


Abstract
The isolation of an opioid receptor-related clone soon led to the isolation and characterization of a new neuropeptide, termed orphanin FQ or nociceptin (OFQ/N). This heptadecapeptide binds to the NOP1 (previously termed ORL1) receptor with exceedingly high affinity, but does not interact directly with classical opioid receptors. Functionally, the actions of OFQ/N are diverse and intriguing. Most work has focused upon pain mechanisms, where OFQ/N has potent anti-analgesic actions supraspinally and analgesic actions spinally. Other OFQ/N activities are less clear. The diversity of responses might reflect NOP1 receptor heterogeneity, but this remains to be established. The actions of this neurochemical system may also be uniquely dependent on contextual factors, both genetic and environmental. This review will address the molecular biology and behavioral pharmacology of OFQ/N and its receptor.
I. Introduction
The use of molecular biological approaches has led to extraordinary advances in our understanding of opioid action in the last decade. Soon after the cloning of the δ opioid peptide receptor (DOP1, originally termed DOR-1) (Evans et al., 1992; Kieffer et al., 1992), a number of laboratories identified clones corresponding to the μ opioid peptide (MOP1, originally termed MOR-1) and κ opioid peptide (KOP1, originally termed KOR-1) receptors. A meeting held in Washington, DC, as a tribute to the memory of Dr. William Martin,2documented these advances in opioid receptor pharmacology (Uhl et al., 1994). At this meeting, several laboratories first described a fourth receptor clone closely homologous to the traditional opioid receptors (Table 1). These clones were isolated from a number of species and were typical G-protein-coupled receptors with the expected predicted seven transmembrane domains. These novel clones displayed approximately 50% identity with the traditional opioid receptors overall, with the transmembrane regions showing even higher homologies of up to 80%.
Initial reports of the cloning of the NOP1 receptor
Despite their close homology to the other opioid receptors, the novel clones were difficult to characterize and were considered by many to be an orphan receptor. Few opioids labeled these novel clones, and their affinities were markedly lower than those seen with the cloned opioid receptors. Functionally, the ability of opioids to modulate adenylate cyclase with these clones also was markedly limited (Mollereau et al., 1994; Pan et al., 1995). Several early papers uncovered a close relationship between this clone and the κ3receptor but concluded that they were not identical (Pan et al., 1994,1995). The evidence for a relationship between them came from several lines of investigation. A monoclonal antibody capable of neutralizing κ3 opioid binding in brain tissue and κ3 analgesia in vivo recognized the expressed receptor generated through in vitro translation in Western blot analysis. Furthermore, in antisense mapping studies a number of antisense probes directed against the second and third coding exons of the murine clone (KOR-3) blocked the analgesic activity of κ3 analgesic naloxone benzoylhydrazone in mice without affecting the analgesic actions of traditional μ, δ, and κ1 drugs. Yet, the inability of a range of antisense probes targeting the first coding exon and the markedly different binding profile of the new clone indicated that the novel clone was not identical to the κ3 receptor. The differences between the two were further documented with the identification of the endogenous ligand for this receptor, a heptadecapeptide termed orphanin FQ or nociceptin (OFQ/N3) (Fig.1). Despite an exceedingly high affinity for the cloned receptor, OFQ/N does not compete binding to the traditional μ opioid receptors.

OFQ/N and its related peptides. Sequences of the endogenous opioid peptides and ppOFQ/N peptides are presented. In some situations, sequences are species-dependent. The sequence of ppOFQ/N160–187 is from the mouse and nocistatin from human.
The nomenclature in this field is confusing, particularly for the receptor. In the early papers each laboratory used its own nomenclature for the receptor (Table 1), but as the years went by the term ORL1 gained favor. Recently, it has been suggested that all the opioid receptor family of receptors be renamed, with the term NOP1 (nociceptin/orphanin FQ peptide) receptor referring to the ORL1 clone in all species. Similarly, the ligand is known as nociceptin or orphanin FQ, having been isolated by two groups independently (Meunier et al., 1995; Reinscheid et al., 1995). Nociceptin was chosen by one group to denote its presumed pronociceptive activity. The term orphanin FQ refers to its affinity for the “orphan” opioid receptor, while the F and Q refer to the first and last amino acids, phenylalanine and glutamine. Neither term predominates and most laboratories use the two together, as is done in this review: orphanin FQ/nociceptin, or OFQ/N.
This field has burgeoned enormously since the cloning of the receptor and the identification of its peptides. Aspects of this field have been reviewed previously (Henderson and McKnight, 1997; Meunier, 1997;Civelli et al., 1998; Darland et al., 1998; Taylor and Dickenson, 1998;Zaki and Evans, 1998; Yamamoto et al., 1999; also see a special issue of the journal Peptides, volume 21, number 7, 2000). The current review will focus rather comprehensively upon the behavioral pharmacology of OFQ/N, with an attempt to understand it from the molecular perspective.
II. Molecular Biology of the Orphanin FQ/Nociceptin Receptor
A. Cloning NOP1 and Its Gene
NOP1 was cloned from various species by a number of laboratories at about the same time. NOP1 is a typical G-protein-coupled receptor with seven predicted transmembrane domains (Fig.2A) and is localized to murine chromosome 2 (Chen et al., 1994; Nishi et al., 1994). It was readily detected by Northern analysis, where a major band of 3.4 kb was detected in mice (Pan et al., 1995). Several laboratories found a similar band in rats at approximately 3.4 kb, as well as additional bands (Chen et al., 1994; Wang et al., 1994; Lachowicz et al., 1995). One group reported additional bands of 7.5 and 10 kb (Chen et al., 1994), another observed a single additional band of approximately 7.6 kb (Lachowicz et al., 1995), and a third group reported two additional bands of 13 and 23 kb (Wang et al., 1994). The significance of these additional larger bands is not clear, particularly with the differences noted among groups. However, the differing ratios of these larger bands to the 3.4-kb band among regions raising interesting questions regarding regional processing (Wang et al., 1994; Lachowicz et al., 1995).
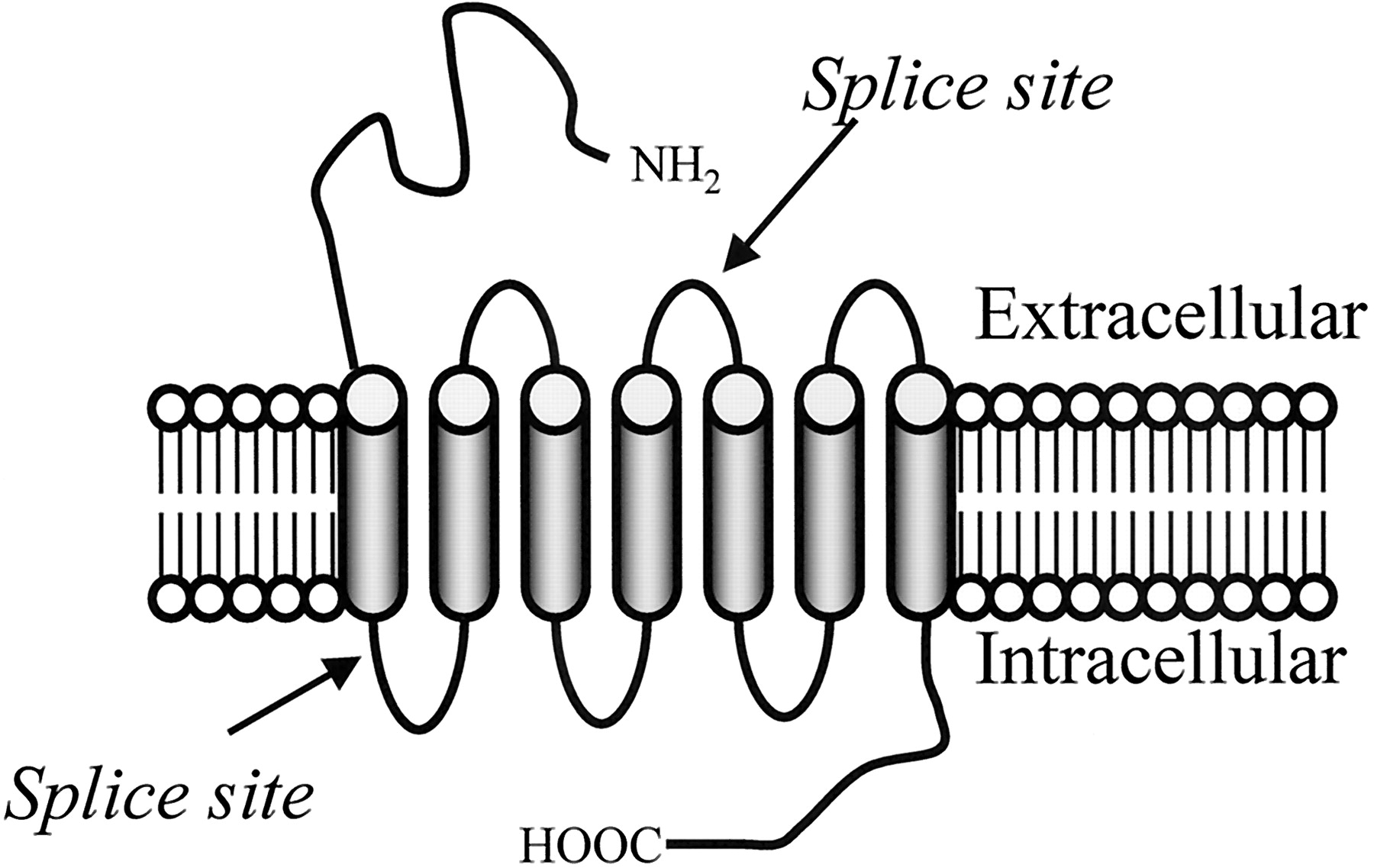
Schematic of the NOP1 receptor and its gene, Oprl1. A, schematic of the NOP1receptor is presented with the sites of the protein corresponding to the splice sites in the mRNA indicated. B, schematic of the structure of the murine Oprl1 gene.
Southern analysis from a number of groups implies a single copy of the gene for the NOP1 receptor, which is termedOprl1. The murine NOP1 gene structure was elucidated soon after the initial reports of the receptor (Pan et al., 1996b). The receptor has three coding exons, similar to the other opioid receptors. The first coding exon yields the amino terminus and the first transmembrane domain (Fig. 2A). The second coding exon is responsible for the next three transmembrane domains. The splice site between the second and third coding exons is located in the second extracellular loop, and the third coding exon is responsible for the remainder of the protein, including the last three transmembrane domains and the intracellular carboxyl tail (Fig. 2A). The binding pocket has been proposed to comprise several of the transmembrane domains (Topham et al., 1998; Mouledous et al., 2000). The initial gene structure identified five exons, with a noncoding exon preceding and following the three coding exons (Fig. 2B) (Pan et al., 1996b). More recent work has identified two mini-exons between the first and second coding exons that are alternatively spliced for a total of seven. The numbering of the exons has changed, as new ones have been uncovered. In the current review, exon 2 corresponds to the first coding exon of the original clone, exon 5 to the second coding exon and exon 6 to the last one.
B. Alternatively Splicing NOP1
Like other members of the opioid receptor family, NOP1 undergoes alternative splicing. The first variant, NOP1d, was identified in lymphocytes and contains a 15-bp deletion from the 3′ end of the first coding exon (exon 2) (Halford et al., 1995; Wick et al., 1995). Similar variants were obtained from mouse (Pan et al., 1998b), rat (Wick et al., 1994;Xie et al., 1999), and human (Peluso et al., 1998) brains. An intron retention variant, NOP1e, containing the intron between the second and third coding exons (exons 5 and 6) was reported in mice (Pan et al., 1998b), rats (Chen et al., 1994; Xie et al., 1999,2000), and human brain (Xie et al., 1999). The initial report in rats found an 84-bp insertion that could be translated through to generate a full-length receptor. The mouse version, however, had only 81 bp, and the presence of a stop codon prevented translation of the last three transmembrane domains (Pan et al., 1998a). A more recent report in rats found a similar 81-bp insertion with a stop codon (Xie et al., 1999). It is not clear which of the two rat variants predominates, but this is an important issue since one has the potential of being a functional variant whereas the other does not.
An additional three NOP1 variants have been described that contain mini-exons located between the first and second coding exons (exons 2 and 5) (Fig. 2B) (Xie et al., 1999). NOP1a contains a 34-bp insertion (exon 3) between the first two coding exons. Due to a frameshift, it gives a predicted stop codon in exon 5, yielding a truncated protein lacking the seven transmembrane domains typically associated with G-protein-coupled receptors. NOP1c contains a different mini-exon insertion of 139 bp (exon 4) and predicts a truncated protein due to a frameshift and a predicted stop-codon. The other variant, NOP1b, shows a different splice site within exon 4, the same mini-exon as NOP1c, and contains only the 3′ portion of the mini-exon (98 bp). Like the others, it also gives a predicted truncated protein. The significance of these truncated proteins is still not fully understood. Clearly, they do not function like traditional G-protein-coupled receptors. Yet, this does not necessarily imply that they have no functional significance. It is interesting that similar truncated variants have been reported for all the other opioid receptor genes.
C. Molecular Modifications of NOP1
Although NOP1 itself has little affinity for traditional opioids, it can be converted into an opioid-like receptor either by simple mutations or by generating chimeras. In the rat NOP1, a series of mutations within the transmembrane regions that had little effect upon the affinity of OFQ/N itself markedly transformed the affinity of the receptor up to 50-fold for dynorphin A and several of its analogs, although the mutants still did not show appreciable affinity for β-endorphin (Meng et al., 1996). These mutations were dispersed throughout the protein, including an A213K mutation in TM5, a triple mutation VQV276–278IHI in TM6, and a T302I mutation near the top of TM7. When the T302I and VQV276–278IHI mutations were combined, the affinity of dynorphin A increased even further, with a K i under 1 nM.
Chimeras also illustrate the close relationship between the NOP1 receptor and the traditional opioid receptors. Chimeras combining the NOP1 and KOP1 (originally termed KOR-1) receptors were able to maintain high affinity for OFQ/N and dynorphin A (Lapalu et al., 1998; Mollereau et al., 1999). The first coding exon of the NOP1 receptor encodes the first transmembrane domain, just as in the traditional opioid receptors. Exchanging the first coding exon of the NOP1 receptor with the first exon of the μ opioid receptor MOP1(originally termed MOR-1) or the δ opioid receptor DOP1 (originally termed DOR-1) did not appreciably affect the affinity of the receptor for OFQ/N, but it did enhance the affinity of the κ3 ligand naloxone benzoylhydrazone (NalBzoH) approximately 5-fold and diminished the affinity of the truncated OFQ/N derivative OFQ/N(1–11) (Pan et al., 1996c). More interesting, however, the exchange with the first exon of DOP1 converted NalBzoH from an agonist into an antagonist without changing its affinity. Opiates such as morphine still did not show appreciable affinity for any of the chimeras.
III. Receptor Binding
Structurally, the NOP1 receptor has the anticipated seven transmembrane domains expected from members of the G-protein-coupled class of receptors. NOP1binding is sensitive to sodium and divalent cations (Ardati et al., 1997), as first described with opioid receptors (Pert and Snyder, 1973;Pasternak et al., 1975a,b; Wilson et al., 1975), and OFQ/N and its analogs modulate guanosine 5′-3-O-(thio)triphosphate binding in a pertussis toxin-sensitive manner (Reinscheid et al., 1995,1996; Knoflach et al., 1996; Shimohira et al., 1997; Sim and Childers, 1997; Meis and Pape, 1998; Narita et al., 1999). The first description of OFQ/N binding to NOP1 utilized an iodinated analog, 125I-[Tyr14]OFQ/N (Reinscheid et al., 1995). Although many laboratories have used this ligand, others have used 3H-OFQ/N (Dooley and Houghten, 1996), which yields results virtually identical to those of the iodinated ligand (Ardati et al., 1997). More recently, a novel radioligand for the NOP1 receptor,3H-ac-RYYRWK-NH2, was reported (Thomsen et al., 2000).
Most groups found a high affinity of OFQ/N for the transfected NOP1 receptor, typically around 50 pM, although there is a moderately wide range of values that likely reflect differences in assay techniques, buffers, and cell lines (Dooley and Houghten, 2000) and ligand stability (Quigley et al., 2000). Yet, there is good agreement regarding the specificity of the labeling, which clearly distinguished the NOP1 receptor from traditional opioid receptors. Of a wide variety of opiates, only NalBzoH showed a significant affinity for the site (K i = 310 nM; Table2), and even this was far less than its potency at opioid sites (K i < 10 nM). Morphine and other alkaloids have K ivalues well above 1000 nM.
Competition of [Tyr14]OFQ/N binding to the cloned NOP1 receptor in CHO cells
The initial characterization of NOP1 binding studies examined the interactions of OFQ/N and its analogs with the cloned receptor. Subsequent studies on brain tissue have yielded more diverse results. Although several laboratories foundK D values in brain similar to those in transfected cell lines (Albrecht et al., 1998; Nicholson et al., 1998;Thomsen et al., 2000), a number of other groups report far lower affinities (Dooley and Houghten, 1996, 2000; Wu et al., 1997; Mathis et al., 1998, 1999). Wide variations of binding levels also were reported. For example, reports of binding in rat cortex range from 22 fmol/mg of protein (Thomsen et al., 2000) to 291 fmol/mg of protein (Albrecht et al., 1998). The reasons underlying these differences are not clear. However, there are a number of variables that may play a role that include the choice of radioligand and binding conditions (Dooley and Houghten, 2000).
IV. NOP1 Receptor Heterogeneity
A major question regarding the NOP1 receptor involves binding site heterogeneity. Evidence raising the possibility of multiple OFQ/N receptors comes from both pharmacological and binding sources. As noted earlier, the Oprl1 gene that encodes the NOP1 receptor undergoes alternative splicing, but none of the additional variants identified to date encode a full-length receptor. Although the combined evidence for multiple classes of OFQ/N binding sites is suggestive, none of the studies provides conclusive evidence for NOP1 receptor heterogeneity. Yet, it is worthwhile reviewing the evidence.
The behavioral pharmacology of OFQ/N is reviewed in detail in subsequent sections. However, several issues played a major role in exploring the possibility of multiple OFQ/N receptors. As discussed below, OFQ/N reportedly has both hyperalgesic and analgesic activities. The hyperalgesic activity was insensitive to opioid antagonists, as expected based upon their poor affinity for the OFQ/N binding sites. However, several laboratories have observed that opioid antagonists reverse the analgesic responses of OFQ/N (Rossi et al., 1996b, 1997,1998b; Jhamandas et al., 1998; Kolesnikov and Pasternak, 1999). Many assumed that OFQ/N activated a neural circuit with a downstream opioid link, which could be blocked by the antagonists, and this is still a consideration. However, the opioid antagonist Win44,441 blocks the inhibition of cAMP accumulation produced by OFQ/N in mouse brain (Mathis et al., 1997). Since this assay directly measures the effects of the receptor coupled to cyclase in membrane fragments, it is difficult to envision how the antagonist could act other than directly blocking the receptor activated by OFQ/N. Thus, this OFQ/N action in brain membranes argues for an opioid antagonist-sensitive OFQ/N receptor.
Antisense mapping studies also differentiated between OFQ/N analgesia and hyperalgesia. Whereas probes targeting the second and third coding exons of the NOP1 receptor down-regulated OFQ/N and NalBzoH analgesia (King et al., 1997; Rossi et al., 1997), a probe targeting the first coding exon was ineffective. Yet, the same antisense probe based upon the first coding exon blocked the hyperalgesia and anti-opioid activity of OFQ/N, whereas the antisense probes targeting exons 2 and/or 3 that were active against OFQ/N analgesia had no effect against hyperalgesia (King et al., 1997; Rossi et al., 1997, 1998). Although these pharmacological assays are suggestive, behavioral approaches have many potential subtleties and alternative explanations. Thus, they do not provide conclusive evidence for multiple OFQ/N receptors.
Binding studies also suggest binding site heterogeneity. Early studies with 125I-[Tyr14]OFQ/N in mouse brain revealed curvilinear Scatchard plots, suggestive of sites of differing affinity (Mathis et al., 1997). However, this does not necessarily imply different receptors. In NOP1-transfected HEK293 cells with high levels of expression, 3H-OFQ/N binding also yielded biphasic Scatchard plots, presumably reflecting two conformations of the receptor (Ardati et al., 1997). Alternatively, curvilinear Scatchard plots can result from radioligand degradation. Saturation studies alone cannot distinguish among these possibilities and must be interpreted in conjunction with other types of studies. However, other approaches also implied NOP1 receptor binding heterogeneity.
OFQ/N(1–11) is a truncated peptide derived from OFQ/N. In vivo, it is functionally active, eliciting analgesia (Rossi et al., 1997) and inhibiting cAMP accumulation in brain membranes (Mathis et al., 1997). These actions were not anticipated based upon its very poor affinity for the cloned NOP1 receptor in binding assays. To more accurately assess the possibility of a novel OFQ/N(1–11) binding site, tyrosine-containing analogs were generated that could be iodinated and used to examine binding sites directly (Mathis et al., 1998), as done earlier with OFQ/N itself (Reinscheid et al., 1995). Of the analogs, [Tyr10]OFQ/N(1–11) proved most valuable. Like OFQ/N(1–11), [Tyr10]OFQ/N(1–11) is analgesic when given supraspinally in mice and it competes125I-[Tyr14]OFQ/N binding in mouse brain more potently (K i = 79 nM) than OFQ/N(1–11) itself (K i = 262 nM). Iodinating the analog to iodo[Tyr10]OFQ/N(1–11) further enhanced its potency (K i = 39 nM).
In mouse brain,125I-[Tyr10]OFQ/N(1–11) labeling strongly suggested a novel binding site distinct from the binding of125I-[Tyr14]OFQ/N. Binding parameters of125I-[Tyr10]OFQ/N(1–11) revealed an affinity (K D) of 0.24 nM, which is over 100-fold lower than itsK i against125I-[Tyr14]OFQ/N binding in mouse brain and more than 10-fold lower than itsK D determined in CHO cells transfected with the NOP1 receptor. Furthermore, in brain it displayed a B max of only 43 fmol/mg of protein, which is far fewer sites than observed in companion assays with 125I-[Tyr14]OFQ/N (Table 3) or from the literature (Dooley and Houghten, 1996; Albrecht et al., 1998; Nicholson et al., 1998). It also is interesting that the capacity of the125I-[Tyr10]OFQ/N(1–11) site is similar to the higher affinity (K D = 4 pM) site observed in mouse brain for125I-[Tyr14]OFQ/N. A possible association of the two is further suggested by saturation studies with125I-[Tyr14]OFQ/N in which the inclusion of OFQ/N(1–11) appeared to selectively reduce the higher affinity binding component of125I-[Tyr14]OFQ/N.
Saturation analysis of 125I-[Tyr14]OFQ/N and125I-[Tyr11]OFQ/N(1–11) binding in transfected CHO cells and mouse brain
The difference in selectivity between OFQ/N(1–11) and the standard OFQ/N radioligands was quite revealing (Mathis et al., 1999). OFQ/N and its analogs labeled the125I-[Tyr10]OFQ/N(1–11) site with very high affinity, confirming its classification as an OFQ/N site. The affinity of OFQ/N(1–11) increases about 30-fold, with itsK i dropping to only 8.7 nM. It is interesting, however, that the affinity of OFQ/N(1–7) is unchanged and remains quite poor.
As previously noted,125I-[Tyr14]OFQ/N binding is insensitive to opioids. In contrast,125I-[Tyr10]OFQ/N(1–11) binding is competed by a wide variety of opioid ligands. Although the affinities of most of the opioids examined remain lower than against traditional opioid receptors, a number of compounds showed high affinity for this site (Table 4). Among the opiates, NalBzoH was the most impressive. In brain membranes, its affinity against125I-[Tyr10]OFQ/N(1–11) binding (K i = 3.9 nM) is similar to that seen with traditional opioid binding sites and almost 100-fold greater than125I-[Tyr14]OFQ/N binding. The affinity of fentanyl is increased over 100-fold against the OFQ/N(1–11) site. Some opioid peptides also show high affinity for the125I-[Tyr10]OFQ/N(1–11) site, particularly dynorphin A and α-neoendorphin. Indeed, dynorphin A labels the125I-[Tyr10]OFQ/N(1–11) site as potently as μ and δ opioid receptors.
Competition of 125I-[Tyr10]OFQ/N(1–11) binding in mouse brain membranes
Together, along with the dramatic anatomical differences between125I-[Tyr14]OFQ/N and125I-[Tyr10]OFQ/N(1–11) binding (see below), these studies suggest the possibility of OFQ/N receptor heterogeneity. If multiple OFQ/N sites exist, they may correspond to splice variants of NOP1 receptor. Alternatively, they might correspond to post-translational modifications of the receptors, result from modulation of the receptor by additional proteins, or be expressed by a totally different gene. However, without more definitive biochemical evidence, OFQ/N binding site heterogeneity remains tentative.
V. Orphanin FQ/Nociceptin
A. Structure of Orphanin FQ/Nociceptin
The unusual properties of the “orphan opioid receptor” soon led to the identification of an endogenous peptide, termed orphanin FQ (Reinscheid et al., 1995) or nociceptin (Meunier et al., 1995) (OFQ/N), that labeled the cloned receptor with very high affinity (Fig. 1). OFQ/N is a heptadecapeptide with some interesting structural homologies to the classical opioid peptide dynorphin A (Fig. 1). Both peptides are comprised of 17 amino acids bounded by pairs of basic amino acids important in their production from their precursors. Furthermore, both have internal pairs of basic amino acids, raising the possibility of further processing. The opioid peptides share a YGGF motif, where the fifth amino acid is either leucine or methionine. The amino terminus of OFQ/N is a phenylalanine instead of a tyrosine, followed by GGF. Finally, both peptides contain the same last two amino acids at the carboxyl terminus. Despite these similarities, the peptides are functionally quite distinct. OFQ/N has no appreciable affinity for any of the opioid receptors. Alanine scanning reveals the importance of the amino acids in positions 1, 2, 4, and 8 (Dooley and Houghten, 1996). Of these, the phenylalanine in position 1 is particularly important in establishing the selectivity of binding since replacing it with a tyrosine yields analogs with far greater affinity at traditional opioid receptors, although the [Tyr1] analog still can induce naloxone-insensitive actions presumably mediated through NOP1 receptors (Champion and Kadowitz, 1997a,b). The basic structure can be modified and even truncated at its carboxyl terminus without major loss of activity, but the initial FGGF motif is required for activity (Guerrini et al., 1997).
B. Orphanin FQ/Nociceptin Analogs and Antagonists
Full descriptions of all the structure-activity relationships of OFQ/N is beyond the scope of this review. However, several analogs are important. The first analog, [Tyr14]OFQ/N, was developed to enable the detection of receptor binding and has been particularly important (Reinscheid et al., 1995). Replacing the leucine at position 14 yielded a peptide that could be iodinated and still maintain affinity for the receptor similar to the parent compound (Reinscheid et al., 1995). This analog has proven extremely valuable in the characterization of the receptor in both transfected cell lines and in the brain.
OFQ/N has two pairs of basic amino acids within its structure, raising the possibility of further processing to yield OFQ/N(1–11) and OFQ/N(1–7). Although there are studies showing the activity of these peptides and suggesting that their pharmacology may differ from that of OFQ/N itself (Rossi et al., 1997), as described below, the physiological significance of these truncated peptides has not been fully established. The possibility that the truncated peptides also might be relevant led to the development of a tyrosine-containing analog of OFQ/N(1–11) suitable for iodination (Mathis et al., 1998). Analogs were synthesized with tyrosine at positions 1, 10, or 11. The placement of tyrosine at position 1 lowered its affinity against NOP1 binding in transfected cells, but enhanced its potency against the traditional opioid receptors. [Tyr11]OFQ(1–11) and its iodinated version, iodo[Tyr11]OFQ/N(1–11), on the other hand, were devoid of activity against traditional opioid receptors and more potent against NOP1 binding in transfected cells than OFQ/N(1–11) itself. Both [Tyr11]OFQ(1–11) and iodo[Tyr11]OFQ/N(1–11) were pharmacologically active, eliciting analgesia in mice. As discussed earlier, these agents have proven valuable in binding studies.
The evaluation of the pharmacology of OFQ/N was hindered for a number of years by the lack of an effective antagonist. The first proposed antagonist, [Phe1ψ(CH2-NH)Gly2]-nociceptin(1–13)-NH2, was subsequently found to be a partial agonist, with many groups observing OFQ/N-like actions in a variety of models. A recently described small molecule antagonist, J-113397 (1-[3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one) (Kawamoto et al., 1999; Ozaki et al., 2000a,b), has proven valuable in a number of models (see Section VIII.G.2.).
C. Orphanin FQ/Nociceptin Precursors and Their Processing
The OFQ/N sequence contains pairs of basic amino acids that might imply additional processing of the peptide to OFQ/N(1–11) and/or OFQ/N(1–7). Both of these truncated peptides are functionally active when administered in vivo (Rossi et al., 1997), producing analgesia that is reversed by opioid antagonists. Neither peptide shows appreciable affinity for any of the traditional opioid receptors, but OFQ/N(1–11) does label cloned NOP1 receptors moderately well (K i = 55 nM), although its affinity still is far lower than OFQ/N itself. OFQ/N(1–7) does not compete with binding to the NOP1 receptor at doses as high as 1 μM. The true significance of these peptides remains to be demonstrated.
Like most neuropeptides, OFQ/N is generated from a larger precursor peptide, prepro-OFQ/N (ppOFQ/N) that has been cloned from mouse, rat, and human (Fig. 3) (Meunier et al., 1995;Pan et al., 1996a; Reinscheid et al., 2000) and that has been localized in man to chromosome 8 (8p21) (Mollereau et al., 1996). Overall, there is high interspecies homology, with 80% identity among the three organisms. Within the precursor, there are several additional peptides suggested by the presence of pairs of basic amino acids. Nocistatin has been examined most extensively (see Section IX.A.). Nocistatin possesses analgesic actions and presumably acts through a distinct receptor since it has no appreciable affinity for any of the traditional opioid receptors or NOP1. It is interesting that the nocistatin sequence shows the most variability of the putative peptides within ppOFQ/N among species. The mouse version is the longest, containing 41 amino acids, whereas the rat peptide has 35 and the human form only 30. The mouse sequence has an interesting DAEPGA motif that is repeated three times. The rat form has a similar double repeat, but the human form does not. The differences between the species rests primarily over the length of this repeat, with the human form lacking 10 of the amino acids of the mouse version at this location.

Schematic of the prepro-OFQ/N gene.
Another peptide was predicted from the sequence of ppOFQ/N based upon the presence of pairs of basic amino acids suggesting sites of peptide processing. Orphanin FQ2 is a heptadecapeptide, like OFQ/N and dynorphin A, with a phenylalanine (F) and glutamine (Q) at the first and last position, leading to its name, OFQ2 (also called NocII; and hereinafter called OFQ2/NocII). The placement of OFQ2/NocII within ppOFQ/N is interesting in that OFQ2/NocII is immediately downstream from OFQ, much like dynorphin B is immediately downstream of dynorphin A in preprodynorphin. When administered centrally, OFQ2/NocII is pharmacologically active, raising the possibility that it is physiologically relevant (Rossi et al., 1998a; Florin et al., 1999) (see Section IX.B.). A longer peptide containing the OFQ2/NocII sequence at its amino terminus, ppOFQ/N(180–187), has been described and it also is functionally active in mice (Mathis et al., 2001). It is still an open question as to whether ppOFQ/N(180–187) is active itself or whether it is further processed to OFQ2/NocII.
VI. Anatomy of Orphanin FQ/Nociceptin and Its Receptor
The regional distribution of OFQ/N and the NOP1 receptor have been well described (Bunzow et al., 1994; Fukuda et al., 1994; Mollereau et al., 1994; Wick et al., 1994; Lachowicz et al., 1995; Nothacker et al., 1996; Riedl et al., 1996; Houtani et al., 2000; Neal et al., 1999a,b; Letchworth et al., 2000; O'Donnell et al., 2001). These series of publications provide detailed descriptions of the distribution of the NOP1 receptor mRNA and binding in the brain which are beyond the scope of this review. Overall, they report a good correlation between receptor binding distributions and those seen with in situ hybridization. Regions with NOP1 receptor binding typically express NOP1 mRNA as well, although the levels of mRNA and binding do not always match very closely. Regions with high levels of NOP1binding/mRNA include the cortex, anterior olfactory nucleus, lateral septum, hypothalamus, hippocampus, amygdala, central gray, pontine nuclei, interpeduncular nucleus, substantia nigra, raphe complex, locus coeruleus, and spinal cord. The distribution patterns have suggested the involvement of the NOP1 receptor system in “motor and balance control, reinforcement and reward, nociception, the stress response, sexual behavior, aggression and autonomic control of physiological processes” (Neal et al., 1999a).
The distribution of OFQ/N also has been well described in the literature (Dickenson, 1996; Riedl et al., 1996; Kummer and Fischer, 1997; Mitsuma et al., 1998; Neal et al., 1999b; Houtani et al., 2000;O'Donnell et al., 2001). In brief, the localization of OFQ/N corresponds reasonably well with the NOP1receptor. As with the receptor, OFQ/N immunoreactivity and mRNA levels detected using in situ hybridization are closely correlated. OFQ/N is found in lateral septum, hypothalamus, ventral forebrain, claustrum, mammillary bodies, amygdala, hippocampus, thalamus, medial habenula, ventral tegmentum, substantia nigra, central gray, interpeduncular nucleus, locus coeruleus, raphe complex, solitary nucleus, nucleus ambiguous, caudal spinal trigeminal nucleus, and reticular formation, as well the ventral and dorsal horns of the spinal cord (Neal et al., 1999b).
The distribution of125I-[Tyr10]OFQ/N(1–11) in the brain also is distinct autoradiographically (Fig.4) (Letchworth et al., 2000). The distribution of125I-[Tyr14]OFQ/N binding was described earlier.125I-[Tyr10]OFQ/N(1–11) binding also shows intense labeling of the cortex, but far lower levels of labeling in deeper structures. Compared with125I-[Tyr14]OFQ/N,125I-[Tyr10]OFQ/N(1–11) labeling is far less intense in the olfactory tubercle, nucleus accumbens, striatum, lateral and medial septum, hypothalamus, as well as a number of brain stem structures such as the periaqueductal gray, medial raphe, and locus coeruleus.

Autoradiographic studies of125I-[Tyr14]OFQ and125I-[Tyr10]OFQ(1–11) in rat brain. Sections of rat brain were incubated with either125I-[Tyr14]OFQ or125I-[Tyr10]OFQ(1–11) and opposed to film. The exposure time was longer for125I-[Tyr10]OFQ(1–11) than for125I-[Tyr14]OFQ due to the lower binding levels. Reprinted from Letchworth et al., 2000.
VII. Range of Effects of Orphanin FQ/Nociceptin
Befitting its particularly wide distribution in the nervous system (see above), there are a myriad of proposed functional roles for OFQ/N. Receiving by far the most attention is the involvement of this peptide in the mediation and modulation of pain in the supraspinal, spinal, and peripheral compartments of the nervous system. Related proposed functions for OFQ/N include roles in opiate tolerance, dependence/withdrawal, and adaptive responses to anxiety and stress. However, studies based on direct injection of the peptide, measurement of peptide levels, administration of antagonist/antisense compounds and/or the evaluation of the phenotype of transgenic “knockout” mutants have implicated OFQ/N in the mediation of biological phenomena ranging from learning and memory to hearing to water balance to reproductive physiology. A list of OFQ/N-associated systems-level phenomena is presented in Table 5. Some of the more well studied and noteworthy phenomena will be discussed presently, starting with pain processing.
Systems-level biological phenomena in which OFQ/N has been implicated
VIII. Effects of Orphanin FQ/Nociceptin on Pain
A. Effects of Supraspinally Administered Orphanin FQ/Nociceptin
The first in vivo action of OFQ/N reported by both its discoverers was a reduction in latency to respond to noxious thermal stimuli on the tail-flick (Reinscheid et al., 1995) and hot-plate tests (Meunier et al., 1995) after supraspinal (intracerebroventricular) injection in the mouse. Both groups interpreted these data as reflective of a hyperalgesic action; i.e., a decrease in nociceptive threshold (increase in nociceptive sensitivity) produced by the peptide. This was very much a surprise, since classical opioids, with the possible exception of dynorphin (see Caudle and Mannes, 2000), produce analgesic and/or antihyperalgesic effects (see Pasternak, 1993). The apparent hyperalgesia produced by supraspinal OFQ/N inspired Meunier and colleagues (1995) to dub the peptide nociceptin.
Exogenous administration of an endogenous compound is not an ideal method for gleaning its true physiological role. When injected intracerebroventricularly, OFQ/N will be widely dispersed throughout the ventricular system, possibly affecting populations of ORL1 receptors that would not normally be activated by endogenously released peptide. Tissue levels are dependent upon diffusion of the agent from the cerebrospinal fluid into the brain, which results in a decreasing gradient of drug concentrations in deeper structures. The drug even can diffuse to spinal sites, particularly with high injection volumes. This makes it difficult to judge the concentration of peptide in relevant brain loci and thereby assess whether its concentration is appropriate or grossly supraphysiological. Finally, this approach entirely ignores contextual elements accompanying OFQ/N release under usual circumstances. Nonetheless, in the absence of an ORL1 antagonist and with the vast majority of the studies reviewed herein conducted before any such antagonist was available, direct injection of OFQ/N was one of only a handful of feasible experimental approaches.
In contrast to the conclusions from the initial descriptions of OFQ/N, we now recognize that there is no widely accepted “role” of OFQ/N in supraspinal pain-modulatory circuits. In fact, even the effects of supraspinal projection of OFQ/N on nociceptive sensitivity remain highly contentious. As detailed in Table6, reports in the literature have suggested six different “effects” of supraspinal OFQ/N on nociception: 1) hyperalgesia, 2) analgesia, 3) hyperalgesia followed by analgesia, 4) neither hyperalgesia nor analgesia, 5) anti-analgesia but not hyperalgesia, and 6) anti-analgesia plus hyperalgesia. The only uncontested observation is the anti-analgesic activity of OFQ/N, first documented in 1996 (Mogil et al., 1996a)
Parameters and results of published studies investigating the effect on nociceptive sensitivity of supraspinally (i.c.v.)-injected OFQ/N in rodents
OFQ/N blocks analgesia from a wide variety of exogenous and endogenous opioid compounds. Since OFQ/N has negligible affinity for any of the traditional opioid receptors, it must act through neural circuits as a “functional antagonist”, rather than through direct molecular interactions with opioid receptors. Given intracerebroventricularly, OFQ/N can reverse and/or prevent analgesia from drugs acting at supraspinal μ-opioid receptors, including morphine (Grisel et al., 1996; Mogil et al., 1996a; Tian et al., 1997b;Zhu et al., 1997; Calo' et al., 1998; King et al., 1998; Lutfy et al., 1999; Citterio et al., 2000), DAMGO (Mogil et al., 1996b), fentanyl (Zhu et al., 1998), acetorphan (Suaudeau et al., 1998), endomorphin-1 (Wang et al., 1999a,c), and morphine-6β-glucuronide (King et al., 1998). It has similar effects against supraspinal δ-opioid agonists, like DPDPE (Mogil et al., 1996b; King et al., 1998) and DSLET (Zhu et al., 1998; Wang et al., 1999a), κ1-opioid agonists like U50,488 (Mogil et al., 1996b; Zhu et al., 1998; Wang et al., 1999a), and dynorphin A (Citterio et al., 2000), and the κ3-opioid agonist, naloxone benzoylhydrazone (King et al., 1998).
Direct injections of OFQ/N into specific brain loci also induce anti-analgesic actions. OFQ/N placed into the periaqueductal gray (PAG) blocks morphine analgesia (Morgan et al., 1997) and its administration into the rostral ventromedial medulla (RVM) reverses DAMGO analgesia (Heinricher et al., 1997; Pan et al., 2000) (see Section VIII.H.). Importantly, OFQ/N also blocks analgesia from endogenous opioid-mediated manipulations, including electroacupuncture (Zhu et al., 1996; Tian et al., 1997a; Zhang et al., 1997) and mild stressors (Mogil et al., 1996a; Suaudeau et al., 1998; Rizzi et al., 2001). The latter phenomenon may be responsible for much of the confusion surrounding the actions of OFQ/N (see Section VII.D.3.).
The anti-opioid effect of OFQ/N against morphine analgesia is long-lasting, persisting for up to 4 to 6 h (Candeletti and Ferri, 2000). Repeated OFQ/N dosing induces tolerance, with a decreasing response over time (Lutfy et al., 1999). Although it is tempting to only assume a functional interaction between OFQ/N and other members of the opioid gene family, the anti-analgesic actions of this peptide are by no means restricted to opioid analgesia. OFQ/N equally efficaciously blocks analgesia from the α2-adrenergic receptor agonist, clonidine (King et al., 1998), the GABAB receptor agonist, baclofen (Citterio et al., 2000), and naloxone-insensitive forms of swim stress (Rizzi et al., 2001).
This ability to block non-opioid analgesia sets OFQ/N apart from other known functional anti-opioid peptides, including adrenocorticotrophic hormone (ACTH), cholecystokinin (CCK), dynorphin, FMRFamide (and its analogs), α-melanocyte-stimulating hormone (α-MSH), MIF-1/Tyr-MIF-1, neurotensin- and tyrosine-releasing hormone (Rothman, 1992), and ς1 receptor systems (e.g.,Chien and Pasternak, 1993). Anti-opioid systems are thought to play important roles in a number of pain-relevant phenomena, including the mediation of individual differences in analgesic sensitivity (Chien and Pasternak, 1993; Tang et al., 1997), the induction of tolerance and dependence (Rothman, 1992) (see Section X.D.), and in plastic changes underlying neuropathic pain (Wiesenfeld-Hallin et al., 1997). Elucidation of the precise actions of OFQ/N vis-à-vis these other anti-opioid peptides will be a major research challenge for the future.
These anti-analgesic actions of OFQ/N are the most robust activities observed following supraspinal administration, having been seen by all groups examining this question. The two contentious issues, regarding OFQ/N actions, that remain involve direct analgesia and hyperalgesia. In one study, for example, higher OFQ/N doses induced analgesia in mice, although this action is not easily detected in all strains (Rossi et al., 1997). In this study, an initial hyperalgesic response was followed by analgesia. The analgesic response was reversed by opioid antagonists, but the hyperalgesic actions were not. Indeed, the biphasic hyperalgesic/analgesic activity seen with OFQ/N alone reverted to only a monophasic hyperalgesia in the presence of the opioid antagonist. Others, of course, see neither hyperalgesia or analgesia. Factors relevant to interpreting the conflicting data presented in Table 6 are discussed below.
A final comment concerns not the effect of OFQ/N on pain, but the effect of pain on OFQ/N. A recent study by Rosen and colleagues (2000)examined OFQ/N-like immunoreactivity in various nociception-related brain areas 2 weeks after the induction of a neuropathic state usingBennett and Xie's (1988) surgical model or a carrageenan inflammatory model. Both injuries increased OFQ/N levels in the cingulate cortex, and carrageenan increased levels also in the hypothalamus and the dorsal horn of the spinal cord. OFQ/N levels did not change in the PAG or RVM (in contrast to levels of dynorphin B and met-enkephalin-Arg-Phe), prompting the authors to conclude that OFQ/N is involved in the modulation of ascending nociceptive transmission pathways rather than descending nociceptive modulation pathways (but see Section VIII.H.). OFQ/N has also been identified in human cerebrospinal fluid but not at higher levels in women with ongoing labor pain compared with those presenting for elective Caesarean section (Brooks et al., 1998). Thus, any clinical relevance of supraspinal OFQ/N remains to be demonstrated.
B. Effects of Spinally Administered Orphanin FQ/Nociceptin
Although the seminal investigations of OFQ/N featured supraspinal administration of the peptide, opioids play an equally crucial role in pain modulation in the spinal level (see Yaksh, 1999). Although OFQ/N injected intrathecally (10 nmol, i.t.) was initially reported to have no effect on thermal nociception (Reinscheid et al., 1995), a subsequent study reported a trend (p = 0.053) toward enhanced morphine analgesia by intrathecal OFQ/N (Grisel et al., 1996) followed by additional support for spinal OFQ/N analgesia (Xu et al., 1996; King et al., 1997). The situation has become more complicated since then, as shown in Table 7. Strikingly low OFQ/N doses spinally produce spontaneous pain, as evidenced by caudally directed scratching, biting, and licking (SBL) behaviors, and hypersensitivity to thermal and mechanical stimuli. These SBL behaviors are reminiscent of those elicited by substance P and N-methyl-d-aspartate (NMDA) and are eliminated by pretreatment with morphine and neurokinin-1 (NK1) receptor antagonists, but not neurokinin-2 (NK2) or NMDA receptor antagonists (Sakurada et al., 1999b). At higher OFQ/N doses, a number of laboratories have observed analgesic and anti-hyperalgesic/anti-allodynic effects. However, some have been unable to demonstrate OFQ/N analgesia at presumably effective doses. Still others have demonstrated anti-analgesic effects reminiscent of supraspinal peptide, alone or in combination with hyperalgesia (see Table 7).
Parameters and results of published studies investigating the effect on nociceptive sensitivity of spinally (i.t.)-injected OFQ/N
Despite the many contradictions in the established literature to date, most reviewers have concluded that the dominant spinal action of high doses of OFQ/N is inhibitory—congruent with the findings of all electrophysiological studies—producing behavioral analgesia and/or anti-hyperalgesia/anti-allodynia (Henderson and McKnight, 1997;Meunier, 1997; Harrison and Grandy, 2000; Xu et al., 2000). Wang and colleagues (1996) have arrived at the same conclusion for the trigeminal system. Assuming that spinal OFQ/N is analgesic, the potential role of classical opioid receptors remains a further unresolved issue. Of the eight studies looking at the effects of opioid antagonists on OFQ/N analgesia, only two reported a reversal (King et al., 1997; Hao and Ogawa, 1998). In another study, repeated administration of spinal OFQ/N resulted in the development of tolerance to the peptide's analgesic effects and cross-tolerance to morphine (Jhamandas et al., 1998). This finding, however, is directly contradicted by yet another study that found no cross-tolerance (Hao et al., 1997).
Nociception-relevant elements in the spinal cord undergo anatomical and functional alterations after peripheral nerve injury or inflammation and this plasticity is thought to be important in producing and maintaining chronic pain states (Woolf, 1983). OFQ/N appears to be no exception. OFQ/N levels and binding increase in the dorsal horn of the spinal cord after inflammation (Jia et al., 1998; Rosen et al., 2000). In one study, this increase was bilateral, but restricted to the superficial laminae (I and II) of the cord (Jia et al., 1998). Inflammation also induces expression of the prepro-OFQ/N gene in the dorsal root ganglion, although the increased synthesis of OFQ/N was quite short-lived (<6 h) (Andoh et al., 1997). In contrast to dynorphin, which was increased in the dorsal horn after a Bennett model nerve injury, OFQ/N levels in this study trended lower, although the decrease did not achieve statistical significance. These findings are hard to reconcile with data demonstrating that high-dose OFQ/N′s depressive effect on the flexor reflex is decreased in inflamed rats and increased somewhat in nerve injured rats (Abdulla and Smith, 1998; also see Hao et al., 1998; Xu et al., 1999a). This pattern of functional changes is exactly opposite to that of μ opioids, which exhibit increased efficacy in inflammatory states (Stanfa and Dickenson, 1995) and greatly reduced efficacy against neuropathic pain (Arner and Meyerson, 1988). As pointed out by Xu and colleagues (1999a), however, the effectiveness of exogenously applied OFQ/N is primarily determined by the status of NOP1receptors, not endogenous peptide levels. No data have thus far been collected as to whether NOP1 receptors are altered after injury.
C. Effects of Peripherally Administered Orphanin FQ/Nociceptin
In addition to their effects in the CNS, opioids can produce analgesia in the periphery, especially in the presence of inflammation (Stein et al., 1990; Kolesnikov et al., 1996). This fact, along with the ability of OFQ/N to affect transmitter release in the peripheral nervous system (see Giuliani et al., 2000), suggests that OFQ/N may modulate nociception directly at the site of pain and/or injury. A small number of studies have investigated this possibility, again with somewhat conflicting results. Two elegant studies by Inoue and colleagues (1998, 1999) demonstrated the ability of OFQ/N at remarkably low doses, up to 10,000-fold lower than substance P and 1000-fold lower than bradykinin, to elicit the nociceptive flexor reflex after intraplantar injection into the hindpaw. This effect appears to be secondary to local substance P release in the paw, since the phenomenon can be blocked by inhibition of transmitter release by botulinum toxin, depletion of substance P by capsaicin, by NK1receptor antagonists, and is abolished in tachykinin-1 gene knockout mice (Inoue et al., 1998). In the second study, however, a higher OFQ/N dose (1 nmol) was analgesic, producing a complete blockade of substance P-induced flexor reflexes and SBL (Inoue et al., 1999) (seeSection VIII.D.5.). Another group, also using higher doses, demonstrated the analgesic efficacy of OFQ/N applied subcutaneously to the tail (Kolesnikov and Pasternak, 1999). This analgesia was naloxone-reversible, but insensitive to antagonism by either μ- or κ-specific antagonists.
The modulatory effects of OFQ/N on rat knee joint afferents were very recently studied by McDougall et al. (2000). They found a sensitizing effect of OFQ/N in normal joints, and a desensitizing effect during hyper-rotation in acutely inflamed knees. Interestingly, both these effects may be explained by the OFQ/N-substance P interactions described above (Inoue et al., 1998; Lecci et al., 2000b). However,Kumar and colleagues (1999) were unable to demonstrate [3H]OFQ/N binding in human synovial joint fluid or tissue. OFQ/N has been implicated in fibromyalgia where female sufferers display decreased plasma levels of the peptide (Anderberg et al., 1998).
D. Reconciling the Literature
The preceding descriptions of OFQ/N effects on nociceptive phenomena at the supraspinal, spinal, and peripheral levels (see Tables2 and 3) illustrate the considerable uncertainty that still surrounds the simplest of questions: What are the actions of OFQ/N when injected? The next sections will address a number of factors that may be relevant to reconciling the divergent results found in the literature and thus to illuminating the endogenous role of OFQ/N.
1. Noxious Stimulus Modality.
There is a large literature demonstrating differential processing of different types of pain by neurochemically distinct circuitries (for reviews, see Mogil et al., 1996c, 1999b). It is possible, therefore, that activities of OFQ/N may be dependent upon the nociceptive assay used. The majority of the studies to date have used thermal assays (tail-flick/withdrawal, hot-plate tests) (Tables 2 and 3), which is to be expected since these assays are easily performed and commonly used in the opioid field. Supraspinal OFQ/N anti-analgesia is a robust response and has been demonstrated against thermal, electrical, and chemical assays, of varying durations (acute to chronic). Spinal hyperalgesia/allodynia and analgesia have been demonstrated against thermal, chemical, and mechanical assays. Even some of the less common findings, such as spinal anti-analgesia or anti-hyperalgesia/anti-allodynia, have been observed using multiple nociceptive assays. Overall, there does not appear to be strong evidence at the present time for modality-specific effects of the pronociceptive actions of OFQ/N.
OFQ/N analgesia, however, is less robust and far more controversial and assay differences are more likely to be important. Variations in the performance of specific tests among laboratories and the tests themselves can influence analgesic potency of opioids. However, it remains to be demonstrated whether assays play a major role in the differences among reported observations.
2. Robustness of Various Phenomena.
Not all of the reported phenomena are equally robust. For example, of the 16 studies reporting supraspinal hyperalgesia listed in Table 6, half featured latency decreases, or formalin rating increases, of <40% compared with baseline and/or vehicle values. By contrast, virtually all studies reporting anti-opioid analgesic actions of OFQ/N demonstrated a complete blockade of even profound analgesia. Also, the supraspinal hyperalgesic actions of OFQ/N are quite transient compared with the anti-analgesic actions, with the former lasting only 15 to 30 min in virtually all cases. One can easily point to degradation of the peptide as an explanation of transient effects, but such degradation does not prevent long-lasting anti-analgesic actions (see especially Candeletti and Ferri, 2000). Particularly weak is the phenomenon of supraspinal OFQ/N analgesia defined as a quantal doubling of the baseline tail-flick latency, which can be demonstrated in only 40% of CD-1 mice and was not seen in two other strains (see Section VIII.D.3.) (Rossi et al., 1996b, 1997). It should be noted that blockade of the anti-opioid ς1 receptor system with the ς receptor antagonist, haloperidol, dramatically enhanced the analgesic actions of OFQ/N and its fragments in all strains tested (Rossi et al., 1997). Supraspinal OFQ/N analgesia does seem to be more robust in the rat (Rossi et al., 1998b).
Critically assessing the reliability or importance of weak phenomena is difficult. Although they might represent chance occurrences, a finding may depend on a particular set of organismic and parametric circumstances that would encourage replication within laboratories but not between them. Furthermore, small overall effects may simply reflect the summation of opposing processes, with one canceling out another (see Section VIII.D.6.). Obviously, there is a need for additional attempts by independent laboratories to replicate and extend some of the OFQ/N phenomena reported.
3. Influence of Stress.
The original investigations of the supraspinal OFQ/N quantified the effect of the peptide relative to a control group receiving an isovolumetric injection of vehicle (Meunier et al., 1995; Reinscheid et al., 1995). Although a reasonable control, it alone is not sufficient since mice receiving intracerebroventricular injections are not at “baseline”. This is particularly evident when dealing with nociceptive assays capable of detecting stress analgesia. Employing either “no injection” and/or preinjection baseline control groups, depending on the nociceptive assay, Mogil and colleagues (1996a) demonstrated that the apparent hyperalgesia noted previously could be explained by the reversal of stress-induced analgesia by OFQ/N. In these studies, the apparent hyperalgesia compared with the vehicle group was actually reversal of stress-induced analgesia related to the injection when compared with the no-injection group (Mogil et al., 1996a).
Stress-induced analgesia is a well known, adaptive phenomenon thought to represent the evolutionary impetus for the development of central pain inhibition mechanisms, and thus the neural substrate on which clinical analgesics like morphine act (Kelly, 1986). The phenomenon can be produced by any number of environmental stressors and can be mediated by opioid or non-opioid neurochemistry (Lewis et al., 1980;Watkins and Mayer, 1982). Although it is not widely appreciated that procedures related to nociceptive testing can themselves produce stress-induced analgesia, we have shown that even intraperitoneal injections of saline can produce the effect in some circumstances (Wilson et al., 1998), and others have shown activation of the hypothalamic-pituitary-adrenal (HPA) axis and c-fosinduction from this mild stressor (Ryabinin et al., 1999). Although intracerebroventricular injections performed by any number of modifications of the method of Haley and McCormick (1957) are often performed under light gaseous anesthesia, they may be considered a significant stressor in at least three possible ways. First, the anesthesia itself may be a stressful experience. Second, the injection proceeds directly through the skull at the coronal suture and thus represents a mild trauma. Third, a nontrivial volume of fluid is injected into the cerebral ventricles. In the hands of Mogil and Grisel (Grisel et al., 1996; Mogil et al., 1996a,b), this method of injection can produce measurable analgesia against mild-to-moderate noxious heat (up to 49°C). Intrathecal injections in the mouse performed by the method of Hylden and Wilcox (1980) are equally subject to the phenomenon (Grisel et al., 1996; Mogil et al., 2000b). The probability of encountering this confound increases as the severity of the noxious stimulus decreases. As the pain research community increasingly switches its attention from analgesia to mechanisms underlying hyperalgesia, increasingly mild noxious stimuli are featured in experiments so that “floor” effects can be avoided. Tables 2 and 3document the especially mild noxious stimulus parameters that have been employed in the OFQ/N literature.
Although some have concluded that their prior data may have been confounded in the manner described above (Suaudeau et al., 1998), many laboratories have continued to report supraspinal OFQ/N hyperalgesia, even with the adoption of the recommended controls (e.g., Calo' et al., 1998). Although OFQ/N clearly can reverse stress-induced analgesia, it also may have hyperalgesic activity in models with less of a confound. Also, OFQ/N hyperalgesia has been reported on a number of occasions in the rat, a species in which i.c.v. injections must proceed through an indwelling cannula, minimizing the possibility of stress-induced analgesia during the testing procedure itself. Therefore, other factors must be considered in an attempt to reconcile the entire literature. Stress may, in fact, play a more general role here, especially given the demonstrated ability of OFQ/N to act as an anxiolytic (Jenck et al., 1997; Mamiya et al., 1998; Koster et al., 1999) (see Section X.B.). Perhaps, then, the effect of exogenously administered OFQ/N is wholly dependent on the psychological state of the subject at the time of testing, a state that can be influenced by husbandry (e.g., fighting among group-housed males), test-related stressors, and genetic factors (see below). If so, then the considerable challenge of parsing out such phenomena may yield large rewards in our understanding of individual differences in pain sensitivity.
4. Organismic Factors: Species, Strain, and Sex Differences.
An inspection of Tables 2 and 3 reveal that species differences are not an obvious explanation of discrepancies in this literature, as virtually all categories of OFQ/N effects include both mouse and rat studies. There are some notable exceptions, however. Spontaneous nociception and hyperalgesia/allodynia from spinal OFQ/N is a phenomenon so far demonstrated only in mice of the inbred ddY strain. Similarly, anti-hyperalgesia/anti-allodynia and anti-analgesia from spinal OFQ/N has only been observed in the Sprague-Dawley rat. There are only two examples to our knowledge of a specific within-laboratory species comparison of OFQ/N actions. Rossi and colleagues (1998b) noted that they were unable to demonstrate the supraspinal OFQ/N hyperalgesia in the rat that they had observed repeatedly in the mouse. The systematic examination of Vanderah et al. (1998) of OFQ/N actions used both rats and mice, although they detected no reliable effects of OFQ/N on nociception in either species. It should also be noted that OFQ/N has been administered to a nonmammalian species, the land snail (Cepaea nemoralis), and found (with all appropriate controls) to produce hyperalgesia on the hot-plate test (Kavaliers and Perrot-Sinal, 1996; Kavaliers et al., 1997). Species differences between binding and coupling properties of the mouse versus human NOP1 receptor have demonstrated, and the authors suggested that this, in combination with possible species differences in receptor reserve, may account for some of the contradictions in the established literature (Burnside et al., 2000).
We believe that intraspecies genotypic differences (i.e., strain differences) may be more useful as an explanation of some of the inconsistencies seen thus far. As noted above, Rossi and colleagues (1997) observed a modest analgesia from supraspinal OFQ/N in CD-1 mice that was not seen in outbred Swiss-Webster or inbred BALB/cJ mice. This finding inspired Mogil et al. (1999a) to examine the effects of supraspinal OFQ/N, and supraspinal injections themselves, on thermal nociception in six mouse strains: outbred CD-1 and Swiss-Webster mice, and inbred AKR/J, BALB/cJ, C3H/HeJ, and CBA/J mice. Intracerebroventricular injections per se produced significant increases in 47°C tail-withdrawal latency at 15-min postinjection in four of six strains. This strain dependence is not entirely surprising, since strain-dependent activation of the HPA axis following systemic needle injection has been demonstrated (Ryabinin et al., 1999). OFQ/N reversed the injection-related analgesia in two of the strains, Swiss-Webster and BALB/cJ. In no strain, however, was statistically significant hyperalgesia or analgesia observed, although a strong trend toward the latter was obtained in CBA/J mice (Mogil et al., 1999a). These findings may help explain why only some investigators have observed the phenomenon of vehicle injection stress-induced analgesia in the mouse (e.g., Mogil et al., 1996a;Suaudeau et al., 1998), whereas others have not (Rossi et al., 1997; e.g., Calo' et al., 1998). Also of interest is the fact that strain differences in OFQ/N immunoreactivity have been demonstrated between DBA/2J and C57BL/J mice (Ploj et al., 2000), two strains with highly divergent nociceptive and analgesic sensitivities (see Mogil et al., 1996d; Mogil, 1999). However, those differences are found in the frontal cortex and hippocampus only (C57BL/6J > DBA/2J), and thus it is difficult to see their direct relevance to acute nociception.
Evidence for important sex differences in the mediation and opioid modulation of nociception continues to mount (Berkley, 1997; Kest et al., 2000; Mogil et al., 2000a). However, sexually dimorphic OFQ/N functioning is unlikely to explain contradictions in the existing literature, as this literature has overwhelmingly employed male subjects (see Tables 2 and 3). Furthermore, in those few studies that have tested both sexes (Grisel et al., 1996; Hao et al., 1998; Mogil et al., 1996a,b, 1999a; Tian et al., 1997a,b), no sex differences in the effects of OFQ/N were reported.
5. Dose Dependence.
The OFQ/N dose employed may have a dramatic impact on the effect of the peptide. With respect to supraspinal OFQ/N, both anti-analgesic and hyperalgesic effects have been observed over broad dose ranges (125,000- and 2,000-fold, respectively). By contrast, analgesia has only been observed at high doses (≥5.5 nmol). In many of the studies where anti-analgesiaand hyperalgesia were observed, the former was obtained with lower doses than the latter (Zhang et al., 1997; Zhu et al., 1997,1998; Wang et al., 1999a,c; Citterio et al., 2000).
Spinal and peripheral OFQ/N reveals an even more obvious dose dependence. Extremely low doses (attomolar to picomolar range) of the peptide produce SBL behaviors and hyperalgesia/allodynia, whereas higher doses (picomolar to nanomolar) produce analgesia. In the studies of Xu and colleagues (1996, 1999b), the facilitation of the flexor reflex by low doses of OFQ/N was weak, brief, and unreliable, whereas the inhibition of the reflex caused by higher doses was prolonged and robust. The studies of the Japanese groups, however, have consistently documented inverted U-shaped dose-response relationships between OFQ/N and pronociceptive outcomes, with SBL behaviors, flexor reflex facilitation, and thermal hyperalgesia peaking in the low femtomolar range (Hara et al., 1997; Inoue et al., 1999; Sakurada et al., 1999b). This bell-shaped dose-response curve contrasts with that of other pain-producing peptides, such as substance P. However, Inoue and colleagues (1999) explain the biphasic action of OFQ/N entirely in terms of substance P. At very low doses, OFQ/N is likely stimulating nociceptive primary afferents containing substance P, causing the release of the latter peptide. At higher doses, OFQ/N is still causing the release of substance P but now is able to activate spinal NOP1 receptors (via Gi/o). The activation of NOP1 receptors in the spinal cord produces analgesia via the inhibition of postsynaptic substance P-mediated actions (Inoue et al., 1999). Ito's laboratory has used dose relationships to dissociate mechanisms underlying OFQ/N mechanical allodynia (seen only from 0.55 pmol to 0.28 nmol) and OFQ/N thermal hyperalgesia (seen at all doses tested above 2.75 amol) (Okuda-Ashitaka et al., 1996; Hara et al., 1997). Both phenomena are inhibited by morphine and neonatal capsaicin treatment and mediated by glycine receptors (Hara et al., 1997; Minami et al., 2000). However, only the allodynia is sensitive to blockade by antagonists of the glutamate receptor-nitric oxide pathway and by prostaglandin D2 (PGD2) (Hara et al., 1997; Minami et al., 1997, 2000). In contrast, only the OFQ/N hyperalgesia is mediated by substance P (Minami et al., 2000). This same dissociation between allodynia and hyperalgesia was demonstrated by this group for prostaglandin E2(PGE2) (Minami et al., 1996).
A dose-dependent relationship exists between OFQ/N and release of the endogenous opioid, enkephalin, in the guinea pig myenteric plexus (Gintzler et al., 1997). This in vitro preparation contains strikingly similar proportions of μ, κ, and δ receptors compared with the CNS (Gyang et al., 1964), and has been used to investigate the release of enkephalin by μ-receptor agonists (Glass et al., 1986). With blockade of classical opioid receptors with naloxone, OFQ/N at low concentrations (1–10 nM) inhibited the electrically stimulated release of met-enkephalin by 40%. At higher concentrations (100–1000 nM), OFQ/N facilitated enkephalin release by up to 50% (Gintzler et al., 1997). Note that this biphasic pattern is exactly the opposite of that characterizing the μ-opioid agonist, sufentanil, which facilitates enkephalin release at low doses and inhibits it at higher doses (Xu et al., 1989). This dose-dependent pattern of enkephalin release could be invoked to explain inconsistencies in both the supraspinal and spinal compartments, since enkephalin plays an important role in nociceptive processing at both levels (Millan, 1986). The decreased enkephalin release produced by low concentrations of OFQ/N would tend to be pronociceptive, perhaps partially accounting for the hyperalgesia/allodynia seen after low dose spinal injections and the anti-analgesia/anti-hyperalgesia seen after supraspinal injections. The increased enkephalin release produced by high concentrations of OFQ/N would tend to be antinociceptive, perhaps accounting for the high dose spinal analgesia seen by many and/or the supraspinal analgesia seen by Pasternak's laboratory. These possibilities are purely speculative, of course, since OFQ/N has not yet been shown to release enkephalin in either the spinal cord or the brain. Most intriguing is the fact that while OFQ/N was shown to modulate the evoked release of enkephalin, it did not affect the basal release of the peptide, prompting the authors to conclude that “the hyperalgesic actions of centrally administered nociceptin should be expected to be particularly robust when pain thresholds are elevated due, in part, to augmented enkephalin neurotransmission” (Gintzler et al., 1997). One way of elevating pain thresholds via augmented enkephalin neurotransmission is to expose animals to environmental stressors (e.g., Christie et al., 1981; Schmidt et al., 1991; but see Konig et al., 1996; Mizoguchi et al., 1997). The blockade of enkephalin release by OFQ/N may thus provide a mechanistic explanation for the behavioral hyperalgesia produced by supraspinal OFQ/N in situations involving stress-induced analgesia (test-related or otherwise).
A final issue related to dose is the possibility that the exogenous administration of OFQ/N at different doses may lead to the differential production of metabolites. Such metabolites may be bioactive and may functionally interact with OFQ/N, changing its apparent effect. This has been explicitly demonstrated by Sakurada et al. (2000), who observed that N-terminal fragments of OFQ/N, specifically OFQ/N(1–7), OFQ/N(1–9), and OFQ/N(1–13), fully blocked the SBL behaviors induced by low OFQ/N doses. OFQ/N(1–13) was actively antagonistic at doses equimolar to OFQ/N, suggesting an endogenous role. In a separate study by the same group, OFQ/N(1–7) was shown to block supraspinal OFQ/N hyperalgesia but not spinal OFQ/N analgesia (Sakurada et al., 1999c). Supraspinal OFQ/N(1–7) and OFQ/N(1–11) produce analgesia in mice without evidence of hyperalgesia (Rossi et al., 1997) and these fragments were unable to reverse morphine analgesia (King et al., 1998). Finally, the hexapeptide OFQ/N(1–6) exhibits a biphasic pattern of responses on the hot-plate test, but not the hot water tail-withdrawal test, whereby a short-lived analgesia was replaced by a modest hyperalgesia (Suder et al., 1999). Intriguingly, this pattern is entirely opposite of the pattern observed by Rossi and colleagues (1997) using the full peptide. The hyperalgesia but not the analgesia was reversed by noncompetitive antagonists at the NMDA receptor: MK-801, a pore blocker, and L-701,324, an allosteric glycine site blocker (Suder et al., 1999). Experiments in the land snail also implicate a role for NMDA receptors in OFQ/N hyperalgesia (Kavaliers et al., 1997).
OFQ/N fragments appear to have poor affinity for NOP1 (Dooley and Houghten, 1996; Mathis et al., 1997). Even though OFQ/N(1–11) labels NOP1 sites modestly well (K i ∼50 nM), its affinity does not compare to OFQ/N (K D∼50 pM), so competitive antagonism of NOP1receptors is unlikely. These peptides do affect cAMP accumulation, however, suggesting the existence of functionally heterogeneous receptors related to NOP1, possibly via alternative splicing (see Section VI.) (Mathis et al., 1997). Given that the vast majority of radiolabeled [Tyr14]OFQ/N administered supraspinally is metabolized within 15 min, it is possible that OFQ/N analgesia, which exhibits a delayed onset, may be due entirely to its conversion to bioactive metabolites (Rossi et al., 1997; Suder et al., 1999).
6. Opioid Tone.
A recent study by Lutfy and Maidment (2000)demonstrated that a hyperalgesic effect of high doses of OFQ/N (15 or 30 nmol) could be revealed by pharmacological blockade of μ receptors by naloxone or CTOP. These investigators, using the hot-plate test, observed no effect on nociception of OFQ/N unless μ receptors were previously blocked by μ antagonists. They argue that in addition to OFQ/N exerting an anti-opioid effect, μ receptors might be regarded as anti-OFQ/N, counteracting a pronociceptive action of OFQ/N. That is, the failure to observe OFQ/N hyperalgesia is due to a counteracting opioid “tone”, and not to stress-induced analgesia. The tone could not exist prior to the OFQ/N injection, or else naloxone pretreatment would have produced an apparent hyperalgesia, which was not seen (Lutfy and Maidment, 2000). Therefore, the OFQ/N injection must have released classical endogenous opioids acting at μ receptors. Since there was no evidence in their study of injection-related analgesia, one must conclude that OFQ/N itself caused the release of the opioid, as has been shown to be the case in the guinea pig myenteric plexus at high OFQ/N concentrations (Gintzler et al., 1997) (see Section VIII.D.5.). OFQ/N has also been shown to cause the release of stress hormones (ACTH and corticosterone) after supraspinal injection (Devine et al., 2001), suggesting another mechanism by which endogenous opioids may be indirectly released. If this result is replicated, it may represent a major advance in our understanding of OFQ/N actions. It still remains unclear, however, how i.c.v. OFQ/N could activate a μ receptor-mediated analgesia and simultaneously exert anti-analgesic actions at the same doses. This observation may be consistent with the earlier report showing both an opioid antagonist-sensitive OFQ/N analgesia, perhaps mediated by the release of endogenous opioids, and hyperalgesia (Rossi et al., 1997).
E. Effects of Other NOP1 Receptor Agonists
The actions of a number of peptidergic ligands other than OFQ/N with affinity for the NOP1 receptor have been examined to shed light on the role of the system in nociception. For example, the amide form of OFQ/N, NCNH2, binds to NOP1 receptors with equal or greater affinity than the natural peptide (Calo' et al., 2000a). Calo' et al. (1998)observed an identical and equimolar hyperalgesic and anti-analgesic effect of supraspinal NCNH2 compared with OFQ/N on the hot water tail-withdrawal assay. Bertorelli et al. (1999)observed supraspinal NCNH2, but not OFQ/N, hyperalgesia in a rat arthritis model using complete Freund's adjuvant on the test of Hargreaves et al. (1988) in both the arthriticand the contralateral hindpaw. This finding points out the utility of using stabilized derivatives with reduced susceptibility to peptidases, like NCNH2, as functional probes (Calo' et al., 2000a). NC(1–13)NH2 is another amidated fragment retaining full agonist potency when injected supraspinally. In contrast, NC(1–9)NH2 was entirely without effect (Calo' et al., 1998). The hexapeptide ac-RYYRWK-NH2, identified from a combinatorial library (Dooley et al., 1997), displays full agonist properties in behavioral assays (Berger et al., 2000), but so far its effect on nociception has not been reported.
Ro 64–6198 [(1S,3aS)-8-(2,3,3a,4,5, 6-hexahydro-1H-phenalen-1-yl)-1-phenyl-1,3,8-triaza-spiro[4.5]decan-4-one] is a nonpeptidergic NOP1 receptor agonist with full activity. It is particularly appealing since it can be used systemically due to its ability to traverse the blood-brain barrier. Despite its OFQ/N-like anxiolytic effects (see X.B), Ro 64–6198 did not alter thermal or mechanical nociceptive thresholds in the same dose range (Jenck et al., 2000). Its effects on other nociception-related phenomena have not yet been tested. Finally, in vitro data raise the possibility that the clinically important opiate, buprenorphine (Temgesic), is a partial agonist of NOP1 receptors (Bloms-Funke et al., 2000). Although its lack of selectivity among the opioid and NOP1 receptors precludes its use as a research tool to explore the pharmacology of the NOP1receptors, buprenorphine may have interactions with NOP1 receptors that may be of considerable value in explaining the complexities of buprenorphine's pharmacology, such as biphasic or even triphasic dose-response curves and anti-opioid activity (Dum and Herz, 1981; Pick et al., 1997).
F. Phenotypes of Knockout Mice
The absence of a NOP1 receptor antagonist until recently, led researchers in the OFQ/N field to a useful alternative to pharmacology, gene deletion. Transgenic “knockout” mice lacking functional expression of the NOP1receptor gene (Oprl1; chromosome 2, 110 cM) and the ppOFQ/N gene (Npnc1; genomic location unknown) have been constructed and tested for pain-related phenotypes.
The NOP1 receptor mutants were developed first, and as expected, were insensitive to OFQ/N. Neither supraspinal OFQ/N hyperalgesia nor spinal OFQ/N flexor reflex facilitation were found in the knockouts (Nishi et al., 1997; Noda et al., 1998; Ueda et al., 2000). Far more important, however, was the attempt to determine whether basal nociceptive thresholds or analgesic sensitivity to opioids were altered in these animals. The answer in both cases appears to be no; knockout mice are equivalently sensitive to their wildtype counterparts on the thermal tail-flick and hot-plate tests, the mechanical tail-clip test, the electric foot-shock test and the acetic acid abdominal constriction test (Mamiya et al., 1998; Nishi et al., 1997; Ueda et al., 1997; 2000). In addition, morphine's analgesic potency was unchanged in knockout animals following systemic injection over a range of doses (Nishi et al., 1997; Noda et al., 1998; Ueda et al., 1997; 2000). Knockout studies of this type (see Mogil and Grisel, 1998 for review), can be very useful, but always face the potential problem of compensation by other genes. In these NOP1 receptor knockout studies, the authors concluded that the lack of differences between wildtype and knockout mice indicates that NOP1 receptors are not essential for the determination of nociceptive threshold (Nishi et al., 1997). This implies the absence of a “basal tone” (seeDiscussion in sections VIII.D.3 and VIII.D.5 above), much like the enkephalins have little “basal tone”, as shown by the limited actions of naloxone in naı̈ve animals. This leaves the important question of the status of stress-induced analgesia in these mutants.
Two separate groups have generated mice lacking the gene for the precursor of OFQ/N, ppOFQ/N. Koster and colleagues (1999) reported a decreased basal sensitivity on the radiant heat tail-withdrawal (i.e., classic tail-flick) test in the knockout animals. This difference between the ppOFQ/N and the NOP1 knockout mice might reflect differences between the models: a) by eliminating ppOFQ/N, all the peptides within the precursor also are lost, including nocistatin and OFQ2/NocII peptides; b) receptors other than NOP1 may mediate the effects of OFQ/N and related peptides; and c) different embryonic stem cell lines were used in the two projects, with concomitantly different genetic backgrounds (Simpson et al., 1997; see Mogil and Grisel, 1998). A fascinating aspect of this study is restriction of the decreased sensitivity of ppOFQ/N mutants to male mice that were group housed; isolated knockout male mice were equivalently sensitive to isolated wildtypes. This is purported to be a stress-related effect, since knockouts were found to be diminished in their ability to adapt to stress (Koster et al., 1999) (seeSection X.B.). Essentially, the authors argue that mice lacking OFQ/N and other products of the ppOFQ/N gene are tonically stressed, and thus exhibiting tonic stress-induced analgesia. The implication, therefore, is that OFQ/N may serve endogenously to ameliorate stress, and thus stress-induced analgesia.
Although this is a very attractive and powerful hypothesis, its implications for understanding OFQ/N′s effects on nociception are complicated by findings from Pintar's laboratory, in which independently derived ppOFQ/N knockouts exhibited anincreased sensitivity in the hot water tail-withdrawal test (Chen et al., 1999). The contrasting phenotypes are not due to any differences between the closely related radiant heat and hot water versions of the assay, since the increased sensitivity of the Pintar mutants is seen in both versions (J. S. Mogil, unpublished data).
G. Effects of NOP1 Down-Regulation or Blockade
Another major approach to studying the role of OFQ/N and NOP1 receptors in nociception has been to down-regulate available receptor sites with antisense treatment or block them acutely with antagonists. This strategy has a number of advantages compared with the exogenous administration of drugs in interpreting the constitutive role of OFQ/N.
1. Antisense Studies.
Antisense studies can be used alone to explore the tonic activity of OFQ/N systems or in conjunction with a drug to confirm the specificity of the drug's actions. In the opioid field, investigators typically have used short oligodeoxynucleotide probes consisting of approximately twenty bases targeting a region of the mRNA near the translational start site. However, antisense probes can effectively down-regulate proteins virtually anywhere along the mRNA, provided an appropriate sequence is available (Kolesnikov et al., 1997; Pasternak and Pan, 2000; Standifer et al., 1994). This ability to target individual exons has led to the concept of antisense mapping, which has proven valuable in the elucidation of the role of alternative splicing (Pasternak and Standifer, 1995; Kolesnikov et al., 1997).
Earlier, the role of antisense mapping in classifying the NOP1 receptor was briefly discussed (seeSection IV.). The initial cloning studies with the NOP1 receptor pointed out the poor affinity of traditional opioids for this site and raised questions regarding its pharmacological significance. In these initial antisense studies against NOP1, six different antisense probes targeting the second and third coding exons all blocked the analgesic actions of naloxone benzoylhydrazone, strongly implying a role of NOP1 in κ3 analgesia (Pan et al.,. 1994; 1995). However, the inactivity of antisense against the first coding exon, as well as other factors, also pointed out that NOP1 was not the same as the κ3 opioid receptor. Thus, the use of antisense approaches must be interpreted cautiously. Some probes may be active while others based upon a different exon of the same gene are not. Although subtle, these issues must always be considered when assessing antisense paradigms. Optimally, each exon should be individually targeted, but this is not always feasible.
Antisense mapping NOP1 revealed interesting patterns for OFQ/N analgesia and hyperalgesia (Rossi et al., 1997). Antisense targeting exon 1 blocks OFQ/N hyperalgesia in the radiant heat tailflick assay, as well as its anti-opioid actions, while antisense oligodeoxynucleotides targeting the second and third coding exons are inactive (Rossi et al., 1997; King et al., 1998). Zhu and colleagues (1996; 1997) also demonstrated the utility of a NOP1 antisense against the first coding exon in blocking the anti-opioid effect of OFQ/N on morphine and electroacupuncture analgesia. Conversely, antisense based upon the first coding exon of NOP1 was inactive against OFQ/N analgesia, while probes targeting the second and third coding exons effectively blocked OFQ/N analgesia. Thus, these mapping studies imply that both OFQ/N analgesia, hyperalgesia and anti-opioid actions are mediated through receptors generated by the gene producing NOP1 receptors. Yet, it appears that the receptors responsible for analgesia are distinct from those important in hyperalgesia and anti-opioid actions.
One of the seminal OFQ/N papers demonstrated that an antisense oligonucleotide against the first coding exon of the murine NOP1 clone injected daily for four days produced analgesia on the hot-plate test while a missense control was inactive (Meunier et al., 1995). Zhu and colleagues (1996; 1997) replicated this finding on the tail-shock and formalin tests, and also demonstrated the utility of NOP1 antisense in blocking the anti-opioid effect of OFQ/N on morphine and electroacupuncture analgesia.
Finally, in the first study looking at the effects of antisense directed toward ppOFQ/N mRNA, Candeletti and Ferri (2000) demonstrated that repeated intracerebroventricular antisense treatment potentiated morphine analgesia, supporting an anti-opioid role of the peptide.
2. Pharmacological Antagonists.
Both transgenic knockout and antisense approaches have limitations (see, e.g., Mogil and McCarson, 2000), and thus the search for a selective and competitive NOP1 antagonist has been one of the major priorities in this field since 1996. One group found that ς receptor ligands (e.g., carbetapentane, rimcazole) blocked inward potassium currents induced by OFQ/N acting on recombinant NOP1 receptors (Kobayashi et al., 1997), but these compounds are nonselective and of low potency. The κ3 ligand, NalBzoH, has also been proposed as a competitive NOP1 antagonist based on data obtained from transgenic receptor knockouts and one group's success in blocking OFQ/N hyperalgesia, hypolocomotion, and memory impairment with the compound (Noda et al., 1998; Mamiya et al., 1999). However, we have collected data suggesting that NalBzoH does not block supraspinal OFQ/N′s anti-analgesic actions but rather enhances them (J. S. Mogil and J. E. Grisel, unpublished data) and in biochemical assays NalBzoH has at least partial agonist activity in cyclase assays with the cloned murine NOP1 receptor (Pan et al., 1994, 1995). The dextrorotatory enantiomer of the nonpeptidic δ-opioid ligand, TAN-67, blocks spinal OFQ/N analgesia in two different assays, but this antagonism was probably not competitive (Kamei et al., 1999b). The peptide retro-nociceptin methylester also noncompetitively antagonized OFQ/N in the in vitro guinea pig ileum assay and elicited a naloxone-insensitive analgesia on the tail-pinch test, although it did not affect OFQ/N hyperalgesia (Jinsmaa et al., 2000).
Considerable excitement surrounded the publication of the properties of the pseudopeptide [Phe1ψ(CH2-NH) Gly2]-nociceptin(1–13)-NH2, which selectively antagonized NOP1 receptors in the guinea pig ileum and mouse vas deferens assays (Guerrini et al., 1998). Subsequent studies showed, however, that [Phe1ψ(CH2-NH)Gly2]-nociceptin(1–13)-NH2is a partial agonist with low efficacy, explaining its actions as an antagonist, partial agonist, or even full agonist depending on the assay. In vitro, the peptide is an antagonist in cells expressing low levels of NOP1 receptors and is a partial or full agonist in cells expressing high levels of the receptor (Burnside et al., 2000). In most in vivo assays and all nociceptive tests the peptide shows full agonist properties, producing supraspinal hyperalgesia (Calo' et al., 1998; Bertorelli et al., 1999; Wang et al., 1999b; Candeletti et al., 2000), anti-opioid effects (Calo' et al., 1998; Grisel et al., 1998), and intrathecal analgesia (Xu et al., 1998; Wang et al., 1999b; Candeletti et al., 2000). In an electrophysiological study of spinal cord dorsal horn neurons, [Phe1ψ(CH2-NH)Gly2]-nociceptin(1–13)-NH2inhibited C-fiber evoked responses just like OFQ/N (Carpenter and Dickenson, 1998). Unlike the endogenous peptide, however, the effect was partially naloxone-reversible, prompting the authors to suggest that [Phe1ψ(CH2-NH)Gly2]-nociceptin(1–13)-NH2may have some activity at spinal μ or δ receptors as well.
The next year, the same Italian group identified another peptide in a structure-activity study, [Nphe1]nociceptin(1–13)NH2, with uniformly antagonistic properties in all assays thus far examined (Calo' et al., 2000b; Polidori et al., 2000a; Rizzi et al., 2000). Not only is [Nphe1]nociceptin(1–13)-NH2fully able to reverse the supraspinal hyperalgesic and anti-opioid actions of OFQ/N, but the antagonist can potentiate morphine analgesia (Calo' et al., 2000b; Rizzi et al., 2000). This corresponds to the original prediction by Mogil and colleagues (1996a) regarding the effect of an antagonist once developed, and presages a clinical role for such drugs as adjuncts to opiate pharmacotherapy. Interestingly, whereas [Nphe1]nociceptin(1–13)-NH2very efficaciously potentiated the analgesic effects of morphine given supraspinally, it was only marginally effective upon systemic morphine analgesia unless animals were previously rendered tolerant to morphine (Rizzi et al., 2000). This finding supports the notion that OFQ/N′s anti-opioid actions are exerted supraspinally and also contributes to the evidence implicating OFQ/N in the expression of morphine tolerance (see Section X.C.). Less clear is the critical issue of whether [Nphe1]nociceptin(1–13)-NH2can produce analgesia per se. In one study, using the moderately noxious 48°C hot water tail-withdrawal test, a significant (up to 150% increase in latencies), dose-dependent (3–30 nmol), but relatively short-lasting, analgesic effect was demonstrated (Calo' et al., 2000b). In another study, using the highly noxious 55°C test, no analgesia could be elicited from a 30-nmol dose of the antagonist (Rizzi et al., 2000).
Two nonpeptidic NOP1 antagonists were developed and tested for their effects on nociceptive responses. JTC-801 [N-(4-amino-2-methylquinolin-6-yl)-2-(4-ethylphenoxymethyl)benzamide hydrochloride], having at least 12.5-fold selectivity over other opioid receptor types, blocked spinal OFQ/N-induced allodynia and produced naloxone-insensitive, but not dose-dependent, analgesia in the 55°C hot-plate and formalin tests (Shinkai et al., 2000). Another compound, J-113397 [1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethol-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one] showed much higher selectivity (>350-fold) over classical opioid receptor types (Ozaki et al., 2000a,b). Injected subcutaneously in mice, which minimizes the potential of stress effects, J-113397 dose dependently reversed supraspinal OFQ/N hyperalgesia in the radiant heat tail-withdrawal test (Ozaki et al., 2000a). However, even though minimally noxious parameters were used, as illustrated by the relatively long baseline latencies of approximately 9 s, no analgesia from this compound was detected at doses up to 30 mg/kg.
Thus, the question of whether tonic activation of NOP1 receptors by OFQ/N contributes to a nociceptive threshold-controlling tone remains unanswered, as summarized in Table 8, despite the unique advantages of transgenic knockouts, antisense, and especially pharmacological antagonist models. For now, at least, the mystery of the true endogenous role of OFQ/N in nociception has failed to yield to simple explanations and technological advances.
Evidence for and against a role for the OFQ/N/NOP1 system in the tonic control of nociceptive thresholds under basal conditions
H. Mechanisms of Orphanin FQ/Nociceptin Actions: Ubiquitous Cellular Inhibition as a Unifying Hypothesis?
Wide interest in this novel neurotransmitter was engendered by the seeming incongruity between its cellular actions that mimicked classical opioids and its behavioral effects, at least supraspinally, that opposed those of classical opioids. Just like μ, κ, and δ receptors, activation of NOP1 receptors by OFQ/N is associated with inhibition of cAMP formation (via Gi/Go-mediated intracellular signaling), closure of voltage-gated N-type calcium channels, enhancement of an inwardly rectifying potassium conductance, and ultimately, reduction of neuronal excitability (i.e., cellular inhibition) (Hawes et al., 2000). The cellular neurophysiological actions of OFQ/N are also identical to those of classical opioid agonists, including the inhibition of transmitter release in the spinal cord (Moran et al., 2000). Such properties are what led both original investigations to search for behavioral analgesia from OFQ/N; it was considered a paradox that they demonstrated supraspinal hyperalgesia instead. Of course, this is only paradoxical if one fails to consider the potential of interactions at the level of neural circuits. This type of analysis has been used successfully, for example, to explain the anti-μ-opioid actions of κ-opioid agonists like dynorphin (Pan et al., 1997). It is to such a systems-level hypothesis that we now turn, although it should be noted that the heterologous desensitization of μ-opioid receptors by NOP1 activation in CHO cells (Hawes et al., 1998) might in theory provide a purely cellular explanation of OFQ/N′s anti-opioid actions. This is somewhat unlikely to be relevant in vivo, however, since μ and NOP1 receptors are not colocalized in nociception-relevant loci (Schulz et al., 1996;Monteillet-Agius et al., 1998).
Heinricher and colleagues (1997) investigated the electrophysiological and behavioral effects of OFQ/N in the nucleus raphé magnus of the RVM of lightly anesthetized rats. The RVM is a brain stem locus critical for analgesia; it receives afferent input from the PAG and sends a nociception-modulating outflow to the dorsal horn of the spinal cord (Basbaum and Fields, 1984; Fields et al., 1991). Previous work has demonstrated the existence of three types of neurons in the RVM: ON cells, which fire immediately prior to the occurrence of nociceptive reflexes (i.e., tail-withdrawals from noxious heat); OFF cells, which pause their firing immediately prior to nociceptive reflexes; and NEUTRAL cells, which do not change their firing (see Fields et al., 1991). Opioid analgesia is mediated by the direct inhibition of ON cells by the opioids that then disinhibit (i.e., excite) OFF cells, whose firing prevents the tail-withdrawal reflex (Heinricher et al., 1994). Thus, an anti-opioid action of OFQ/N could result by the peptide either: a) preventing classical opioid inhibition of ON cells, or b) directly inhibiting OFF cells.
Not surprisingly, given previous in vitro data (Connor et al., 1996;Vaughan and Christie, 1996; Vaughan et al., 1997), OFQ/N profoundly suppressed the firing of all three cell classes in the RVM (Heinricher et al., 1997). Iontophoretically applied OFQ/N blocked the inhibition of tail-withdrawal reflexes by DAMGO applied to the same site, an anti-opioid action, but had no effect when applied alone. The parsimonious explanation of these findings is that OFQ/N suppressed OFF cell firing, preventing opioid disinhibition from activating those same neurons to produce analgesia (Heinricher et al., 1997). ON cell firing would have been inhibited by both OFQ/N and DAMGO, with no net change in effect. Note that this explanation of OFQ/N′s anti-opioid action differs from an analogous study of the anti-opioid peptide, CCK, which was found to specifically attenuate the opioid activation of OFF cells without affecting ON cell firing (Heinricher et al., 2001).
A subsequent study by Pan et al. (2000) replicated the inhibition of ON and OFF cells by OFQ/N, as measured intracellularly with whole-cell patch clamp, and showed that OFQ/N blocked the electrophysiological and analgesic effects of μ and κ agonists. They performed an intriguing additional experiment, however. ON cells have been implicated in opioid withdrawal-associated hyperalgesia (Bederson et al., 1990), and these investigators demonstrated that during naloxone-precipitated acute opioid withdrawal, OFQ/N iontophoretically applied into the RVM produced an anti-hyperalgesic (i.e., analgesic) action (Pan et al., 2000). Thus, under different behavioral conditions, two opposing effects could be elicited from OFQ/N: anti-analgesia and anti-hyperalgesia. It is quite unlikely that those observing supraspinal OFQ/N analgesia were testing opioid-dependent animals, of course, but the authors suggest that other factors may alter behavioral states in an analogous way, leading to qualitatively different OFQ/N effects (Pan et al., 2000).
OFQ/N effects on RVM neurons are not a full explanation, especially since OFQ/N injected into the RVM did not block systemic morphine analgesia in the radiant heat tail-withdrawal assay (Heinricher et al., 1997). The RVM is a sufficient, but not necessary, substrate for opioid analgesia since lesions of this structure do not eliminate systemic opioid analgesia (Proudfit, 1980). It is interesting, therefore, thatMorgan and colleagues (1997) demonstrated that OFQ/N microinjected into the PAG along with either morphine or kainic acid was able to reverse both their analgesic actions. OFQ/N inhibits virtually all neurons studied in a PAG tissue slice (Vaughan et al., 1997), and thus these authors arrived at a conclusion mirroring that of the RVM workers: OFQ/N produced anti-analgesic effects by inhibiting analgesia-producing neurons (presumably PAG output neurons projecting to the RVM) downstream from the opioid-sensitive neurons (Morgan et al., 1997). Consistent with these results is a recent electrophysiological study showing that microinjection of OFQ/N into the PAG increased C-fiber evoked responses and facilitated postdischarge in spinal wide dynamic range neurons (Yang et al., 2001).
Thus, there may really be no paradox at all between the opposing actions of OFQ/N in the spinal versus supraspinal compartments. The analgesic actions of OFQ/N in the spinal cord can be attributed to the direct inhibition of nociceptive transmission, actions similar to those of the classical opioids. By contrast, the supraspinal circuitry, at least in the RVM and PAG, appears to be set up in such a way that opioid analgesia requires disinhibition. Any compound, like OFQ/N, producing ubiquitous cellular inhibition will thus act to oppose opioid analgesia. Ultimately, then, the differential behavioral actions between supraspinal OFQ/N and classical opioids can be attributed solely to the fact that their respective receptors are located on functionally different groups of neurons.
The explanation detailed above has been challenged by the very recent findings of Rady and colleagues (2001), who argue that OFQ/N′s anti-analgesic actions can be explained by the ability of supraspinally injected OFQ/N to activate a descending anti-analgesic system that releases PGE2 in the spinal cord. Their study was the first to demonstrate that supraspinal OFQ/N also blocks spinal morphine analgesia, which cannot be easily explained by a mode of action confined to the supraspinal compartment. Furthermore, they showed that the cyclooxygenase inhibitor, indomethacin, fully reversed this anti-analgesic action of OFQ/N, and that PGE2 could mimic OFQ/N′s anti-analgesia (Rady et al., 2001). Both OFQ/N′s and PGE2's anti-analgesia could be reversed by PGD2, and evidence was provided implicating spinal EP1receptors in the effect. Although the involvement of PGE2 and EP1 receptors and the blockade by PGD2 is reminiscent of Ito's laboratory's work regarding spinal OFQ/N allodynia (Minami et al., 1997) (see Section VIII.D.5.), it appears that different mechanisms are responsible for supraspinal anti-analgesia, since it is very unlikely that supraspinal OFQ/N releases spinal OFQ/N (Rady et al., 2001).
IX. Effects of Related Peptides on Pain
OFQ/N is not the only bioactive peptide derived from the ppOFQ/N gene. We now turn to a consideration of the actions of the other major maturation products, nocistatin and OFQ/NocII, in hopes that they may shed light on the elusive role of this system in nociceptive modulation.
A. Nocistatin
Nocistatin originally was reported to reverse, in a dose-dependent manner, the allodynia and hyperalgesia produced by low doses of spinal OFQ/N or PGE2 (Okuda-Ashitaka et al., 1998). An endogenous role for the peptide was suggested by the further demonstration that the inverted U-shaped dose-response curve for spinal OFQ/N allodynia could be shifted to the left by 500-fold with nocistatin antibody. Although only the data obtained with bovine nocistatin were presented, related mouse, rat, and human sequences were also reported to inhibit OFQ/N and PGE2 allodynia (Minami et al., 1998; Okuda-Ashitaka et al., 1998). Just as OFQ/N appears to functionally antagonize the analgesic actions of opioid peptides, nocistatin appeared to functionally antagonize the actions of OFQ/N. This conclusion is further supported the inability of nocistatin to compete binding to the NOP1 receptor.
Further research reveals a more complex interaction between nocistatin and OFQ/N. Supraspinal nocistatin dose-dependently blocked supraspinal OFQ/N′s blockade of morphine analgesia, despite having no effect on basal radiant heat tail-withdrawal latencies or morphine analgesia itself, further supporting the concept that nocistatin is a functional OFQ/N antagonist (Zhao et al., 1999). However, nocistatin does not oppose all of OFQ/N′s effects. Although spinal nocistatin reversed the hyperalgesic action of spinal OFQ/N on the tonic phase of the 1% formalin test in mice, it was unable to reverse an analgesic action of OFQ/N on the 2% formalin test in mice (Nakano et al., 2000) and 5% formalin test in rats (Yamamoto and Sakashita, 1999a). Thus, in these assays nocistatin appeared to be ineffective against analgesic actions of OFQ/N.
Nocistatin interacted with OFQ/N in altering the flexor reflex of spinalized rats in a highly complex manner, including an enhancement of the facilitatory effect of low dose OFQ/N (Xu et al., 1999b). Although nocistatin blocks OFQ/N′s inhibition of potassium-evoked glutamate release from rat brain slices (Nicol et al., 1998), a number of other non-nociceptive actions of OFQ/N are not reversed by nocistatin (Okuda-Ashitaka and Ito, 2000).
The effects of nocistatin on nociceptive processing also are uncertain. Spinal or supraspinal nocistatin alone has no effect on nociceptive thresholds on the hot-plate, radiant heat tail-withdrawal, or paw-pressure tests (Okuda-Ashitaka et al., 1998; Nakagawa et al., 1999;Zeilhofer et al., 2000; Zhao et al., 1999). Although Nakano et al. (2000) demonstrate non-naloxone-reversible analgesic effects of low doses of spinal nocistatin on both phases of the formalin test, higher doses are ineffective against the tonic phase (Yamamoto and Sakashita, 1999a). Also, Zeilhofer and colleagues (2000) demonstrate enhanced formalin responding by low doses of spinal nocistatin. Supraspinally administered nocistatin is anti-hyperalgesic when administered alone (Nakagawa et al., 1999).
Okuda-Ashitaka and Ito (2000) summarize this literature by suggesting that nocistatin antagonizes OFQ/N at low doses but not at higher doses. Given that the effects of OFQ/N itself may also depend on dose (seeSection VIII.D.5.), they further propose that different receptors may be responsible. To date, of course, only one receptor (NOP1) has been found for OFQ/N, although OFQ/N receptor heterogeneity has been suggested (see above), and none has been identified for nocistatin. Recent studies reveal that nocistatin attenuates transmitter release from inhibitory GABAergic and glycinergic interneurons in the dorsal horn whereas OFQ/N blocks excitatory glutamatergic synaptic transmission (Zeilhofer et al., 2000), which provides additional insights into the actions of these peptides.
Nocistatin has been detected in human brain tissue, and in the cerebrospinal fluid of two chronic pain patients, one with chronic low backache and one with knee pain (Lee et al., 1999). The authors suggest, somewhat speculatively, that chronic pain may induce production of nocistatin in spinal cord; this possibility should be investigated in a controlled manner.
B. Orphanin FQ/Nociceptin 2
OFQ/NocII is a heptadecapeptide immediately downstream from OFQ/N. Only three studies have examined its effects on nociception, but the results are not consistent. Rossi and colleagues (1998a)demonstrated dose-dependent analgesia after both spinal and supraspinal injection. Like the supraspinal analgesia obtained by these investigators using OFQ/N, OFQ/NocII analgesia was greatly enhanced by pretreatment with the ς1 receptor ligand, haloperidol. Although supraspinal OFQ/NocII analgesia was naloxone reversible, spinal OFQ/NocII analgesia was not, suggesting the possibility of separate OFQ/NocII receptors in the spinal cord or different types of neuronal circuits (Rossi et al., 1998a). Another group using similar doses and mice was unable to demonstrate supraspinal OFQ/NocII analgesia on the hot-plate, tail-flick, or acetic acid abdominal constriction tests (Florin et al., 1999). In fact, the authors reported a significant decrease in hindpaw licking latencies on the hot plate at several doses. The authors did not interpret this decrease as reflecting hyperalgesia, although, since rearing and jumping latencies were not similarly affected (Florin et al., 1999). The related peptide, NocIII, corresponding to OFQ/NocII with an additional three arginines at the carboxyl terminus, did not appear to have any biological activity whatsoever. Finally, Okuda-Ashitaka and colleagues (1998) reported that bPNP-4, which is identical to OFQ/NocII, possessed nocistatin-like anti-OFQ/N properties when injected spinally. No spinal analgesia was observed from bPNP-4, although much lower doses were used.
Reconciling these studies is not easy. Several issues may prove important. First, differences in the assay might change the apparent levels of analgesia. Second, the use of haloperidol reportedly increases the analgesic activity of OFQ/NocII significantly; it would have been interesting if the Florin group had examined OFQ/NocII in conjunction with haloperidol. Finally, it should be noted that OFQ/NocII is not an easy compound to work with. It is quite hydrophobic, difficult to keep in solution and “sticky”; given time, it will come out of solution and deposit along the walls of containers. Thus, particular care must be taken to ensure that drug is not lost along the walls of tubes, etc., thereby lowering the effective dose injected.
Finally, the sequence of ppOFQ/N raises the possibility of another, longer peptide containing the OFQ/NocII sequence at its amino terminus. This larger peptide, mouse ppOFQ/N160–187, is present within the brain and is analgesic both spinally and supraspinally; its actions are reversed by opioid antagonists (Mathis et al., 2001). The activity of this larger peptide raises the question of whether it is active by itself or only through the further processing to OFQ/NocII. The longer peptide also is active in rats (G. C. Rossi, J. Mathis, G. W. Pasternak, and R. G. Allen, unpublished observations).
X. Involvement of NOP1 in Other Central Nervous System-Mediated Behaviors
It would be naı̈ve to expect that a peptide with such broad localization would have biological actions restricted to the modulation of nociception. Although the lion's share of the studies has been performed by pain researchers, OFQ/N and its receptor have been implicated in any number of other phenomena (see Table 5). Below, we briefly address the role of OFQ/N in several important behavioral domains featuring mediation by the CNS.
A. Locomotor Activity and Reward
One of the seminal investigations of OFQ/N reported a dose-dependent decrease in locomotor activity (i.e., hypolocomotion) when the peptide was given supraspinally (Reinscheid et al., 1995), as measured by an automated, photocell-based activity monitor. This effect was significant only at the 10-nmol dose for both horizontal activity (i.e., walking) and vertical activity (i.e., rearing) and was accompanied by muscular flaccidity, ataxia, and loss of the righting reflex in two-thirds of the mice tested. This finding was the first to suggest that OFQ/N possessed biological activity in vivo and spurred the investigators to conduct their subsequent nociception experiments.
The demonstration of changes in locomotor activity after injection of a compound may have a number of different interpretations. A decrease in movement, especially when associated with the inability to perform coordinated actions (e.g., ataxia), may indicate that the dose being administered is too high, exerting nonspecific effects. On the other hand, a number of psychoactive drugs can produce alterations in the motivation to move or, conversely, the motivation to stay still. Thus, changes in locomotor activity may derive from psychologically important states such as reward/reinforcement, novelty seeking, and fear/anxiety. Finally, a drug's alteration of locomotor activity might indicate that it affects neural circuitry (e.g., in the striatum) involved in the production and regulation of complex motor patterns.
A number of investigators pursued the initial observation of Reinscheid and colleagues (1995). One early study replicated the hypolocomotion, muscular flaccidity, and ataxia in rats and demonstrated that all of these signs showed tolerance after repeated injections of 10 nmol of OFQ/N (Devine et al., 1996b; Walker et al., 1998; Lutfy et al., 2001). In mice, both 1 and 10 nmol of OFQ/N inhibited locomotion in the open field, as did the high-affinity NOP1 ligand, ac-RYYRIK-NH2 (Noble and Roques, 1997; Berger et al., 2000). In one study, the hypolocomotion from 1 nmol lasted only 15 min compared with prolonged effects from 10 nmol; the low-dose effect could be prolonged, however, by treatment with inhibitors of aminopeptidase N and endopeptidase 24.15, enzymes that hydrolyze OFQ/N (Noble and Roques, 1997). The fact that these metabolic inhibitors were ineffective in altering locomotor activity by themselves allowed the authors to conclude that OFQ/N has very low tonic release, at least in regions relevant to this behavior. When microinjected directly into the hippocampus or ventromedial hypothalamus, but not the nucleus accumbens, high doses of OFQ/N (10–25 nmol) significantly decrease locomotor activity (Sandin et al., 1997; Stratford et al., 1997). Finally, in accordance with these findings, the repeated injection of antisense oligodeoxynucleotides directed against ppOFQ/N mRNA produced significant hyperlocomotion in rats (Candeletti and Ferri, 2000).
This field is not entirely without contradictions, however, becauseFlorin and colleagues (1996), using a much wider dose range (1–10,000 ng) of OFQ/N, observed significant and dose-dependent, although short-lasting, increases in locomotor activity, in both horizontal and vertical counts. In addition, they demonstrated an increase in exploratory behavior in the hole-board test produced by supraspinal OFQ/N injection. These increases were not affected by naloxone, but were blocked by antagonists of both the dopamine D1 and D2 receptors (Florin et al., 1996). This same group obtained similar data using OFQ/NocII in separate studies (Florin et al., 1997, 1999).
The conflicting findings can be reconciled by considering dose. Studies reporting hypolocomotion feature OFQ/N doses 2- to 20-times higher than those producing hyperlocomotion, as seen in the studies by Florin et al. (1996). In general, the lower doses of a compound are more likely to elicit physiologically relevant effects than those seen at higher doses. However, it is always possible that effects at higher doses are produced at alternate, lower affinity binding sites. Indeed, Florin and colleagues (1996) suggested that the depression of locomotion at high OFQ/N doses might be explained by nonspecific binding of the peptide to κ receptors (see below). The locomotor-inhibiting effects of 10 nmol of OFQ/N seen in wild-type animals were not seen in the NOP1 knockout mice, confirming the importance of the NOP1 receptor in the effect (Nishi et al., 1997; Noda et al., 1998). However, the authors did not observe an effect of 1 nmol of OFQ/N in any genotype. The fact that the knockout mice did not display any evidence of basal hyperactivity prompted these investigators to conclude that this system is not a major player in the regulation of locomotion (Nishi et al., 1997; Noda et al., 1998).
However, changes in locomotor activity may be caused by the activity of brain reward/reinforcement systems. Many abused psychostimulants increase the locomotor activity of animals that are placed in empty home cages or open fields (Gold et al., 1989). The increased movement is assumed to be reflective of the induction of a reward state in the animal, which, futilely, seeks a reinforcing object to consume (e.g., food, water, sex partner). The mesolimbic dopamine pathway projecting from the ventral tegmental area (VTA) to the nucleus accumbens has long been known to play a crucial, although controversial, role in the neural processing of reward (Wise and Bozarth, 1987; Berridge and Robinson, 1998), and classical opioid peptides acting at μ and δ receptors can potently release dopamine in the nucleus accumbens (Devine et al., 1993). In contrast, κ-selective compounds including dynorphin are aversive when administered centrally (e.g., Mucha and Herz, 1985) and inhibit dopamine release in the nucleus accumbens (Spanagel et al., 1992; Devine et al., 1993). Struck by the fact that endorphins and enkephalins stimulate locomotor activity whereas dynorphin inhibits it (Chaillet et al., 1983), Murphy and colleagues (1996) reasoned that the reported hypolocomotor actions of OFQ/N may reflect an inhibition of mesolimbic dopamine release. Indeed, they demonstrated that intracerebroventricular or VTA-injected OFQ/N produced a dose-dependent inhibition of dopamine outflow in anesthetized rats as measured by microdialysis (Murphy et al., 1996;Murphy and Maidment, 1999). This effect was blocked by the GABAA antagonist, bicuculline, suggesting its mediation by the same GABAergic interneurons in the VTA that are affected by opioids (Johnson and North, 1992). In subsequent studies in freely moving rats, supraspinal OFQ/N did not produce similar effects on basal dopamine levels, but it completely abolished morphine-induced increases in dopamine release in the (reward-relevant) shell of the nucleus accumbens but not in the (reward-irrelevant) caudate nucleus (Di Giannuario et al., 1999; Di Giannuario and Pieretti, 2000).
These microdialysis findings strongly suggested that OFQ/N might block the rewarding properties of opiates and other drugs of abuse. Accordingly, OFQ/N (3–10 nmol but not 30 nmol) abolished the acquisition of a morphine conditioned place preference (CPP) (Murphy et al., 1999), in which animals are conditioned to associate a drug with environmental cues and subsequently assessed for their altered preference for that environment. In studies by another laboratory, even lower OFQ/N doses abolished morphine CPP (Ciccocioppo et al., 1999,2000). The effect of OFQ/N was independent of the peptide's ability to impair spatial learning, and unrelated to the development of sensitization processes (Ciccocioppo et al., 2000). Unlike other anti-opioids (e.g., naloxone and dynorphin) that are aversive per se, OFQ/N has no intrinsic motivational effects since it induces neither a CPP nor a conditioned place aversion when given alone over a wide dose range (Devine et al., 1996a ; Ciccocioppo et al., 1999, 2000).
However, the generalized conclusion that OFQ/N opposes the rewarding properties of all opioids is countered by yet another study. Walker and colleagues (1998) observed that OFQ/N failed to affect the intravenous self-administration of heroin in the rat. Self-administration is clearly the “gold standard” of reward/reinforcement paradigms, but recent evidence points to a dissociation of the opioid mechanisms underlying heroin and morphine analgesia (Rossi et al., 1996a; Brown et al., 1997; Schuller et al., 1999; Walker et al., 1999).
Given the considerable literature implicating the involvement of opioid and mesolimbic dopamine systems in alcohol addiction (Herz, 1997; Cowen and Lawrence, 1999), it is not surprising that OFQ/N affects the rewarding properties of ethanol. Using rats artificially selected to prefer alcohol (Marchigian Sardinian line), daily injections of intracerebroventricular OFQ/N attenuated consumption in a subchronic (7-day) protocol in which rats were offered 10% ethanol for 2 h per day (Ciccocioppo et al., 1999). Blood alcohol levels were unaffected by OFQ/N injection, ruling out a pharmacokinetic explanation of the effect. The peptide also blocked the acquisition of an ethanol CPP in these animals (Ciccocioppo et al., 1999). However, in an acute protocol, a single intracerebroventricular injection of OFQ/N increased ethanol consumption. These seemingly contradictory findings can be reconciled by assuming that the rat attempts to compensate for the blockade of reward in the acute situation by increasing consumption of the reinforcer, but abandons this futile strategy over the longer term. In related studies, antisense knockdown of the NOP1 receptor increased ethanol-induced hyperlocomotion in rats (Pohorecky et al., 1998) and OFQ/N blocked stress-induced ethanol self-administration, a model of relapse (Martin-Fardon et al., 2000). Of course, this latter finding may be better explained by OFQ/N′s well documented anxiolytic actions (Jenck et al., 1997) (see Section X.B.) than by a blockade of ethanol reward.
In the only two published studies (Lutfy et al., 2001; Narayanan and Maidment, 1999) of the direct effect of OFQ/N on psychostimulant (e.g., amphetamine, cocaine) reward, the peptide inhibits cocaine-induced hyperlocomotion. Contrary to predictions, the sensitization of cocaine-induced locomotor activation was not affected by OFQ/N, but OFQ/N induced a sensitized response to subsequent cocaine injection in naı̈ve animals (Narayanan and Maidment, 1999). In the study byLutfy and colleagues (2001), the attenuation of cocaine hyperlocomotion by OFQ/N was accompanied by an attenuation of cocaine-induced dopamine release in the nucleus accumbens. OFQ/N also blocked hyperlocomotion from the direct dopamine receptor agonist, apomorphine, however, which suggests an additional mechanism of action independent of extracellular dopamine levels. Finally, in contrast to the blockade of footshock stress-induced ethanol self-administration by OFQ/N, the peptide did not block stress-induced cocaine self-administration (Martin-Fardon et al., 2000).
B. Anxiety, Fear, and Stress
Of all the behavioral actions of OFQ/N, its apparent anxiolytic role may be the most fundamental. Furthermore, this particular action may help explain the effects of OFQ/N on other phenomena. For example, OFQ/N′s effects on locomotor activity, reward, feeding, pain modulation, and tolerance may be secondary to changes in stress levels, at least to some extent. The peptide and its receptor are found in a number of CNS loci involved in emotion and stress regulation, including the amygdala, septal region, locus coeruleus, PAG, and hypothalamus (Herman and Cullinan, 1997). A number of standard behavioral assays reveal the ability of supraspinal OFQ/N to block fear and anxiety in both rats and mice (Jenck et al., 1997). For example, on the elevated plus maze, a test based on the natural aversion of rodents for open spaces, OFQ/N in subsedating doses produced a diazepam-like increase in the time spent in the anxiety-provoking “open arms”. Similar findings reminiscent of the effects of benzodiapines were observed in the light-dark aversion, open field, and operant conflict tests (Jenck et al., 1997). Like benzodiazepines, the anxiolytic effects of OFQ/N generally revealed inverted U-shaped dose-response curves, likely due to increasing sedation and ataxia at the highest doses tested. Ro 64–6198, a synthetic agonist of the NOP1receptor, also was anxiolytic in a large number of behavioral assays (Jenck et al., 2000). Again, in every case the effects were comparable to conventional benzodiazepines such as alprazolam or diazepam, except in a test of panic (Griebel et al., 1995; Jenck et al., 2000). However, the actions of NOP1 agonists are not identical to those of benzodiazepines, as illustrated by the absence of anticonvulsant properties in the former (Jenck et al., 2000).
Griebel and colleagues (1999) extended these findings using the “mouse defense test battery”, a screening test for anxiolytics that quantifies behaviors associated with exposure of a mouse to a rat, a natural predatory threat stimulus (Griebel et al., 1995). Intracerebroventricularly administered OFQ/N produced significant effects on some, but not all, of the dependent measures of anxiolysis in this test. OFQ/N was effective against “terminal defense” reactions (e.g., defensive attack, escape attempts), seen when stressful stimuli are unavoidable, but not against cognitive “risk-assessment” reactions (Griebel et al., 1999). The authors interpreted the pattern of results to suggest that OFQ/N was primarily involved in situations of particularly high stress.
ppOFQ/N gene knockout mice also have provided intriguing data regarding the role of OFQ/N in stress. In addition to their high basal plasma corticosterone levels and high basal anxiety in three standard behavioral assays (open field, plus maze, and light-dark box), these mutants were unable to adapt to repeated stressors (Koster et al., 1999). In this paradigm, mice were stressed by 10 min of forced swimming in 18°C water. A significant stress-induced analgesia was measured in both wild-types and knockouts, with the latter genotype having significantly, but not profoundly, more stress-induced analgesia than the former. After two more daily sessions of forced swimming, wild-type mice had completely adapted, showing virtually no stress-induced analgesia. In contrast, the stress-induced analgesia in the knockout mice was indistinguishable from the phenomenon on the first day. It may be tempting to interpret such data as reflecting a lack of tolerance to the analgesia rather than a lack of adaptation to the stressor, especially since NOP1 knockouts have been shown to display deficits in tolerance (see Section X.C.). However, stress and/or stress hormone treatment also attenuates tolerance development (e.g., Holaday et al., 1979; Takahashi et al., 1988), and thus the “high stress” status of these mutants may indeed be responsible for any paucity in tolerance. The authors further suggested that a failure to adapt to stress-induced analgesia may be responsible for the significantly higher than normal tail-flick latencies exhibited by group-housed (and thus chronically stressed) male knockout mice (see Section VIII.F.). To our knowledge, NOP1 receptor knockouts have only been tested on one behavioral assay of anxiety, the elevated plus maze, and not found to differ from wild types (Mamiya et al., 1998).
The site(s) mediating the anxiolytic effects of OFQ/N are not clear. In one recent study (Kyuhou and Gemba, 1999), OFQ/N microinjected into the PAG fully inhibited vocalization in the guinea pig elicited by electrical stimulation of the anterior cingulate cortex. This phenomenon is a model of the separation call of guinea pigs isolated from their conspecifics (Berryman, 1976).
C. Tolerance and Dependence
The development of tolerance to and dependence upon exogenously administered opiates limits the utility of these agents clinically. Tolerance exists at many levels within the organism. Many studies investigating the phenomena have focused on cellular changes, either of μ-opioid receptors themselves or of signal transduction elements coupled to these receptors (e.g., see Nestler, 1997). A wide range of other transmitter systems has been implicated. Many groups have documented the importance of the NMDA receptor/nitric oxide cascade in tolerance (Pasternak and Inturrisi, 1995), with more recent work demonstrating the involvement of δ-opioid receptors as well (Abdelhamid et al., 1991; Kest et al., 1996; Zhu et al., 1999). Clearly, tolerance and dependence involve a complex series of events that are all intertwined.
Others have proposed systems-level theories (Harrison et al., 1998). One such theory of tolerance holds that with repeated administration of opioids, the release of anti-opioid peptides in the CNS is increased, counteracting the analgesia produced (tolerance) and contributing to the production of a withdrawal syndrome (dependence) once the opioid administration ceases (Rothman, 1992). Given the strong evidence that OFQ/N is itself an anti-opioid peptide, the involvement of this transmitter/receptor system in the phenomena of tolerance and dependence was predicted very early on (Mogil et al., 1996b).
The first clear evidence that OFQ/N and its receptor may indeed play a role in tolerance came from NOP1 knockout mice, which showed a partial reduction of morphine tolerance development (Ueda et al., 1997). After five daily injections, wild-type and heterozygous mice displayed a profoundly reduced analgesic response to morphine on the tail-pinch test. Mutants, on the other hand, displayed equivalent peak levels of analgesia on day 5 and day 1, although the analgesia was slower to develop and quicker to dissipate (Ueda et al., 1997). Subsequent work by the same group described 12-fold versus 3.3-fold rightward shifts (representing tolerance) in morphine analgesia dose-response curves on the tail-pinch test in wild-type and knockout mice, respectively and replicated the finding on the radiant heat tail-withdrawal test (Ueda et al., 2000). ppOFQ/N knockout animals have not been tested for their tolerance status, although their inability to adapt to stress-induced analgesia after repeated stress exposure might be interpreted as an inability to develop tolerance to this analgesic manipulation (Koster et al., 1999) (see Section X.B.).
Probably the best evidence for a role of OFQ/N and NOP1 in tolerance is provided by the partial reduction of morphine tolerance by systemic administration of the nonpeptidic NOP1 antagonist, J-113397 (Ueda et al., 2000). Intriguing differences were noted when the effects of the antagonist were evaluated separately by route of administration and nociceptive assay. On the tail-pinch test, intracerebroventricular administration of J-113397 produced a partial blockade of tolerance, whereas intrathecal administration produced a complete blockade. On the radiant heat tail-withdrawal test, spinal administration again produced a complete blockade, but supraspinal J-113397 was entirely without effect (Ueda et al., 2000). The authors attribute this dissociation to the involvement of supraspinal mechanisms in the tail-pinch test versus the predominantly spinal mediation of the reflexive radiant heat tail-withdrawal response and suggest that the NOP1 receptors of relevance to tolerance are located in the spinal cord. In support of this contention, they demonstrated by reverse transcription-polymerase chain reaction that NOP1 gene expression in the spinal cord was increased by 50% in tolerant versus nontolerant mice (Ueda et al., 2000). A supporting independent finding is that chronic morphine infusion produced up to a 43% increase in125I-[Tyr14]OFQ/N binding in the superficial layers of the spinal cord dorsal horn (Gouarderes et al., 1999).
It is interesting that spinal NOP1 receptors have been implicated in tolerance whereas OFQ/N exerts anti-opioid actions supraspinally (e.g., Grisel et al., 1996). There is evidence implicating supraspinal OFQ/N in tolerance. For example, tolerance to morphine and electroacupuncture analgesia is partially reduced following intracerebroventricular treatment with OFQ/N antibodies (Tian et al., 1998). The OFQ/N antibody blocked both chronic (given 30 min a day for 6 days) and acute (given continuously for 6 h) electroacupuncture analgesic tolerance, and chronic (5–60 mg/kg, 3 times a day for 6 days) morphine analgesic tolerance, but not acute (5 mg/kg every 2 h for 16 h) morphine analgesic tolerance. The authors had no explanation for the failure of OFQ/N to affect this latter phenomenon. Further work by this group (Yuan et al., 1999) showed increases in OFQ/N immunoreactivity in brain perfusates and also in the amygdala and PAG of morphine-tolerant rats. The increased production and release of OFQ/N occurred more slowly but lasted longer than that of the anti-opioid CCK in analogous experiments, suggesting that the two peptides may play complementary roles in the mediation of tolerance.
The role of OFQ/N and its receptor in dependence is less clear. Many investigators have assumed that direct injection of OFQ/N would be likely to induce withdrawal symptoms in morphine-tolerant and -dependent mice. No group has convincingly shown this and several groups have reported that it does not occur (Tian et al., 1997b;Kotlinska et al., 2000). However, Malin et al. (2000) reported recently that supraspinal OFQ/N dose dependently produced withdrawal symptoms by itself in nontolerant rats. Most puzzling, although, is the reported ability of both supraspinal OFQ/N and the NOP1antagonist, J-113397, to attenuate withdrawal symptoms in morphine-dependent animals (Kotlinska et al., 2000; Ueda et al., 2000). It should be noted that Kotlinska and colleagues (2000) observed a dose-dependent blockade of naloxone-precipitated “wet-dog shakes” in rats by OFQ/N, whereas Ueda et al. (2000) reported the attenuation by J-113397 of five other common withdrawal symptoms (jumping, paw tremor, backward locomotion, sniffing, and defecation) in mice. Consistent with the notion that activation of the NOP1 receptor contributes to rather than blocks the expression of withdrawal is the finding of the latter group that these same withdrawal symptoms are reduced or absent in NOP1 knockout mice (Ueda et al., 2000) (seeSection VIII.F.).
D. Learning and Memory
Classical opioids like β-endorphin and dynorphin modulate learning and memory processes. β-Endorphin consistently disrupts memory, whereas dynorphin can either enhance or disrupt it (Noda et al., 2000). The high density of NOP1 receptors in the anterior cingulate, frontal cortex, and hippocampus suggested that OFQ/N may play a role in these phenomena. Indeed, Sandin and colleagues (1997) showed the ability of OFQ/N [but not OFQ/N(1–13); Sandin et al., 1999] microinjected into the hippocampus to severely impair spatial learning in the hippocampally dependent Morris water maze task (Morris et al., 1982). OFQ/N (Yu et al., 1997), like dynorphin (Wagner et al., 1993), also can impair the induction of the electrophysiological phenomenon thought by many to underlie the synaptic plasticity associated with learning, long-term potentiation (LTP) (Stevens, 1998).
Both of these findings have subsequently been replicated and extended. The modulatory effect of OFQ/N on LTP is due to postsynaptic mechanisms, including the inhibition of dentate gyrus granule cells and inhibition of NMDA receptor-mediated currents (Yu and Xie, 1998). In addition to spatial learning on the Morris water maze, OFQ/N impairs: 1) acquisition in the step-down passive avoidance task (Hiramatsu and Inoue, 1999a,b); 2) working memory (spontaneous alteration) in the Y-maze test (Hiramatsu and Inoue, 1999a,b); and 3) “latent” learning in the water-finding task of spatial attention (Noda et al., 2000). Nocistatin has no effect on the two former tasks but effectively blocks the OFQ/N impairment (Hiramatsu and Inoue, 1999a). Nocistatin also blocked the impairment in these tasks produced by the muscarinic receptor antagonist scopolamine, which suggests an interaction between NOP1 receptors and cholinergic mechanisms of memory (Hiramatsu and Inoue, 1999b). The only conflicting data in this literature is the lack of effect of low OFQ/N doses (1–10 pmol) on spontaneous alternation in the Y-maze test (Mamiya et al., 1999).
The remainder of the relevant data derive from transgenic mice. NOP1 mutants displayed facilitated performance on both escape latency and “probe task”-dependent measures in the Morris water maze and the passive avoidance task (Manabe et al., 1998;Mamiya et al., 1999). In addition, hippocampal LTP was up-regulated in these animals (Manabe et al., 1998; Noda et al., 2000). In another study, latent learning on the water-finding task was enhanced in the knockouts (Mamiya et al., 1998). The authors proposed that a dopaminergic mechanism was underlying the genotypic difference since dopamine receptor agonists can impair performance on this assay (Ichihara et al., 1993), and mutant mice displayed low dopamine levels in frontal cortex. The lack of genotype differences on the Y-maze test supported their contention that OFQ/N is not involved in working memory (Mamiya et al., 1999). Finally, mice lacking the ppOFQ/N gene did not differ from wild-type animals on the Morris water maze (Koster et al., 1999).
E. Feeding
Classical opioid peptides have well documented effects on feeding behavior, with μ, δ, and κ agonists increasing food intake in rodents (Glass et al., 1999). Soon after its isolation, Pomonis and colleagues (1996) showed that supraspinal OFQ/N (1–10 nmol) increased food intake in the satiated rat. OFQ/N′s effects are short-lasting, surprisingly specific to food intake with neither water intake nor 1% sucrose intake affected, and accompanied by transient hypolocomotion (e.g., Polidori et al., 2000a,b). OFQ/N hyperphagia can be blocked by antisense treatment to NOP1 mRNA (Leventhal et al., 1998), competitive NOP1 antagonism (Polidori et al., 2000a), and functional antagonism by nocistatin (Olszewski et al., 2000b). Surprisingly, naloxone/naltrexone pretreatment also blocks OFQ/N′s effects on food intake (Pomonis et al., 1996; Leventhal et al., 1998), although this is probably due to classical opioid receptors being involved in feeding control at a distal site or affecting motivational processes related to food intake (see Section X.A.) (Polidori et al., 2000b). The NOP1agonist, [Phe1ψ(CH2-NH) Gly2]-nociceptin(1–13)-NH2, also increases food intake more potently than OFQ/N when injected supraspinally (Polidori et al., 2000a).
A microinjection study suggested that the sites of action of OFQ/N hyperphagia include the ventromedial hypothalamus (VMH), a locus of crucial but controversial importance in the regulation of feeding and/or weight control (see King, 1980), and the shell of the nucleus accumbens (Stratford et al., 1997). A more recent study (Polidori et al., 2000b) using lower doses was unable to replicate the VMH finding, but it did implicate the hypothalamic arcuate nucleus as the most sensitive site tested. The lack of effect of OFQ/N injected into the fourth ventricle in this same investigation appeared to rule out the involvement of brain stem loci. However, in a study of 18 brain regions, supraspinal OFQ/N elevated Fos-like immunoreactivity in six relevant loci, including the brain stem nucleus tractus solitarius, which had the largest (5-fold) increase (Olszewski et al., 2000a).
Regarding the mechanism of OFQ/N′s hyperphagic effect, Polidori and colleagues (2000b) point to the fact that OFQ/N, like opioids, exerts an inhibitory action on arcuate nucleus neurons (Wagner et al., 1998). This alone could explain its hyperphagic action via the inhibition of the release of POMC peptides like α-MSH and ACTH, which in turn inhibit feeding. It should be noted, however, that OFQ/N interacts with any number of other feeding-relevant neurotransmitter systems, including serotonin, glutamate, and GABA (Polidori et al., 2000b). The hyperphagic effect of OFQ/N does appear to be entirely independent from that of neuropeptide Y (Polidori et al., 2000a).
Competitive and noncompetitive OFQ/N antagonists reduce deprivation-induced food intake (Olszewski et al., 2000b; Polidori et al., 2000a), which implicates this system in the physiological mediation of this phenomenon. A strong role for OFQ/N in basal feeding regulation remains to be demonstrated, and alterations in food intake of antisense-treated or transgenic animals have not been reported. Of potential clinical interest is the abstracted report that rats, provided with a fat-rich diet, became hyperphagic and obese and displayed significantly higher OFQ/N binding density in the arcuate nucleus and VMH than rats fed a standard diet (Malek et al., 1999).
XI. Conclusions and Future Directions
Neuropeptides have become increasingly important in our understanding of brain function since the early description of the enkephalins. OFQ/N offers another example of a neuropeptide discovered as a ligand for a previously described receptor. Despite its structural similarities to opioid peptides, particularly dynorphin A, OFQ/N does not label opioid receptors. Yet, its actions are intimately intertwined with those of the opioids. As noted above, its most robust action remains a functional reversal of opioid analgesia, but OFQ/N also elicits analgesia and has been implicated in a wide range of additional actions as well. Much confusion remains despite the extensive literature that has arisen over the past 6 years since its original isolation. Differences among laboratories may arise from any number of potential factors, but they illustrate the complexity of the OFQ/N system. It is possible, if not likely, that the wide range of divergent results are each reproducible. If so, the differences among the studies implies that OFQ/N actions are highly dependent upon additional factors that may, or may not, be appreciated. This makes their evaluation difficult indeed.
The future of OFQ/N and NOP1 receptor research remains unclear. There seems little doubt that OFQ/N and its receptor(s) are important. They are associated with many actions and have a number of potential therapeutic utilities. One major question to address in the future is the potential of ligand and receptor heterogeneity. Are OFQ/N(1–11) or OFQ/N(1–7) or other fragments physiologically important? Is there truly NOP1receptor heterogeneity and if so, can it be exploited in the design and synthesis of selective agonists and antagonists? What is the interaction between OFQ/N functioning, genotype, and stress? These are the questions to answer. Only when we understand these issues is it likely that we will finally be able to comprehend and integrate the results described in this review.
Acknowledgments
J.S.M. is supported by U.S. Public Health Service Grants DA11394 and DE12735. G.W.P. is supported by a Senior Scientist Award (DA00220) and research grants (DA02615, DA07242, and DA06142) from the National Institute on Drug Abuse. We thank Dr. Alan Gintzler and Dr. Kabir Lutfy for useful discussions regarding this manuscript.
Footnotes
-
↵1 Address for correspondence: Dr. Gavril W. Pasternak, Department of Neurology, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY. E-mail: pasterng{at}mskcc.org
-
↵2 NIDA Technical Review “The Molecular Neurobiology and Pharmacology of Opiate Receptor Subtypes: A Tribute to William Martin” held in Washington, DC, November 6–7, 1993.
Abbreviations
- OFQ/N
- orphanin FQ/nociceptin
- NalBzoH
- naloxone benzoylhydrazone
- kb
- kilobase(s)
- bp
- base pair(s)
- CHO
- Chinese hamster ovary
- ppOFQ/N
- prepro-OFQ/N
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- DPDPE
- [d-Pen2,d-Pen5]-enkephalin
- DSLET
- [d-Ser2,Leu5]-enkephalin-Thr
- PAG
- periaqueductal gray
- RVM
- rostral ventromedial medulla
- GABA
- γ-aminobutyric acid
- ACTH
- adrenocorticotrophic hormone
- CCK
- cholecystokinin
- MSH
- melanocyte-stimulating hormone
- SBL
- scratching, biting, and licking
- NMDA
- N-methyl-d-aspartate
- NK
- neurokinin
- CNS
- central nervous system
- HPA
- hypothalamic-pituitary-adrenal
- PG
- prostaglandin
- VTA
- ventral tegmental area
- CPP
- conditioned place preference
- LTP
- long-term potentiation
- VMH
- ventromedial hypothalamus
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Molecular Biology of the Orphanin FQ/Nociceptin Receptor
- III. Receptor Binding
- IV. NOP1 Receptor Heterogeneity
- V. Orphanin FQ/Nociceptin
- VI. Anatomy of Orphanin FQ/Nociceptin and Its Receptor
- VII. Range of Effects of Orphanin FQ/Nociceptin
- VIII. Effects of Orphanin FQ/Nociceptin on Pain
- IX. Effects of Related Peptides on Pain
- X. Involvement of NOP1 in Other Central Nervous System-Mediated Behaviors
- XI. Conclusions and Future Directions
- Acknowledgments
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters