ) is an orally active ethyl ester prodrug of a selective inhibitor of viral neuraminidase glycoprotein and used in the treatment of influenza types A and B (Fig. 3). After absorption, oseltamivir undergoes rapid bioconversion to its parent drug mostly by the action of carboxylesterase (McClellan and Perry, 2001; Shi et al., 2006). The bioavailability of the more lipophilic oseltamivir is almost 80%, whereas the corresponding value for free carboxylate is as low as 5%. Likewise, adefovir dipivoxil (Hepsera; Gilead Sciences, Inc., Foster City, CA) represents a more recent example of a prodrug, in which the ester promoiety is attached to a phosphoryl group on the parent drug via a spacer (Fig. 3). This pivaloyloxymethyl phosphoric acid ester of the nucleotide reverse transcriptase inhibitor adefovir (PMEA) is given orally to treat retro-, herpes-, and hepadnaviruses and has almost four times greater bioavailability than adefovir itself (Benzaria et al., 1996; Noble and Goa, 1999).

Bioactivation of ester prodrugs oseltamivir, adefovir dipivoxil, and dabigatran etexilate.
The latest clinical example of a more lipophilic oral prodrug is dabigatran etexilate (Pradaxa; Boehringer Ingelheim Pharma GmbH, Ingelheim, Germany), a direct thrombin inhibitor for stroke prevention, which has been available in Europe and many other countries since 2008. However, it was not until the October 2010 when dabigatran etexilate became the first oral alternative to warfarin to be approved in the United States (Hughes, 2010). The active drug dabigatran is a very polar, permanently charged molecule with a log P of −2.4 (n-octanol/buffer, pH 7.4) and therefore it has zero bioavailability after oral administration (Eisert et al., 2010). In the more lipophilic bifunctional prodrug dabigatran etexilate, the two polar groups, the amidinium moiety and carboxylate, are masked by carbamic acid ester and carboxylic acid ester groups, respectively, which results in better absorption with bioavailability of 7% after oral administration (Stangier et al., 2007; Blech et al., 2008). Unlike its predecessor, the bifunctional prodrug ximelagatran [Exanta (AstraZeneca, Pharmaceuticals LP, Wilmington, DE); withdrawn from the market in 2006] (Sorbera et al., 2001), dabigatran etexilate has not shown any long-term adverse effects, such as liver toxicity.
Another method to increase oral absorbtion is to design prodrugs, which have structural features similar to substrates that are absorbed by carrier-mediated transport. This strategy is particularly important when passive transcellular absorption is negligible. Good examples of carrier-mediated of prodrugs are midodrine (ProAmatine, Gutron; Shire Llc, Florence, KY) and valacyclovir (Valtrex; SmithKline Beecham, Philadelphia, PA; Glaxo Wellcome Inc., Research Triangle Park, NC) (Fig. 4). Midodrine is a glycine prodrug of desglymidodrine [1-(2',5'-dimethoxyphenyl)-2-aminoethanol (DMAE)], a selective α1-receptor agonist used in the treatment of orthostatic hypotension, in which the glycine promoiety is attached to an amine group of DMAE. Midodrine is absorbed via the intestinal proton-coupled peptide transporter 1 and is bioconverted by as-yet-unknown peptidases, mainly in the liver and systemic circulation (Cruz, 2000; Tsuda et al., 2006). It has an oral bioavailability of 93%, which is significantly more than the corresponding value for DMAE (50%).
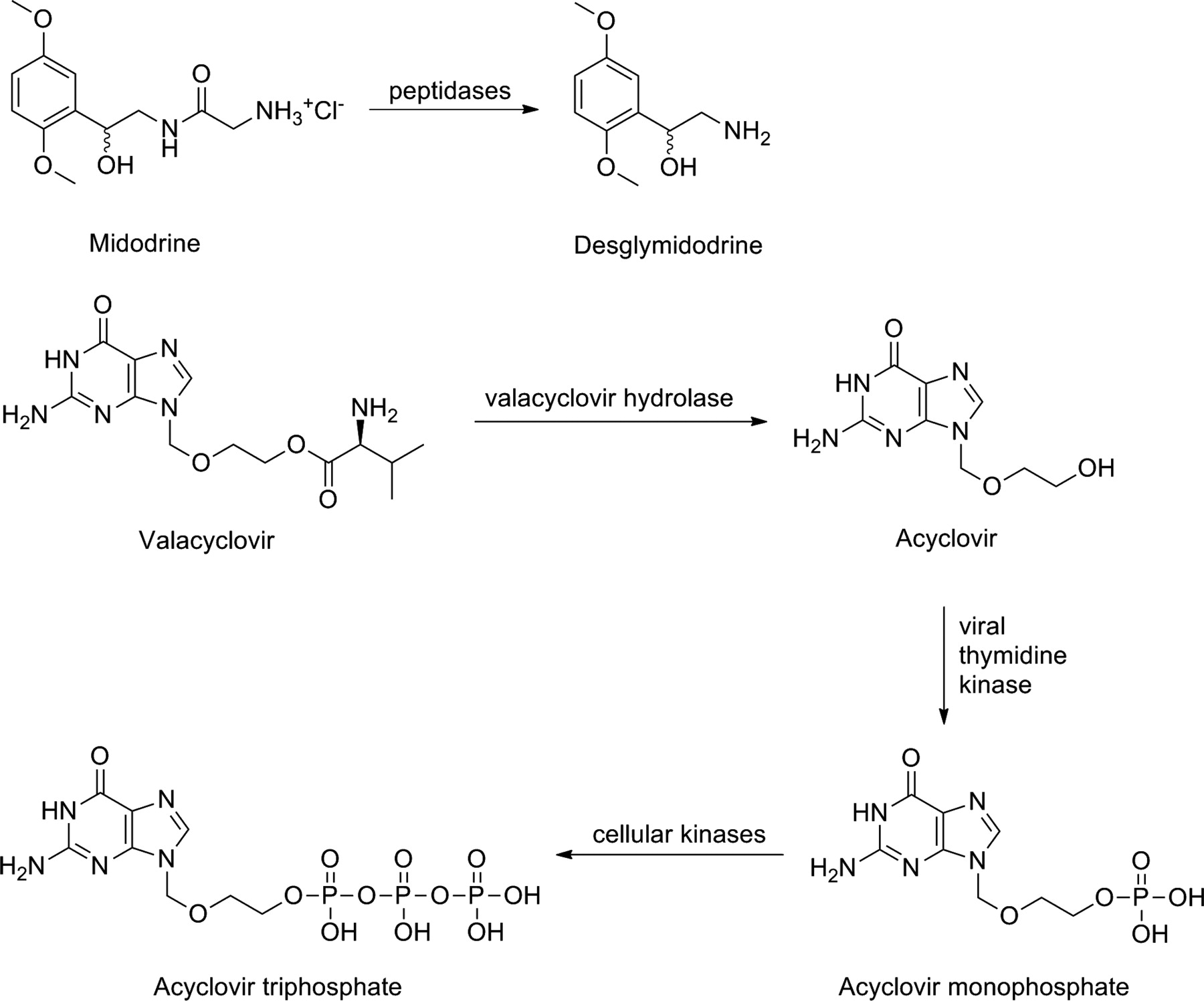
Bioactivation of amino acid prodrugs midodrine and valacyclovir.
Valacyclovir is a l-valyl ester prodrug of acyclovir, a purine nucleoside used for the treatment of herpes virus infections. Valacyclovir is a substrate, not only for peptide transporter 1 (Balimane et al., 1998; Sinko and Balimane, 1998) but also for Na+-dependent neutral amino acid transporter, ATB0,+ (Ganapathy and Ganapathy, 2005). After absorption, it is bioactivated by valacyclovir hydrolase (Burnette et al., 1995). The bioavailability of this prodrug is more than 50%, which is 20 to 35% better than the bioavailability of acyclovir (Granero and Amidon, 2006). Valacyclovir is actually a preprodrug or double prodrug, because after the hydrolysis of the valine promoiety, acyclovir, like all nucleosides, requires phosphorylation before it forms the active nucleotide triphosphate. The first phosphorylation reaction is mediated by viral thymidine kinase, which is far more effective than the endogenous enzyme in uninfected cells. This further improves the selectivity of acyclovir. Cellular kinases subsequently phosphorylate the nucleotide monophosphate to its active triphosphate form, which then can act as a selective inhibitor of viral DNA polymerase (Elion, 1983; Darby, 1993).
Prodrugs with lipophilic promoieties have also been used to improve topical absorption for transdermal and ocular drugs (Sloan and Wasdo, 2003; Sloan et al., 2006). The stratum corneum, the outermost layer of the epidermis, represents a high resistance barrier against topical drug delivery. Only the drugs with balance of both water and lipid solubilities can efficiently penetrate through the layers of the skin. These optimal features can be achieved by prodrug technology. Tazarotene (Tazorac; Allergan, Inc., Irvine, CA) is an example of a marketed ethyl ester prodrug with enhanced transdermal drug delivery (Foster et al., 1998). Tazarotene is used for psoriasis and acne treatment and causes less skin irritation than the parent drug tazarotenic acid (Fig. 5). For more examples of investigational transdermally administered prodrugs, see the review by Fang and Leu (2006).

Bioactivation of topically administered ester prodrugs tazarotene and dipivefrin.
In the case of topically administered ophthalmic drugs, the corneal epithelium limits the permeation of drugs into intraocular tissues. However, more lipophilic ester prodrugs as well as transporter-mediated amino acid prodrugs have improved ocular drug delivery with greatly reduced adverse effects, especially on the surface of the eye (Duvvuri et al., 2003, 2004). The first prodrug to improve ocular absorption was the dipivalyl diester of epinephrine (adrenaline), dipivefrin (Propine; Allergan, Inc.), introduced to the market over 15 years ago to lower the intraocular pressure in glaucoma (Fig. 5). Dipivefrin penetrates the human cornea 17 times more rapidly than epinephrine and is therefore not only more effective but also has fewer systemic adverse effects (Hussain and Truelove, 1976; Kaback et al., 1976; Mandell et al., 1978; Kohn et al., 1979). Some recently marketed counterparts of dipivefrin are the prostaglandin analogs latanoprost (Xalatan; Pfizer Health AB, Stockholm, Sweden), bimatoprost (Lumigan; Allergan, Inc.), travoprost (Travatan; Alcon, Inc., Hünenberg, Switzerland), and unoprostone isopropyl (Rescula; Sucampo AG, Zug, Switzerland) (Netland et al., 2001; Susanna et al., 2002; Hellberg et al., 2003).
C. Changing the Distribution Profile
After administration, a drug molecule has to bypass several pharmaceutical and pharmacokinetic barriers before it can reach its physiological target and exert the desired effect. For decades, attempts have been made to harness different macromolecular strategies and nanotechnologies to achieve site-selective drug delivery. The lack of clinical success of these methods, however, has focused interests on other approaches. Today, one of the most promising site-selective drug delivery strategies is the prodrug approach, which exploits target cell- or tissue-specific endogenous enzymes and transporters. A great many prodrugs have increased efficacy and safety profiles because of their targeting properties. Prodrugs that have been designed to overcome toxicity issues will be discussed more detailed in section III.E.
However, there are only a few examples of prodrugs that have been designed to accumulate into a specific tissue or organ only to improve the efficacy of the drug. One of these is the antiparkinson agent l-DOPA (various trade names). Because of its hydrophilic nature, the neurotransmitter dopamine is not able to cross the blood-brain barrier and distribute into brain tissue. However, the α-amino acid prodrug of dopamine, l-DOPA, enables the uptake and accumulation of dopamine into the brain via the L-type amino acid transporter 1 (Stella et al., 2007; del Amo et al., 2008). After L-type amino acid transporter 1-mediated uptake, l-DOPA is bioactivated by aromatic l-amino acid decarboxylase to hydrophilic dopamine, which is concentrated in dopaminergic nerves (Fig. 6). Because l-DOPA is extensively metabolized in the peripheral circulation (90–95%), DOPA decarboxylase inhibitors, such as carbidopa, benserazide, or methyldopa, and/or catechol-O-methyltransferase inhibitors, such as entacapone, tolcapone, or nitecapone, are coadministered with levodopa to prevent the unwanted metabolism (Khor and Hsu, 2007; Hornykiewicz, 2010).

Bioactivation of an amino acid prodrug of dopamine.
D. Protecting from Rapid Metabolism and Excretion
Presystemic metabolism (the so-called first-pass effect) in the gastrointestinal tract and liver may greatly reduce the total amount of active drug reaching the systemic circulation and ultimately its target. This problem has been bypassed by sublingual or buccal administration or by modified or controlled release formulations. Rapid metabolic breakdown of the drug can also be protected by a prodrug structure. This is usually carried out by masking the metabolically labile but pharmacologically essential functional group(s) of the drug. In the case of the bronchodilator and β2-agonist terbutaline, sustained drug action has been achieved by converting its phenolic groups, which are susceptible to rapid and extensive presystemic metabolism, into bis-dimethylcarbamates (Fig. 7). This prodrug, bambuterol (Bambec, Oxeol; AstraZeneca Pharmaceuticals LP, Wilmington, DE), is slowly bioactivated to terbutaline predominantly by nonspecific butyrylcholinesterase mainly outside the lungs (Svensson and Tunek, 1988; Tunek et al., 1988; Sitar, 1996). As a result of the slower release and prolonged action, once-daily administration of bambuterol provides relief of asthma with a lower incidence of adverse effects than terbutaline, which needs to be taken three times a day (Persson et al., 1995).
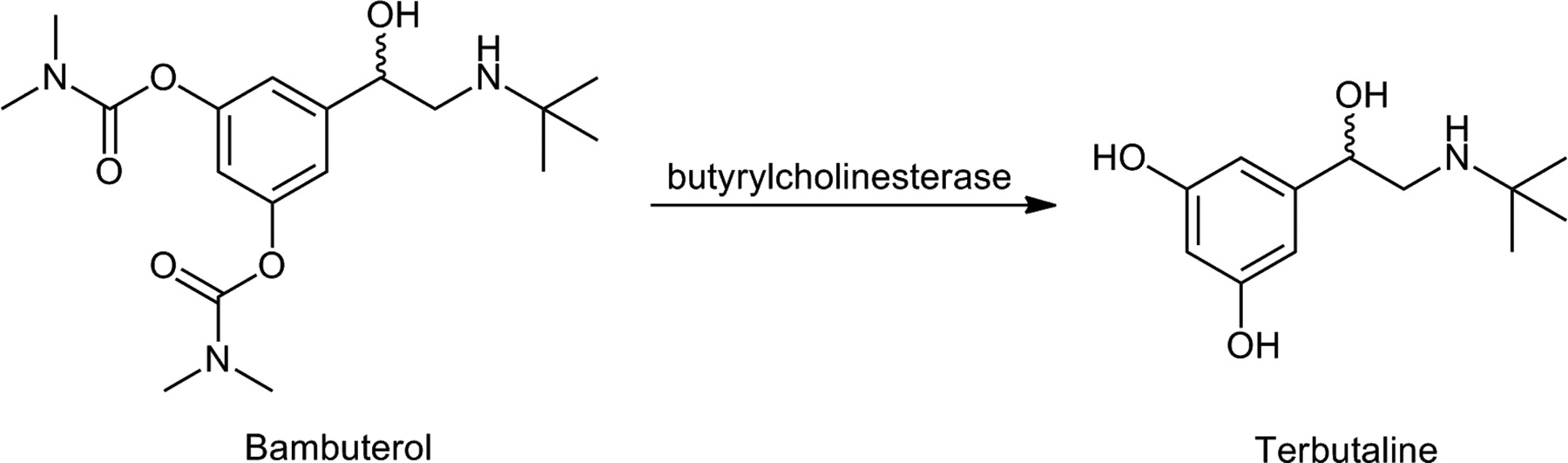
Bioactivation of a carbamate prodrug of terbutalin.
In addition to metabolic pathways, the beneficial effects of drugs can be impaired as a result of extensive excretion. The kidneys, which are the principal organs of excretion, efficiently eliminate water-soluble substances from the body. Therefore, the use of lipophilic promoieties to decrease the solubility is one way to prolong the duration of action of very water-soluble drugs. Good examples of these prodrugs are anticancer and antiviral nucleoside phosphates and phosphonates, such as adefovir dipivoxil (see section III.B).
E. Overcoming Toxicity Problems
Adverse drug reactions can change the structure or function of cells, tissues, and organs and can be detrimental to the organism. Reduced toxicity can sometimes be accomplished by altering one or more of the ADME barriers discussed earlier but more often is achieved by targeting drugs to desired cells and tissues via site-selective drug delivery. A successful site-selective prodrug must be precisely transported to the site of action, where it should be selectively and quantitatively transformed into the active drug, which is retained in the target tissue to produce its therapeutic effect (Ettmayer et al., 2004; Stella et al., 2007). The ubiquitous distribution of most of the endogenous enzymes that are responsible for bioactivating prodrugs diminishes the opportunities for selective drug delivery and targeting. Therefore, exogenous enzymes selectively delivered via monoclonal antibodies (antibody-directed enzyme prodrug therapy) or as genes that encode prodrug activating enzymes in the target-specific nonviral or viral vectors (gene-directed enzyme prodrug therapy and virus-directed enzyme prodrug therapy) are widely used, especially with highly toxic compounds, such as anticancer drugs, to reduce the toxicity of the drugs at other sites in the body (Denny, 2001; Shanghag et al., 2006).
Cytochrome P450 enzymes (P450s) can be exploited for liver-targeted drug delivery, because these enzymes are present at the highest concentrations in hepatocytes. However, when targeting the P450 enzymes, special attention needs to be paid during the drug development process to potential species and patient related variations, genetic polymorphisms, as well as the potential for drug-drug interactions (Ettmayer et al., 2004) (see section IV.B). Despite the presence of numerous P450-activated prodrugs, only cyclic phosphate and phosphonate prodrugs, called HepDirect prodrugs, are liver-specific (Erion et al., 2004, 2005). Pradefovir was the first HepDirect prodrug that advanced to clinical trials. It was developed to improve the therapeutic potential of the adefovir dipivoxil (pivaloyloxymethyl phosphoric acid ester prodrug of PMEA; see section III.B), because the phase III clinical studies had revealed that the presence of PMEA in the kidneys and the subsequent kidney toxicity limited the use of adefovir dipivoxil (Dando and Plosker, 2003). In contrast, the HepDirect prodrug of PMEA (Fig. 8) displayed 12-fold higher liver/kidney and 84-fold higher liver/intestine targeting ratio relative to adefovir dipivoxil in vivo in rats (Reddy et al., 2005; Stella et al., 2007). Moreover, the prodrug has good therapeutic efficacy in patients with hepatitis B but still achieves low systemic adefovir levels. Although the nonclinical carcinogenicity studies revealed increased incidence of cancer at higher doses, pradefovir has completed the phase II clinical trials and its clinical development continues (Erion et al., 2006; Tillmann, 2007).

Bioactivation of cyclic phosphonate prodrugs pradefovir and MB07133.
4-Amino-1-(5-O-(2-oxo-4-(4-pyridyl)-1,3,2-dioxaphosphorinan-2-yl)arabinofuranosyl)-2(1H)-pyrimidinone (MB07133), a cyclic phosphonate of the anticancer agent arabinofuranosyl cytidine [cytarabine (araC)], is another HepDirect prodrug currently undergoing clinical evaluation for the treatment of hepatocellular carcinoma (Fig. 8) (MacKenna et al., 2009). In vivo studies with rodents have shown that the araC triphosphate levels are over 19-fold greater in the liver than in plasma representing a 120-fold improvement over araC administration (Boyer et al., 2006). Furthermore, the initial data from the phase Ib clinical trial have indicated that MB07133 is well tolerated in patients with no overt effects on liver function (MacKenna et al., 2009). In both cases, the prodrug is bioactivated by CYP3A4-catalyzed hydroxylation that occurs selectively in the benzylic carbon atom adjacent to a phosph(on)ate oxygen. This results in irreversible ring opening and the formation of a transient intermediate. The anionic intermediate and the generated negatively charged phosphate have poor diffusion properties across cell membranes and are therefore retained in the hepatocytes, which augments their liver specificity.
Site-selective prodrug activation can also be achieved by exploiting the physiological conditions of the target tissue if that differs from conditions elsewhere in the body. Many solid tumors, for example, have selective enzyme expression, hypoxic cells, and lower extracellular pH (Denny, 2001). Hypoxia, the deficiency of oxygen as a result of an insufficient vasculature, imposes an environmental stress on the cells, which contributes to tumor progression, invasion, and metastasis. Moreover, a low oxygen content increases the resistance to radiotherapy and chemotherapy (Denny, 2010). However, the hypoxic state of solid tumors can be turned to a therapeutic advantage by designing cytotoxic prodrugs, which are bioactivated to their active species selectively in the hypoxic environment of solid tumors by endogenous reductive enzymes. Although these enzymes are also present in normal aerobic cells, the action of the cytotoxic species is limited to tumor cells, because the reduced prodrug intermediates are rapidly reoxidized back to the inactive prodrugs in healthy cells.
Tirapazamine (TPZ, SR 4233), a heteroaromatic N-oxide, is a well known example of a hypoxia-activated anticancer prodrug, which has served as a lead compound in the development of a number of newer prodrugs with improved anticancer properties (Fig. 9). TPZ is bioactivated by a one-electron bioreduction, primarily by NADPH:cytochrome P450 reductase, to a highly DNA-reactive radical (Lloyd et al., 1991; Fitzsimmons et al., 1994; Patterson et al., 1995; Anderson et al., 2003) and has 50- to 200-fold hypoxic selectivity in cell suspension cultures (Zeman et al., 1986). Currently, TPZ is undergoing several phase I to III clinical trials in both single and combination therapies for lung and cervical cancers, as well as head and neck cancers. The gene-directed enzyme prodrug therapy approach has also been used to encode NADPH:CYP450 reductase within the hypoxic tumor cells to achieve enhanced TPZ activation (Cowen et al., 2004).

Bioactivation of hypoxia-activated prodrugs tirapazamine and PR-104.
2-((2-Bromoethyl)-2-{[(2-hydroxyethyl)amino]carbonyl}-4,6-dinitroanilino)ethyl methanesulfonate phosphate ester (PR-104) is another fairly recent example of a hypoxia-selective anticancer prodrug (Fig. 9). It is a bifunctional prodrug, because it has a phosphate ester promoiety to improve its aqueous solubility and nitroaromatic group as a hypoxia-selective promoiety. After phosphatase-catalyzed hydroxylation of PR-104 to PR-104A, the dinitrobenzamide mustard is selectively bioreduced to the corresponding hydroxylamine (PR-104H) and amine (PR-104M), which are DNA-crosslinking species able to diffuse to a limited extent into extracellular environment and kill also neighboring cells (Hicks et al., 2007). One-electron bioreduction is catalyzed mainly by cytochrome NADPH:cytochrome P450 reductase (Guise et al., 2007), although in certain tumor cell lines, PR-104 is also known to be bioactivated by aldo-keto reductase 1C3 in the presence of oxygen (Guise et al., 2010). PR-104 is currently undergoing several phase I and II clinical trials in both single and combination therapies for solid tumors, lung cancer, and leukemia.
The widely used proton pump inhibitors (PPIs), such as omeprazole, represent examples of prodrugs that are site-selectively bioactivated to their active species in a unique pH environment. To protect these acid-labile prodrugs from rapid destruction within the gastric lumen, PPIs are formulated for delayed release as acid-resistant, enteric-coated tablets. After passing through the stomach into the alkaline intestinal lumen, the enteric coatings dissolve, and the prodrug is absorbed (Missaghi et al., 2010). Because omeprazole is a basic molecule (pKa of 3.97), it accumulates in the acidic secretory parietal cells after protonation of its pyridyl group (Sachs et al., 1995). Omeprazole is then converted to its active sulfonamide in the acidic conditions of parietal cells, which then binds irreversibly to a cysteine group in the gastric H+/K+ ATPase (Fig. 10). This subsequently prevents the parietal cells from producing and secreting gastric acid (Shin et al., 2004). The favorable safety profile of omeprazole exists because nongastric H+/K+ ATPases lack the highly acidic compartment that is present in the parietal cells and are thus unable to convert omeprazole into its active form. Other PPIs are bioactivated by a similar mechanism as omeprazole.

Bioactivation of omeprazole in acidic conditions.
F. Managing the Life Cycle
If the prodrug structure of an existing drug has not been described previously in the literature, it can be considered a new chemical entity and therefore receive intellectual property protection against unauthorized competition during its development and commercialization. Because the development of a prodrug can be less expensive and faster than optimizing the therapeutic activity and ADME properties of a novel drug candidate at the same time, prodrugs can offer a valuable opportunity to improve the life-cycle management of the parent drug. Fosamprenavir, the water-soluble prodrug developed by GlaxoSmithKline (see section III.A), represents an excellent example of a prodrug with improved life-cycle management (Fig. 11). This phosphate ester of the HIV protease inhibitor amprenavir was originally designed to improve solubility and oral bioavailability (Chapman et al., 2004; Furfine et al., 2004; Wire et al., 2006). However, at the same time, this prodrug will continue the life cycle of amprenavir, because the fosamprenavir patent will last until 2017, whereas the patent on amprenavir will expire in 2013. Therefore, it is very likely that the additional costs that were consumed in the development of fosamprenavir will be fully covered by its extended patent duration (Stella et al., 2007).

Bioactivation of a phosphate prodrug of amprenavir.
IV. Preclinical and Clinical Considerations of Prodrug Bioconversion
Prodrug strategies have arguably been successful for a number of clinically used therapeutic agents. However, prodrug research encounters various challenges and additional work in preclinical and clinical settings, much of which can be attributed to understanding the bioconversion mechanisms of prodrugs. Many enzymes involved in prodrug activation are subject to interindividual variabilities in their activities. The main factors contributing to this variability are intrinsic, especially polymorphisms in the genes encoding the enzymes, but can also be extrinsic (i.e., interactions caused by other drugs and xenobiotics). Both intrinsic and extrinsic factors may cause insufficient or excessive conversion of the prodrugs into their active forms. Moreover, interspecies differences in enzyme activation represent another hurdle to the prediction of human disposition of certain prodrugs. This section focuses on esterases and P450 enzymes, which are particularly widely exploited enzymes in prodrug research.
A. Esterase-Catalyzed Hydrolysis of Prodrugs
The most common approaches for prodrug design are aimed at prodrugs undergoing metabolic bioconversion to the active parent drug by functionally prominent and diversity-tolerant hydrolases such as peptidases, phosphatases, and (in particular) carboxylesterases (Beaumont et al., 2003). Because they are ubiquitously distributed, the potential for carboxylesterases to become saturated or the potential for their substrates to become involved in drug-drug interactions is generally considered to be negligible (Liederer and Borchardt, 2006) although not impossible (Bellott et al., 2008).
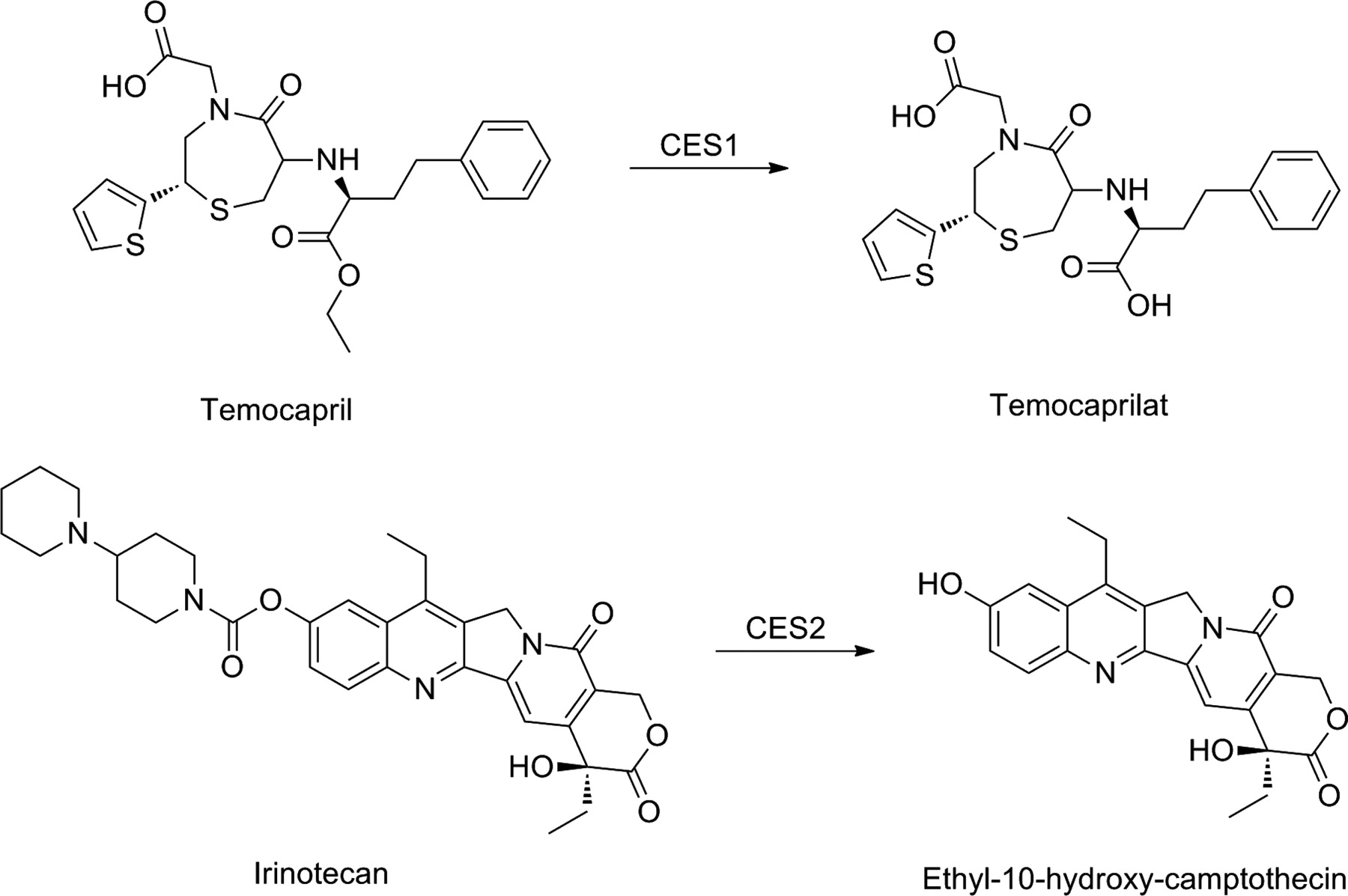
The majority of carboxylesterases can be found in two isozyme families, CES1 and CES2, which are characterized by their substrate specificities, tissue distribution, and gene regulation. In humans, CES1 is highly expressed in the liver, but can be also detected in other tissues, with the notable exception of the intestine. Human CES2, however, is highly expressed throughout the intestine and kidney, although at much lower levels than CES1 in the liver (Satoh and Hosokawa, 2006). The differences in both localization of CES1 and CES2 in humans and their substrate specificities can be taken into account in prodrug design. In general, CES1 prefers the hydrolysis of esters with large acyl and small alcohol groups, whereas CES2 hydrolyzes esters of smaller acyl and larger alcohol groups (Satoh et al., 2002). Therefore, CES1 can hydrolyze, for example, temocapril, with its small alcohol group and large acyl group, with a higher efficiency than CES2, which, on the other hand, is the enzyme almost exclusively hydrolyzing drugs such as irinotecan and heroin, with their bulky alcohol groups (Fig. 12).
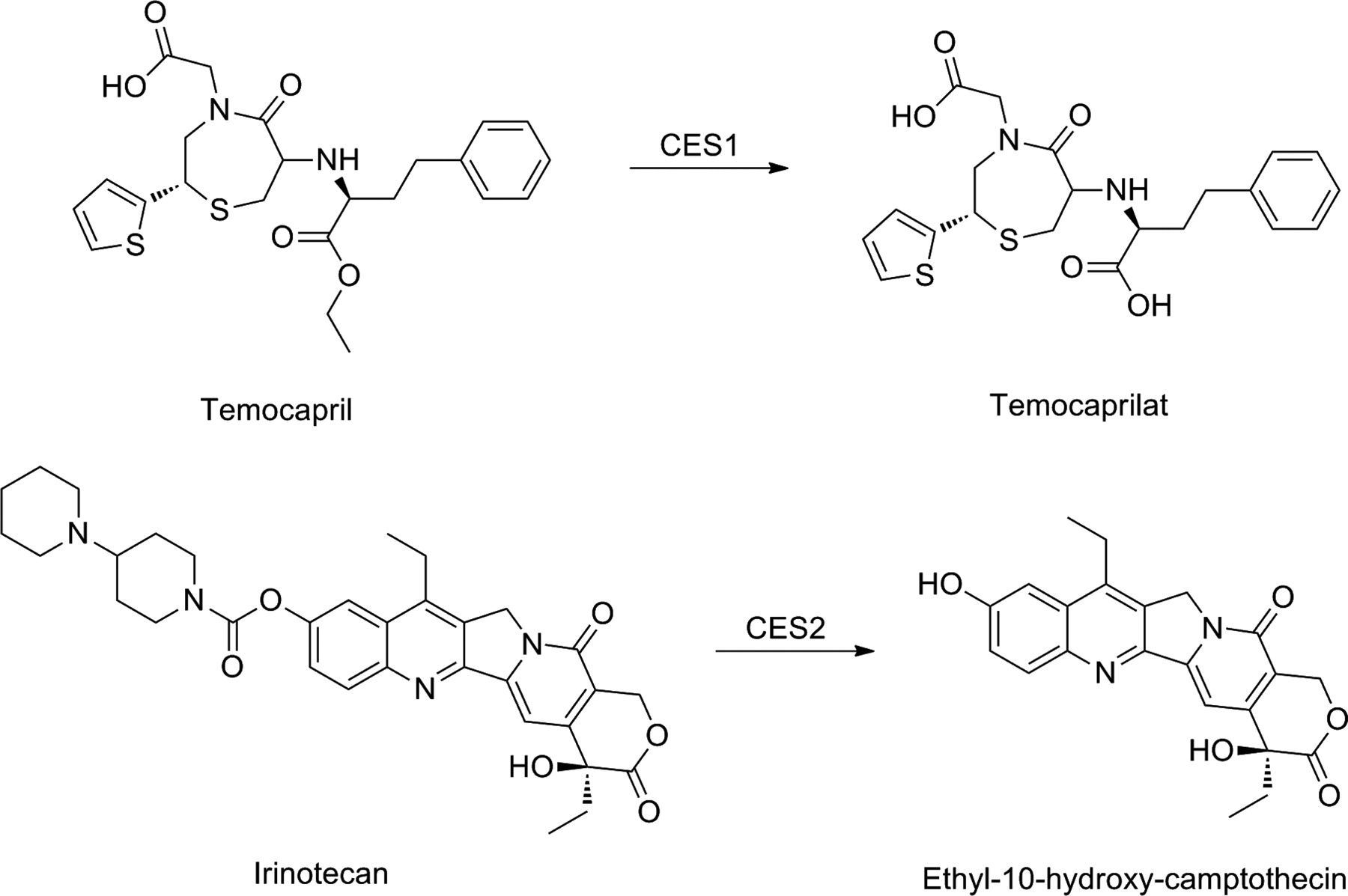
Bioactivation of ester prodrugs temocapril and irinotecan.
Although being an attractive target for various prodrugs, interspecies differences in esterase expression make it difficult to translate preclinical data in humans. For example, there is minimal hydrolase activity in small intestine of dog, but rodent species frequently have higher hydrolase activity in plasma than humans (Imai, 2006). Moreover, screening for the effects of hydrolase activated prodrugs in colon carcinoma tissue (e.g., Caco-2 monolayers) may be somewhat misleading due to differences between carboxylesterase expression in colon carcinoma and the adjacent normal tissues (Imai et al., 2005). Using human recombinant enzymes or human tissue extracts, however, can reduce the interspecies variability and help to indentify tissue specific hydrolysis.
The less than complete absorption observed with several hydrolase-activated prodrugs of penicillins, cephalosporins, and angiotensin-converting enzyme inhibitors highlights yet another challenge with prodrugs susceptible to esterase hydrolysis. These prodrugs typically have bioavailabilities of around 50% because of their premature hydrolysis during the absorption process in the enterocytes of the gastrointestinal tract (Beaumont et al., 2003). Hydrolysis inside the enterocytes releases the active parent drug, which in most cases is more polar and less permeable than the prodrug, and which is more likely to be effluxed by passive and carrier-mediated processes back into the lumen than to proceed into blood, therefore limiting oral bioavailability.
Finally, although more studies are needed to understand whether polymorphisms of CES genes can have a major impact on the clinical variability of certain prodrugs, scattered evidence from several reports indicates that CES variants can indeed cause variable systemic exposure; this has been observed with prodrugs such as angiotensin-converting enzyme inhibitors, as well as capecitabine and irinotecan (Bai et al., 2010).
B. Prodrug Bioactivation by Cytochrome P450 Enzymes
The P450 enzymes are a superfamily enzymes accounting for up to 75% of all enzymatic metabolism of drugs, including several prodrugs. There is accumulating evidence that genetic polymorphisms of prodrug-activating P450s contribute substantially to the variability in prodrug activation and thus to efficacy and safety of drugs using this bioactivation pathway (Gonzalez and Tukey, 2006; Testa and Kramer, 2007). Several drug-metabolizing P450 genes are known to be polymorphic, including CYP2A6, CYP2B6, CYP2C9, CYP2C19, and CYP2D6. In particular, the CYP2D6 gene is highly polymorphic, with 75 different major alleles currently known. Some of these alleles are associated with reduced enzyme activity or even the absence of enzyme function (Krämer and Testa, 2008). On the basis of the combination of CYP2D6 alleles, persons can be categorized into three main genotypes: poor metabolizers (PMs), extensive metabolizers (EMs), and ultrarapid metabolizers (Table 3). All of the variant P540 alleles are described by the Human Cytochrome P540 Allele Nomenclature Committee (http://www.cypalleles.ki.se/).
Simplified genotype-phenotype relationship for CYP2D6
On the other hand, there are numerous cases of drug interactions causing altered activation of prodrugs with deleterious clinical consequences. A drug interaction occurs when the effects of a drug are markedly altered as a result of coadministration of another drug. Drug interactions may be pharmacokinetic, pharmacodynamic, or a combination of the two. Pharmacokinetic interactions result in increased or decreased delivery of drugs to the sites of action. Metabolic drug interactions may occur during the phase I or II biotransformation reactions. They are mainly based on inhibition or induction of the metabolic enzymes. Drug metabolism may be considered in four different outcomes: 1) an active parent substrate forms an inactive metabolite, 2) an active substrate forms an active metabolite, 3) an inactive substrate (prodrug) forms an active compound, or 4) a parent compound is transformed into toxic metabolites. P450s are the main enzymes in drug metabolism and thus the main mediators of metabolic drug interactions.
This section will describe several examples in which genetic polymorphisms of P450 enzymes and drug interactions have been shown to influence the systemic exposure and response to prodrugs. Certain anticancer drugs, drugs for cardiovascular diseases, and opioid analgesics are highlighted.
1. Anticancer Drugs.
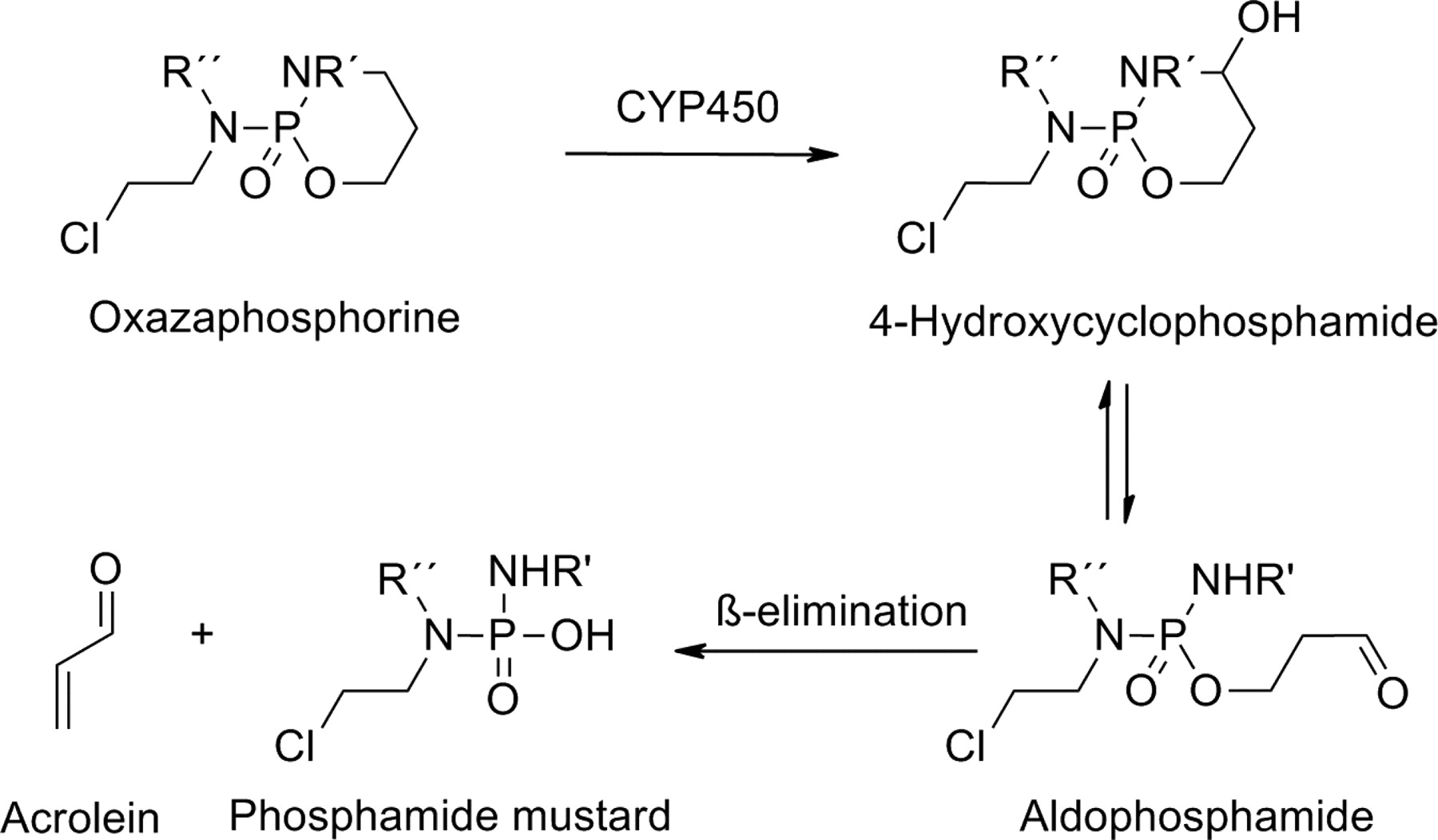
Oxazaphosphorines, including cyclophosphamide, ifosfamide, and trofosfamide, are prodrugs used for treating different types of cancers and autoimmune diseases. The bioconversion of these cyclic phosphate drugs to the corresponding cytotoxic species is initiated by P450 enzymes. Many different P450 enzymes participate in the hydroxylation of a carbon-4 of the oxazaphosphorine ring. The unstable intermediates formed decompose to the corresponding aldehydes, which are degraded further to active nitrogen mustard derivatives (Fig. 13).
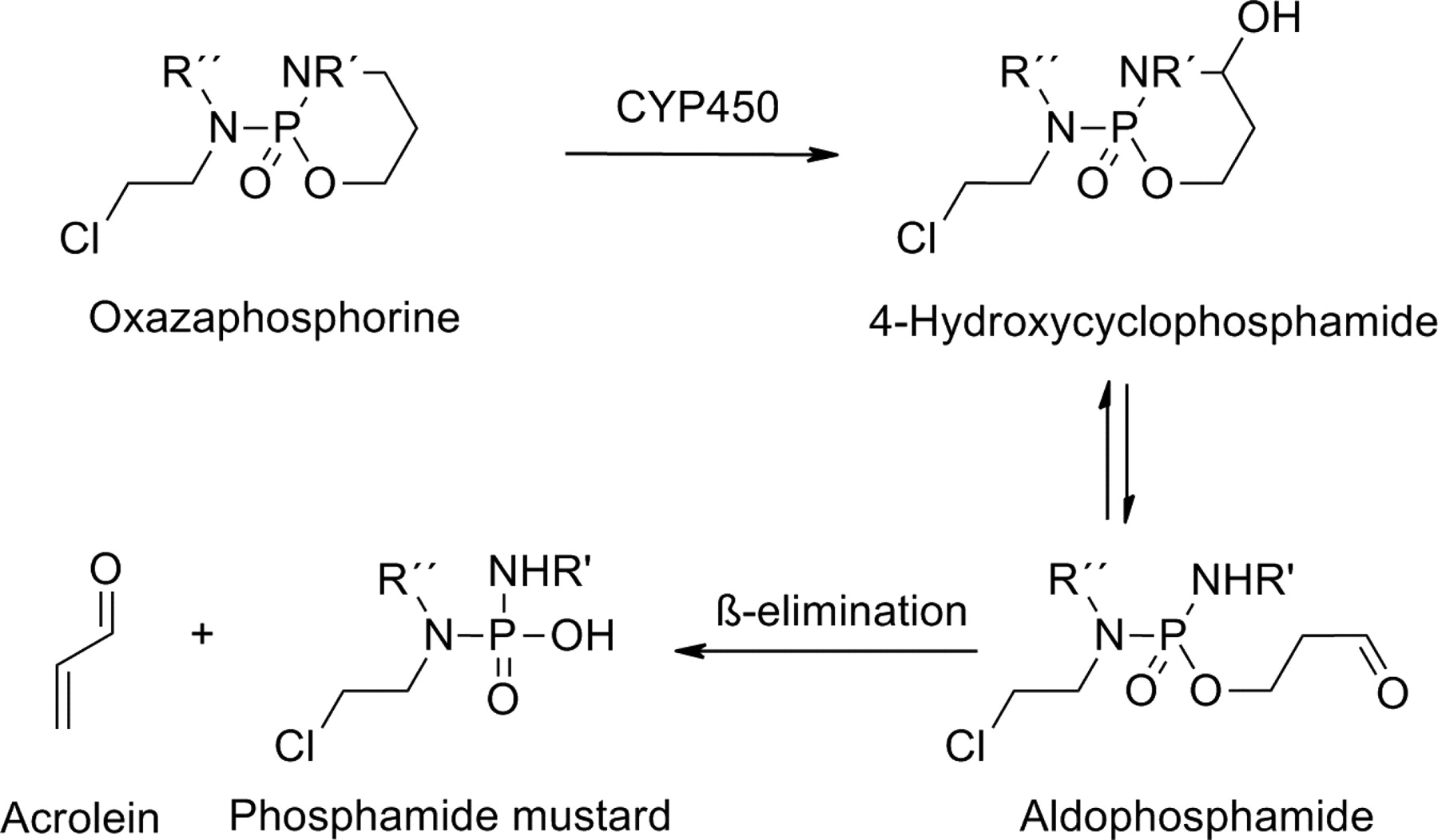
Bioactivation of oxazaphosphorine prodrugs.
Several P450 enzymes, including CYP2B6, CYP3A4, and CYP2C19, have been implicated in the activation of cyclophosphamide to its active metabolites 4-hydroxylcyclophosphamide and aldophosphamide. The use of oxazaphosphorines can be associated with severe toxicity, because their metabolism leads to the formation of the highly reactive metabolite acrolein, which is responsible for urotoxicity, neurotoxicity, and nephrotoxicity (Huttunen et al., 2008; Giraud et al., 2010). CYP2B6 and CYP2C19 are highly polymorphic, such that some of their variant alleles have reduced or no activity compared with the wild-type allele. These and other genetic factors are thought to play a role in the interindividual variation in both response and toxicity associated with cyclophosphamide-based therapies (Pinto et al., 2009).
Tegafur is a prodrug has been used clinically for almost 30 years in the treatment of several common tumors. Tegafur is activated by P450 enzymes, via hydroxylation, to 5-fluorouracil (5-FU) (Fig. 14). 5-FU inhibits thymidylate synthase and is incorporated into DNA and/or RNA. The NADPH-dependent P450-catalyzed hydroxylation of the furan ring produces an intermediate that spontaneously decomposes to the active 5-FU and the succinic aldehyde moiety. CYP2A6 is the principal enzyme responsible for the activation of tegafur in human liver microsomes, but CYP1A2, CYP2C8, and thymidine phosphorylase also contribute to this process (Huttunen et al., 2008). Several clinical studies have indicated that systemic exposure to tegafur is increased in patients with CYP2A6 PM genotype, caused either by deletion of the CYP2A6 gene or the presence of inactive alleles, leading to reduced enzyme activity (Bai et al., 2010).
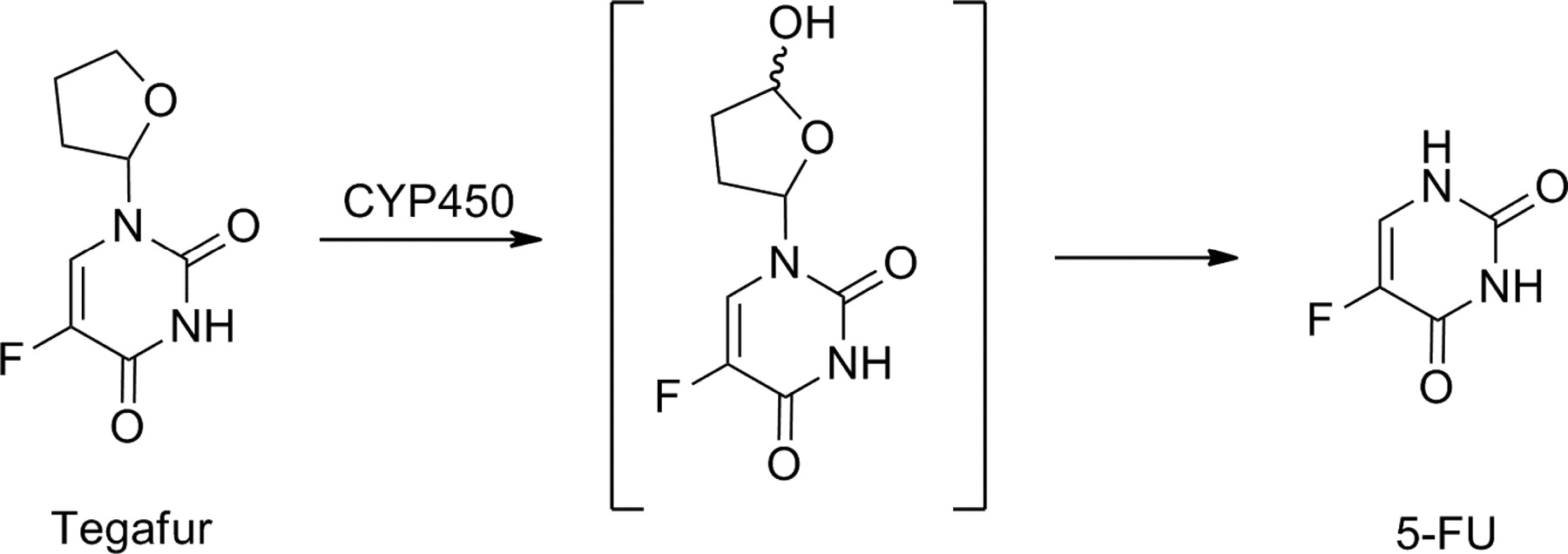
Bioactivation of tegafur.
Tamoxifen is a synthetic antiestrogen widely used for breast cancer therapy and even as a prophylactic agent in healthy women who carry a high risk of contracting breast cancer. Treatment with tamoxifen can reduce the incidence of breast cancer in these high-risk women by almost half. It also reduces by almost 30% the annual risk of death when administered after surgery for invasive breast cancer. In vitro studies have demonstrated that tamoxifen itself has very little affinity for its biological target, the estrogen receptor in breast tissue (Jordan, 2007). As shown in Fig. 15, to exert its antiestrogenic effects in breast, tamoxifen must first be metabolized in the liver by P450 enzymes to two active metabolites, 4-hydroxytamoxifen and 4-hydroxy-N-desmethyltamoxifen (endoxifen). Endoxifen is present at concentrations up to 20-fold higher than 4-hydroxytamoxifen and is pharmacologically distinct from either tamoxifen or 4-hydroxytamoxifen.

Bioactivation of tamoxifen.
Several different P450 forms may be involved in catalyzing the N-demethylation of tamoxifen. The CYP2D6 enzyme is responsible for the oxidation of the most abundant tamoxifen metabolite, N-desmethyltamoxifen, to endoxifen (Jordan, 2007; Huttunen et al., 2008; Sideras et al., 2010). Considerable mechanistic, pharmacologic, and clinical pharmacogenetic evidence supports the concept that CYP2D6 genetic variants and phenocopying effects through drug interactions with CYP2D6 inhibitors can influence plasma concentrations of active tamoxifen metabolites and negatively affect the therapeutic outcome. Women with genetically normal CYP2D6 function have been shown to benefit more from tamoxifen therapy than women who have deficient activity (Brauch and Jordan, 2009). On the basis of these findings, the United States Food and Drug Administration (FDA) Advisory Committee for Pharmaceutical Science in 2006 recommended a label change to include information that CYP2D6 is an important pathway in the formation of endoxifen and that postmenopausal women with estrogen receptor-positive breast cancer who are CYP2D6 PMs, either by genotype or because of drug interactions, are at increased risk for breast cancer recurrence (United States Food and Drug Administration, 2006).
Drugs that inhibit CYP2D6 activity can also affect the concentrations of active tamoxifen metabolites. For example, tamoxifen-treated extensive metabolizers who are coprescribed potent CYP2D6 inhibitors such as paroxetine or fluoxetine displayed endoxifen concentrations similar to patients with the PM CYP2D6 genotype. Several other drugs are known to inhibit CYP2D6 and could in theory lead to insufficient activation of tamoxifen. However, prospective clinical studies addressing this issue thus far are lacking. A recent review recommended that potent CYP2D6 inhibitors should be avoided in women receiving tamoxifen (Sideras et al., 2010).
2. Cardiovascular Drugs.
Losartan is an angiotensin II type 1 receptor antagonist used as for the treatment of hypertension and other cardiovascular diseases. After oral administration, approximately 14% of the losartan dose is converted to the pharmacologically active metabolite called EXP3174 [2-n-butyl-4-chloro-1-((2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl)imidazole-5-carboxylic acid] (Fig. 16). EXP3174 is 10- to 40-fold more potent than losartan and has a longer duration of action. The major metabolic pathway for losartan is through CYP2C9 with some contribution from CYP3A4 (Sica et al., 2005). In patients with the CYP2C9 PM genotype, the metabolism of losartan to EXP3174 and its hypotensive effect are significantly reduced (Sekino et al., 2003).

Bioactivation of losartan.
In healthy volunteers, concomitant use of the CYP2C9 inhibitors fluconazole and bucolone has been shown to prevent the formation of the active metabolite in healthy volunteers (Kaukonen et al., 1998; Kobayashi et al., 2008). A pharmacoepidemiologic study (Tirkkonen and Laine, 2004) indicated that in hospitalized patients, losartan is often used with drugs that inhibit CYP2C9 (19% of all losartan treatment periods). In an attempt to overcome these problems, a novel prodrug of the active metabolite EXP3174 was developed (EXP3174-pivoxil). Initial pharmacokinetic characterization showed that it could be administered at a lower dose than losartan, and it also achieved a more rapid and more consistent therapeutic response compared with that of losartan (Yan et al., 2010).
Statins are HMG-CoA reductase inhibitors used in treatment of dyslipidemias. They are effective in both the primary and secondary prevention of atherosclerotic heart disease. These drugs lower the low-density lipoprotein (LDL) cholesterol concentrations in plasma by blocking cholesterol biosynthesis in the liver, which leads to an increase in the LDL clearance and an up-regulation of LDL receptors. They also decrease very-low-density lipoprotein cholesterol and triglyceride concentrations and increase high-density lipoprotein cholesterol levels slightly. In particular, the reduction in plasma levels of LDL cholesterol diminishes the risk for major cardiovascular events (Delahoy et al., 2009).
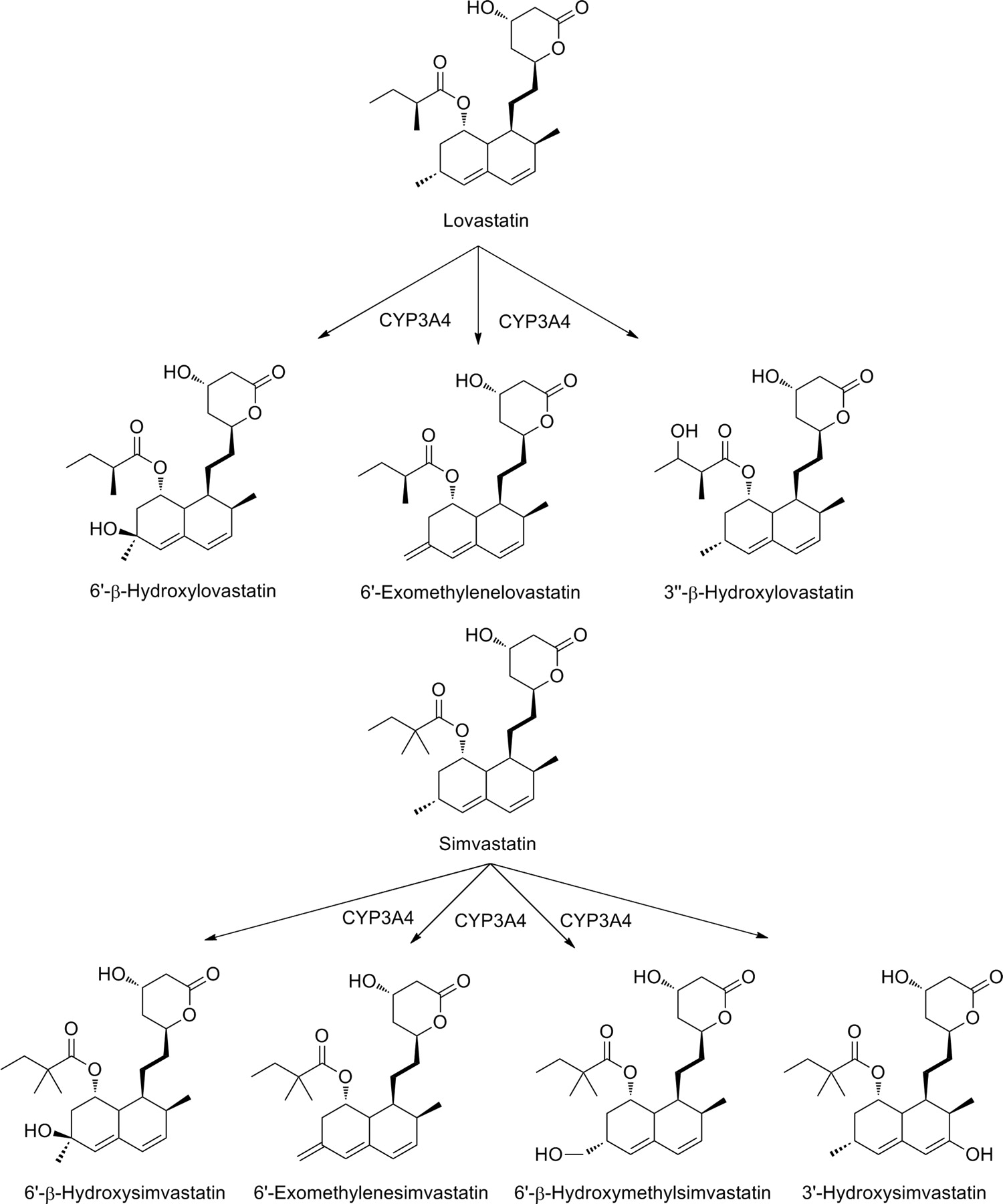
Because lovastatin and simvastatin are lactone prodrug forms, they are less active than the respective β-hydroxy acid metabolites (Fig. 17). Both parent drugs undergo extensive first-pass metabolism. Lovastatin is oxidized mainly by CYP3A4 to three known primary metabolites: 6′β-hydroxylovastatin, 6′-exomethylene metabolite, and 3″β-hydroxylovastatin. All these metabolites are pharmacologically active. It is probable that the first two compounds derive from a single metabolic intermediate. Simvastatin is metabolized to at least four primary metabolites: 6′β-hydroxysimvastatin, 6′-exomethylene metabolite (which may derive from a common metabolic precursor), 6′β-hydroxymethyl metabolite, and 3′-hydroxysimvastatin (Fig. 17) (Neuvonen et al., 2008).

Bioactivation of lovastatin and simvastatin.
CYP3A4 inhibitors (e.g., clarithromycin, erythromycin, telithromycin, itraconazole, ketoconazole, diltiazem, and verapamil) have been shown to increase the exposure and toxicity of lovastatin and simvastatin. The increase in plasma concentrations can be up to 20-fold. Because they have such marked effects on the statin plasma concentration, these kinds of drug interactions increase the incidence of skeletal muscle toxicity, an adverse drug reaction concerning the entire class of statins, as well as the risk of potentially fatal rhabdomyolysis. On the other hand, the potent CYP3A4 inducers rifampicin and carbamazepine have been shown to reduce simvastatin concentrations (Neuvonen, 2010).
A few pharmacoepidemiologic studies have attempted to assess the clinical consequences of interactions between lovastatin or simvastatin and CYP3A4 inhibitors or inducers. The risk of developing myopathy during concomitant simvastatin and CYP3A4 inhibitor treatment was increased 9.1-fold over a low baseline incidence of 0.025% (Gruer et al., 1999). A later study (Tirkkonen et al., 2008) showed that 8.8 and 3.9% of hospital inpatients had received lovastatin or simvastatin concomitantly with a CYP3A4 inhibitor or inducer drug, respectively. However, no major effect was observed on serum lipid concentrations or creatinine kinase activity. This lack of clinical effect is explained at least in part by the low doses of lovastatin and simvastatin taken by the patients in this study. It thus seems that lovastatin and simvastatin can be used rather safely with CYP3A4 inhibitors if the statin doses are low and the patients are monitored carefully.
Clopidogrel is an antithrombotic drug belonging to the thienopyridine group. The addition of clopidogrel to aspirin achieved significant reductions in major cardiovascular events in patients with acute coronary syndrome in general and particularly among those treated invasively by percutaneous coronary intervention. A barrier to clopidogrel absorption in the duodenum is its active intestinal transport by the P-glycoprotein efflux pump (ABCB1). Upon absorption, approximately 85% of the dose is hydrolyzed by esterases to an inactive carboxylic acid derivative. Multiple P450 isoforms metabolize the thiophene ring of the remaining 15% to produce the intermediate 2-oxoclopidogrel, which is then further metabolized to the pharmacologically active metabolite (Fig. 18). Upon release into the systemic circulation, the active metabolite's thiol group forms an irreversible disulfide bond with the platelet ADP P2Y12 receptor (P2RY12), altering its shape to prevent ADP-mediated platelet activation. Consequently, activation of the glycoprotein IIb/IIIa receptor, which binds fibrinogen and initiates platelet aggregation, is inhibited. Although multiple P450 isoforms can catalyze the conversion of clopidogrel to its active metabolite in vitro, CYP2C19 seems to be the primary P450 enzyme responsible for the bioactivation of clopidogrel in humans (Wallentin, 2009; Farid et al., 2010).

Bioactivation of clopidogrel.
Numerous genes are involved in clopidogrel pharmacokinetics and pharmacodynamics, and the potential contribution of functionally relevant polymorphisms in these genes to the interindividual variability in clopidogrel response has been evaluated in a series of recent studies. Several studies have reported that, compared with wild type, CYP2C19 PM allele carriers exhibit a significantly lower capacity to metabolize clopidogrel into its active metabolite and inhibit platelet activation. Therefore, the poor metabolizers are at significantly higher risk of suffering adverse cardiovascular events, such as myocardial infraction, stent thrombosis, and death. Many studies have demonstrated a consistent association between CYP2C19 genotype, clopidogrel pharmacokinetics, and clopidogrel pharmacodynamics in multiple populations, suggesting that the CYP2C19 genotype may be an important predictor of clinical response to clopidogrel (Ellis et al., 2009).
A recent meta-analysis revealed that the hazard is particularly increased among patients undergoing percutaneous coronary intervention for acute coronary syndrome and who carry a CYP2C19 PM allele (Mega et al., 2010). However, confirmation of this finding in larger trials powered to evaluate clinical outcome, which will ultimately be necessary. In March 2010, the FDA approved a new label for clopidogrel with a “boxed warning” describing the diminished effectiveness of the standard drug dosing in persons with impaired metabolic function based on their CYP2C19 genotype. The warning also notes that tests are available to identify patients with genetic polymorphisms and that alternative treatment strategies, either higher clopidogrel dosing regimens or the use of other antiplatelet agents, should be considered in those patients (Holmes et al., 2010).
CYP2C19 is responsible for the metabolism and clearance of many different drugs, including phenytoin, diazepam, proguanil, citalopram, venlafaxine, cyclophosphamide, and the proton pump inhibitors. Because of the increased risk for gastrointestinal tract bleeding associated with concomitant aspirin and clopidogrel antiplatelet therapy, PPIs are often administered to decrease risk of bleeding. Several recent studies have indicated that concomitant PPI therapy could be a possible contributor to clopidogrel nonresponsiveness, presumably via inhibition of the CYP2C19-mediated conversion of clopidogrel to its active metabolite. In addition to being well characterized substrates for CYP2C19, the PPIs are also potent competitive inhibitors of CYP2C19 metabolic activity in vitro (lansoprazole > omeprazole = esomeprazole > rabeprazole > pantoprazole), with pantoprazole having the least potent CYP2C19 inhibitory capacity (Ellis et al., 2009).
In May 2009, the European Medicines Agency issued a public statement on the putative interaction between clopidogrel and PPIs (Wathion, 2009). The Agency recommended that the product information for all clopidogrel-containing medicines should be amended to discourage concomitant use of PPIs and clopidogrel-containing medicines unless necessary. Furthermore, the Agency recommended that more information is needed in relation to the inhibition of clopidogrel metabolism by other medicines and in relation to the implications of CYP2C19 genetic variation.
Very recent data (Bouman et al., 2011) obtained with in vitro liver microsomal assays confirmed the participation of multiple P450 forms (but curiously not CYP2C19) in the first step of clopidogrel activation. In addition, there was evidence that paraoxonase 1, an esterase associated with high-density lipoprotein concentrations in the blood, may be the rate-limiting enzyme for the second step of hydrolytic cleavage of the 2-oxoclopidogrel ring to the thiol active metabolite. Thus, the relative contribution of paraoxonase 1-mediated and oxidative P450-dependent ring cleavage needs to be elucidated before mechanistic interpretation to the epidemiological findings can be given.
3. Opioid Analgesics.
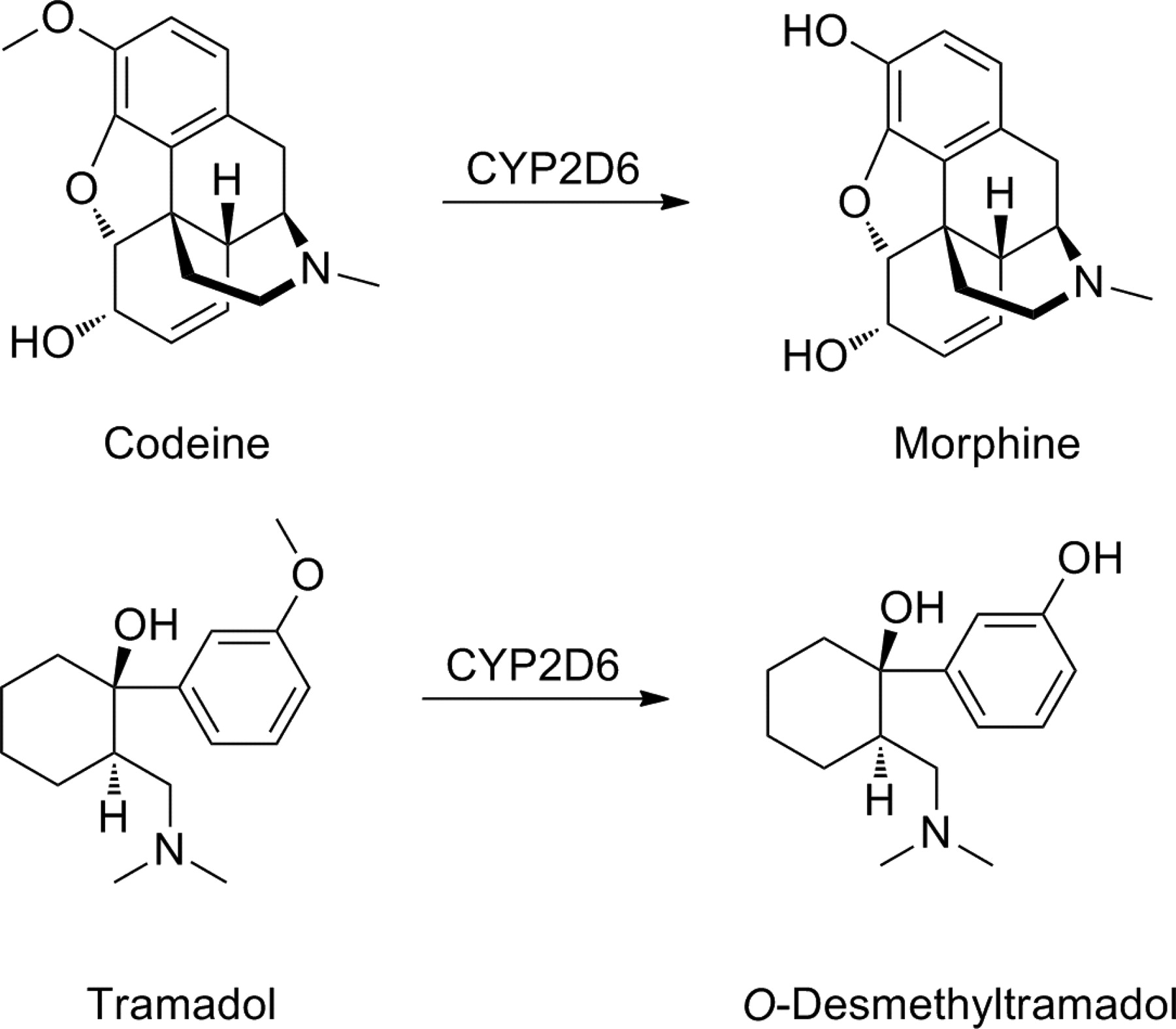
Opiates are the cornerstone of the management of cancer pain and postoperative pain and are used increasingly for the management of chronic noncancer pain. Codeine and tramadol are weak opioid analgesics, and they are both prodrugs. It has been long known that codeine (methylmorphine) is converted into morphine by CYP2D6 (Fig. 19). The O-demethylation is essential for analgesia, because codeine itself has very low affinity for the opioid receptors. The CYP2D6 inhibitor quinidine has been shown to minimize the analgesic effect but also to reduce codeine abuse. In addition, the threshold of experimental pain by codeine is increased in CYP2D6 EMs but not in PMs, who lack the activation machinery. However, evidence for impaired analgesic outcome in CYP2D6 PMs is sparse, and it has been postulated that large-scale studies will be needed to demonstrate genetically determined unresponsiveness to codeine analgesia (Smith, 2009; Stamer et al., 2010).
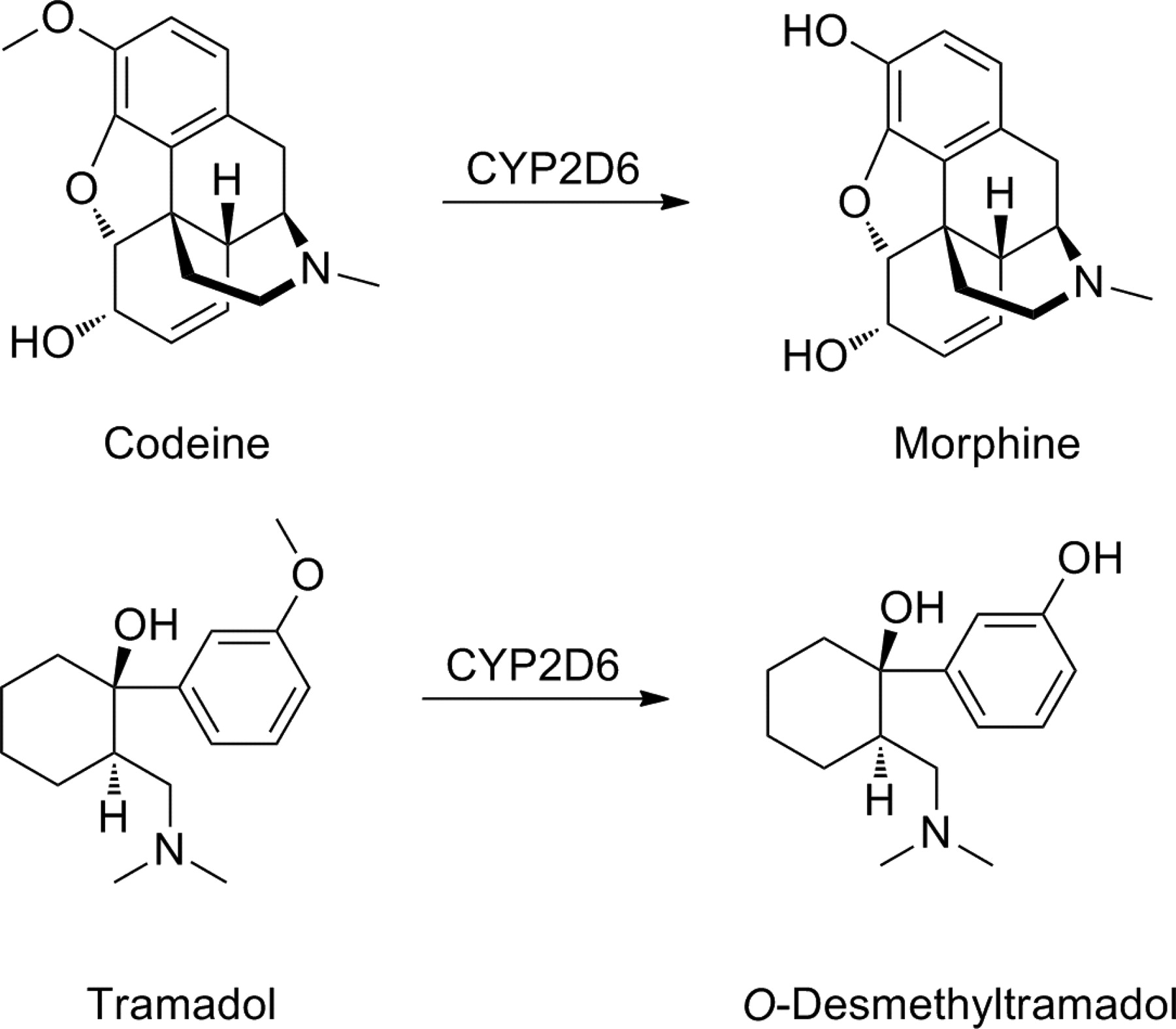
Bioactivation of codeine and tramadol.
On the other hand, subjects carrying a gene duplication or multiduplication may experience exaggerated pharmacologic effects in response to regular doses of codeine. Enhanced CYP2D6 activity in these genotypes can predispose to life-threatening opioid intoxication. Several recent case reports of codeine fatalities highlighted that the use of this weak opioid, particularly in young children, could be attributed to their metabolic profile, especially in subjects displaying the ultrarapid metabolizer genotype (Gasche et al., 2004; Voronov et al., 2007; Ciszkowski et al., 2009).
Tramadol is a codeine analog with weak μ-opioid receptor affinity. It is used as a racemic mixture. The (+)-enantiomer binds to the μ-receptor but also increases serotonin activity; the (−)-enantiomer stimulates α2-adrenergic receptors and inhibits noradrenaline reuptake. Thus, it is believed that at least part of the analgesic effect of tramadol is due to its ability to increase noradrenaline and serotonin activity. Like codeine, tramadol requires CYP2D6-mediated metabolism to an active metabolite, O-desmethyltramadol (M1), to be fully effective (Fig. 19) (Smith, 2009). Studies in healthy subjects have demonstrated that serum concentrations of M1 are lower in PMs than EMs. Thus, PMs lacking CYP2D6 activity demanded and received more tramadol doses via patient-controlled analgesia compared with patients with normal catalytic CYP2D6 activity. Prospective clinical studies have demonstrated that the analgesic effect of tramadol is linked to CYP2D6 genotype. In one study, the proportion of tramadol nonresponders was increased in the CYP2D6 PM group compared with the patients with functionally active CYP2D6 alleles. Thus, there is evidence that CYP2D6 polymorphisms can influence the efficacy of codeine and tramadol analgesia, although in most trials, only a limited number of patients have been monitored (Stamer et al., 2010).
V. Conclusions
Biodegradable prodrugs can offer an attractive alternative to improve undesirable ADMET properties of a wide variety of drugs without losing the benefits of the drug molecule. Because of its versatility, the prodrug approach has enhanced the clinical usefulness of many pharmacological agents in the past, and as many as 10% of all approved small molecular drugs on the market today can be classified as prodrugs. However, the design of prodrugs should be considered at very early stages of the drug research and development process, because changing the ADMET properties may expose other undesired properties of the drug candidates. Bioconversion of prodrugs is perhaps the most vulnerable link in the chain, because there are a great many intrinsic and extrinsic factors that can influence the bioconversion mechanisms. For example, the activity of many of prodrug activating enzymes may be decreased or increased due to genetic polymorphisms, age-related physiological changes, or drug interactions, leading to adverse pharmacokinetic, pharmacodynamic, and clinical effects. In addition, there are wide interspecies variations in both the expression and function of the major enzyme systems activating prodrugs, and these can pose challenges in the preclinical optimization phase. Nonetheless, developing a prodrug can still be more feasible and faster strategy than searching for an entirely new therapeutically active agent with suitable ADMET properties.
Acknowledgments
This work was supported by the Academy of Finland [Grants 135439 (to K.M.H.), 132637 (to J.R.)]; and the Finnish Cultural Foundation (to K.M.H.).
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Huttunen, Raunio, and Rautio.
Footnotes
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.110.003459.
↵1 Abbreviations:
- ADME
- absorption, distribution, metabolism, and excretion
- ADMET absorption
- distribution, metabolism, excretion, and toxicity
- araC
- arabinofuranosyl cytidine (cytarabine)
- CES
- carboxylesterase
- DMAE
- 1-(2′,5′-dimethoxyphenyl)-2-aminoethanol
- EM
- extensive metabolizer
- EXP3174
- 2-n-butyl-4-chloro-1-((2′-(1H-tetrazol-5-yl)biphenyl-4-yl)methyl)imidazole-5-carboxylic acid
- 5-FU
- 5-fluorouracil
- FDA
- United States Food and Drug Administration
- LDL
- low-density lipoprotein
- MB07133
- 4-amino-1-(5-O-(2-oxo-4-(4-pyridyl)-1,3,2-dioxaphosphorinan-2-yl)arabinofuranosyl)-2(1H)-pyrimidinone
- P450
- cytochrome P450
- PM
- poor metabolizer
- PMEA
- 9-(2-phosphonomethoxyethyl)adenine
- PPI
- proton pump inhibitor
- PR-104
- 2-((2-bromoethyl)-2-{[(2-hydroxyethyl)amino]carbonyl}-4,6-dinitroanilino)ethyl methanesulfonate phosphate ester
- TPZ
- tirapazamine (SR 4233).
- © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}