


Abstract
Proteinase-activated receptors are a recently described, novel family of seven-transmembrane G-protein-coupled receptors. Rather then being stimulated through ligand receptor occupancy, activation is initiated by cleavage of the N terminus of the receptor by a serine protease resulting in the generation of a new tethered ligand that interacts with the receptor within extracellular loop-2. To date, four proteinase-activated receptors (PARs) have been identified, with distinct N-terminal cleavage sites and tethered ligand pharmacology. In addition to the progress in the generation of PAR-1 antagonists, we describe the role of thrombin in such processes as wound healing and the evidence implicating PAR-1 in vascular disorders and cancer. We also identify advances in the understanding of PAR-1-mediated intracellular signaling and receptor desensitization. The cellular functions, signaling events, and desensitization processes involved in PAR-2 activation are also assessed. However, other major aspects of PAR-2 are highlighted, in particular the ability of several serine protease enzymes, in addition to trypsin, to function as activators of PAR-2. The likely physiological and pathophysiological roles for PAR-2 in skin, intestine, blood vessels, and the peripheral nervous system are considered in the context of PAR-2 activation by multiple serine proteases. The recent discovery of PAR-3 and PAR-4 as additional thrombin-sensitive PARs further highlights the complexity in assessing the effects of thrombin in several different systems, an issue that remains to be fully addressed. These discoveries have also highlighted possible PAR–PAR interactions at both functional and molecular levels. The future identification of other PARs and their modes of activation are an important future direction for this expanding field of study.
I. Introduction
Seven transmembrane G-protein-coupled receptors comprise the biggest group of receptors in mammalian systems, and a large number of cognate receptors and associated ligands have been identified. A recently described novel subset of this group, the proteinase-activated receptors (PARs), has been shown to have unique mechanism of activation. Rather than being activated by simple ligand occupancy, based on the law of mass action, they are activated enzymatically through proteolysis of the receptor. This proteolytic cleavage is specifically mediated by a well characterized family of enzymes that require serine within the active site, the serine proteases. Classically, serine proteases have been shown to play important roles in diverse biological functions, particularly in relation to clot formation and wound healing. However, proteolytic cleavage of PARs as a mode of receptor activation now represents an increasingly important feature of this family of enzymes. In this review, we will outline the pharmacological characteristics of the four members of the PAR family, PAR-1 through -4, their mechanism of activation by serine proteases, coupling to intracellular signaling pathways, and their potential physiological and pathophysiological roles. Such is the rapidly expansive nature of the field; the reader is directed to a number of excellent recent shorter reviews that will complement this current work (Grand et al., 1996; Hollenberg, 1996;Dery et al., 1998; Cocks and Moffatt, 2000; Coughlin, 2000).
II. Historical Perspectives—Cellular Effects of Thrombin and the Cloning of the Thrombin Receptor, Proteinase-Activated Receptor-1
The role of thrombin as a key intermediate in the coagulation process has been established for a number of decades. Thrombin was originally identified as a trypsin-like serine protease, produced from prothrombin by the action of factor Xa, which mediated the formation of fibrin, the fibrous matrix of blood clots, from fibrinogen (Davie et al., 1991; Stubbs and Bode, 1993). Since the initiating factor in the cascade, tissue factor, was also found primarily on cells that under normal conditions do not access the bloodstream, thrombin was seen primarily as a component of a coagulation process linking tissue damage to wound repair. However, thrombin in the absence of other products of the coagulation cascade, was also found to be a strong activator of platelet aggregation, suggesting the potential of cellular effects in addition to a role in clot formation (Davey and Luscher, 1967). In addition, several studies established thrombin to have direct effects upon a number of other cell types, including monocytes, smooth muscle cells, endothelial cells, and lymphocytes, among others (Chen and Buchanan, 1975; Chen et al., 1976; Bar-Shavit et al., 1983; Bizios et al., 1986; Daniel et al., 1986; Hattori et al., 1989) (seeSection IV.). Significantly, several of these studies used serine protease inhibitors to confirm that the protease activity of thrombin was essential for these cellular effects (Shuman, 1986). Although classical radioligand binding studies with modified thrombin had identified several candidate thrombin-binding proteins (Okamura et al., 1978; Gronke et al., 1987), up until 1990 a functional thrombin receptor had not been identified.
A. Cloning of a Thrombin Receptor
In 1991, Coughlin and colleagues (Vu et al., 1991a) used a dilution cloning approach in an attempt to isolate the cDNA encoding the thrombin receptor. Initially, mRNA, from cells highly responsive to thrombin, was injected into45Ca2+-labeledXenopus oocytes and thrombin-stimulated45Ca2+ release assayed. The mRNA transcript encoding the receptor was fractionated and used to construct a size-specific cDNA library, which was then plated into 50 pools of estimated 20,000 clones each. By injecting in vitro transcribed cRNA from each pool into Xenopus oocytes, and functionally assaying each pool for thrombin-stimulated45Ca2+ release, positives were identified and could then be progressively subdivided and plated into fractions containing fewer cDNAs. Eventually, a single cDNA species was isolated which, when assayed in oocytes, displayed 100-fold higher calcium release and chloride entry when compared with oocytes injected with a similar quantity of nonpurified Dami cell mRNA. In oocytes expressing the receptor, thrombin was found to be an extremely potent agonist (EC50 = 50 pM), whereas trypsin, although effective, was considerably less potent and efficacious. Furthermore, responses to thrombin were found to be blocked by the thrombin antagonists hirudin and hirugen, a hirudin-derived peptide (Vu et al., 1991a) indicative of the thrombin-specific nature of the cloned receptor.
B. Receptor Structure and Mode of Activation
Sequencing of the functional clone revealed a 3.5-kb insert, containing an open reading frame encoding a 425 amino acid protein. Hydropathy analysis of the sequence revealed the protein to be a member of the seven transmembrane domain receptor superfamily, being most closely related to the peptide (e.g., substance P) and glycoprotein hormone receptor subfamilies. The protein was found to contain an N-terminal hydrophobic signal sequence with potential cleavage sites at Thr24 and Ala26. The remainder of the 75 amino acid N terminus is extracellularly disposed and contains several asparagine-linked glycosylation sites. Crucially, a putative thrombin cleavage site (LDPR/S), similar to the activation cleavage site in the zymogen protein C, was identified in the amino terminus, suggesting that receptor activation involves proteolytic cleavage. Mutation of this residue, Arg41 to Ala, rendered the receptor insensitive to stimulation when expressed in oocytes (Vu et al., 1991a), whereas a peptide mimicking the new amino terminus created by cleavage at Arg41, SFLLRNPNDKYEPF (TRAP-14), was able to activate both wild-type and mutant receptors. In addition, Northern blotting of mRNA revealed high levels of receptor in HEL and DAMI cells, both known to be highly responsive to thrombin, and also in platelets and endothelial cells by RT-PCR.
In the same year, Rasmussen et al. (1991) cloned the hamster thrombin receptor from CCL39 hamster lung fibroblasts. Functional expression inXenopus oocytes indicated a functional thrombin receptor, and the deduced amino acid sequence revealed a thrombin consensus cleavage site in the extracellular N terminus, followed by a negatively charged cluster of residues comprising a binding region for the anion exosite found in thrombin (see below). Cellular and tissue-specific expression was also consistent with that observed for the human receptor. Other thrombin receptors cloned to date include those from rat (Zhong et al., 1992), mouse (Tanaka et al., 1993), Xenopus laevis (Gerszten et al., 1994), and bovine (Ma et al., 1996) sources. Taken together, these findings confirmed the identification of PAR-1 and a novel, proteolytic, mechanism of activation (see Figs.1 and2).
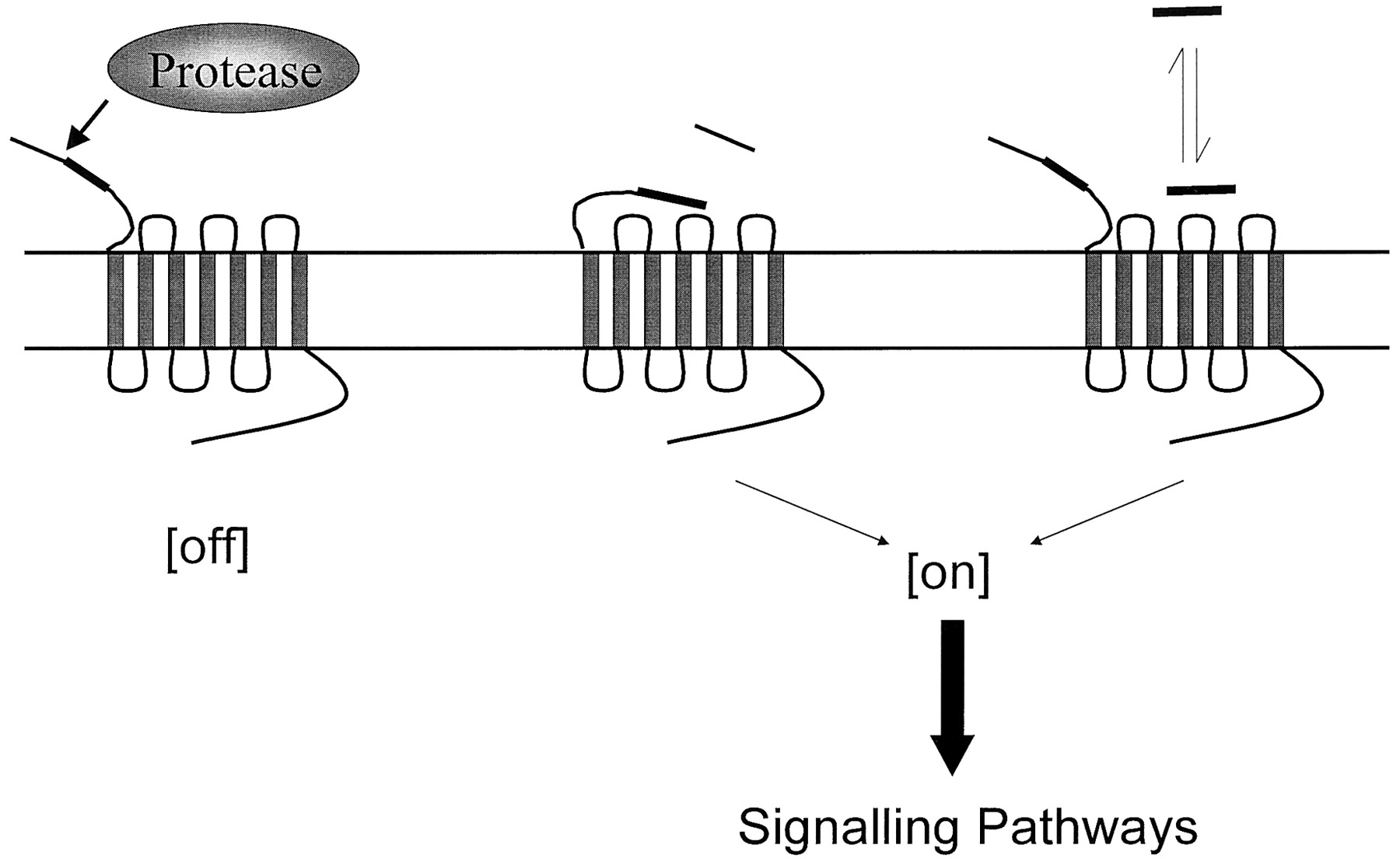
Protease activation of PARs. Proteolytic cleavage the N terminus generates a new tethered ligand designated by the filled section that interacts with the extracellular loop-2 of the receptor. A peptide sequence derived from the N terminus is able to activate the receptor in the absence of protease-mediated cleavage of the N terminus.

Structural features of PARs 1–4. The key areas of PAR receptor activation are highlighted. The N terminus cleavage domain and the hirudin-like binding domain, the ECL-2 where tethered ligand/receptor interactions occur, and the C-terminal tail that is involved in desensitization and some aspects of intracellular signaling.
C. Thrombin/Receptor Interactions
Several further studies identified additional features regarding the mechanism of the interaction between thrombin and the receptor. Initially, the crucial role of the N terminus was confirmed. A mutant receptor with the LDPR/S site replaced by an enterokinase site was fully responsive to enterokinase, suggesting no requirement for an additional mechanism of activation other than that initially proposed (Vu et al., 1991b). Subsequently, a mutant lacking the N terminus was found to be both inactive and unresponsive to thrombin (Chen et al., 1994a). The findings of this study not only confirmed the crucial role for this region of the receptor, but also provided an argument against the idea that the role of the N terminus was to prevent receptor activation, and that cleavage left the receptor free to form an active conformation. Additionally, the differences in the potency between enterokinase and thrombin in their ability to activate wild-type or mutant receptors suggested additional binding sites for thrombin within the N terminus. Mutation of the N terminus identified the presence of a hirudin-like domain within region 51–63 that was essential for high affinity binding and the potent effects of thrombin (Vu et al., 1991b). Peptides not susceptible to thrombin cleavage but which encompass this region, or other exosite ligands, such as thrombomodulin and fibrinogen, blocked the actions of thrombin in whole cells or thrombin-stimulated cleavage of a GST/N-terminal receptor fusion protein expressed in Escherichia coli (Bouton et al., 1995). Furthermore, γ-thrombin, which lacks the anion exosite, was found to be considerably less (100-fold) potent than thrombin in activating the receptor (Bouton et al., 1995; Seiler et al., 1995). Subsequent studies have confirmed the importance of the N-terminal DKYEPF hirudin-like domain in reducing the kinetic barrier to thrombin/receptor complex formation. These studies have also provided evidence to suggest that thrombin binding at this site initiates a conformational change in the active center of the enzyme that accommodates the LDPR cleavage sequence and facilitates binding (Ishii et al., 1995).
The ability of thrombin receptor activating peptide (TRAP) to activate a thrombin receptor lacking the amino terminal exodomain implicated a site, or sites, of interaction with the other extracellular loops. Experiments utilizing chimeras generated from human andXenopus receptors or antibodies directed against different segments of the thrombin receptor (Bahou et al., 1994) showed that both the N-terminal exodomain and the second extracellular loop determine SFLLRN binding to the receptor. Subsequent studies using PAR-1/PAR-2 chimeras (see below) confirmed the role of ECL-2 in determining the specificity of this interaction (Lerner et al., 1996). Similar studies also generated detailed information regarding the molecular basis of thrombin/receptor interactions. The N terminus and the ECL-2 regions of the receptor were shown to dictate the selectivity of eitherXenopus or human thrombin receptor for stimulation by human and Xenopus TRAPs. Point mutation at only two residues within the Xenopus receptor, Phe for Asn87 in the N-terminal exodomain and Glu for Leu260 in the second extracellular domain, conferred human receptor-like specificity (Nanevicz et al., 1995). Additional experiments using substituted TRAPs showed that Arg5 of the peptide was involved in binding to Glu260 since a human receptor with Glu260 mutated to arginine lost the ability to signal to SFLLRN. This mutation was also complementary for activation in response to SFLLEN, normally inactive at the wild-type receptor, indicating the importance of Arg5-Glu260 in defining the specificity for thrombin activating peptides for human PAR-1. This interaction is also likely to be important in initiating a conformational change in the receptor and subsequent intracellular signaling since a human receptor containing a Xenopus ECL-2 domain encompassing region 259–268 is constitutively active (Nanevicz et al., 1996). However, no subsequent studies utilizing only the human thrombin receptor have confirmed this hypothesis.
III. Pharmacology of Proteinase-Activated Receptor-1
A series of studies utilizing substituted TRAP analogs representing the cleaved N terminus were undertaken to derive information regarding the structure-function relationship for activation of the thrombin receptor (see Table1). Initial studies using a number of functional assays, in particular platelet aggregation and [3H]IP accumulation, showed that the pentapeptide SFLLR-NH2 was a minimum requirement for full agonist activity, although the hexapeptide SFLLRN was 2- to 3-fold more potent, suggesting it to be the preferred functional sequence. Peptides truncated from the amino terminus displayed substantially reduced potency, whereas a series of peptides with extended C termini showed similar or reduced potency to the hexapeptide (Chao et al., 1992; Sabo et al., 1992; Scarborough et al., 1992a;Vassallo et al., 1992). A series of single amino acid substitutions indicated that, although Ser1 was essential for binding, changes could be tolerated as long as the free amino group was maintained (Scarborough et al., 1992a; Sakaguchi et al., 1994;Shimamoto et al., 1995). Removal or acetylation of the amino group at Ser1 reduced potency considerably (Sakaguchi et al., 1994). Phe2 was found to be essential for agonist activity and tolerated substitution poorly, displaying complete loss of activity with alanine replacement (Scarborough et al., 1992a), but allowed substitution with tyrosine (Nose et al., 1993; Natarajan et al., 1995). Leu3 was noted to be relatively unimportant in that it could be substituted with many different residues. However, some loss in potency was recorded following alanine substitution at Leu4 and, in particular, Arg5 (Chao et al., 1992; Scarborough et al., 1992a; Vassallo et al., 1992; Natarajan et al., 1995). A bulky aliphatic residue at position 4 and either a basic or aromatic residue at position 5 are moderately important for activity. Positions 1 and 3 tolerate proline substitution, while scanning through positions 1–5 with D- or N-Me amino acids has been shown to cause a major loss of agonist potency (Feng et al., 1995; Natarajan et al., 1995). More recently, reduced amide ψ(CH2N) and ester ψ(COO) scans have revealed the importance of the amide nitrogen between residues 1 and 2 for agonist recognition and the potential involvement of carbonyl groups along the backbone in hydrogen bonding with the receptor (Shimamoto et al., 1995; Ceruso et al., 1999). From these and other studies, a consensus peptide structure has been developed that has provided a template from which additional compounds have been synthesized.
Structure/activity relationships for TRAPs
Additional consideration has also been given to the favored bioactive conformations of TRAP-5. Information derived from NMR and other modeling techniques has suggested an extended structure for the active form of the peptide. These studies also suggest a limited conformation for Phe2, a φ torsional angle similar to Pro, a ψ torsional angle close to that of a β-sheet for Leu3, and a trans configuration for the amide bonds of S-F and F-L (Shimamoto et al., 1995). Furthermore, despite the finding that Leu3 can tolerate a wide variety of substitutions, the peptide bond itself is sensitive to conformational changes possibly due to a hydrophobic contact between Phe2 and Leu4 side chains (Ceruso et al., 1999). Thus, this region may play a crucial role in changes in conformation during interaction with the receptor.
Although the aforementioned studies have indicated an extended structure for the peptide, another group has proposed a curved cyclic backbone structure for the active form of TRAP-5. This hypothesis is based on the potential of weak contacts between the Arg5 side chain and the Ser1 and Phe2 residues (Matsoukas et al., 1997). Consistent with this is the finding that a 19-membered-ring macrocyclic SFLLR, linked from the P1 side chain to the C terminus, is nearly equipotent with SFLLR in induction of gastric smooth muscle contraction (Matsoukas et al., 1996). However, it has been subsequently shown that these compounds are generally less potent than SFLLRN in platelet aggregation assays (McComsey et al., 1999). The contradictions between studies may be further exacerbated by the fact that the respective conclusions, despite utilizing sophisticated modeling techniques, are based substantially on extrapolation of data derived from experiments using the untethered ligand rather than the tethered bioactive form. Future development of studies using X-ray crystallography allowing direct examination of peptide/receptor interactions will represent a vital step forward in this area.
Recent studies have further refined the structure activity relationships for PAR-1 and its ligand, leading to the synthesis of a number of penta- and tetrapeptides with enhanced agonist potency (Table1). Substitution of Phe2 withp-fluorophenylalanine, but not other larger halogen derivatives, increases agonist potency by approximately 5-fold (Nose et al., 1993), possibly by enhancing the π-π bonding between the ligand and the receptor (Nose et al., 1998). Replacement of Leu3 with residues containing either neutral or basic side chains, such as (2-napthyl) alanine (Natarajan et al., 1995;Seiler et al., 1996) p-guanidinophenylalanine (Bernatowicz et al., 1996) or arginine (Feng et al., 1995; Natarajan et al., 1995), also results in enhanced agonist potency. Introducing a hydrophobic cyclohexylalanine in place of Leu4 increases potency a further 2-fold (Feng et al., 1995). One synthetic peptide combining some of these modifications with an additional tyrosine substitution in position 6, H-Ala-(pF-)Phe-Arg-Cha-hArg-Tyr-NH2, has been shown to give an EC50 value of 10 nM in platelet aggregation assays and a K d of 15 nM when a tritium-labeled form is used in radioligand binding assays (Feng et al., 1995; Ahn et al., 1997). Despite these findings, peptides such as these show only moderate selectivity (100-fold) over the recently described PAR-2 in both activation and desensitization assays (Kawabata et al., 1999b), and more selective PAR-1 agonists, such as Ala-(pF)Phe-Arg-Cha-Cit-Tyr-NH2 (Kawabata et al., 1999b) with increased potency, are still required.
Rational drug design methodology has also been utilized to generate a series of substituted peptides displaying partial agonist and antagonist properties. The peptide 3-mercapto-propionyl-Phe-Cha-Cha-Arg-Lys-Pro-Asn-Asp-Lys amide (C186–65), initially designed from agonist peptides (Scarborough et al., 1992b), was found to inhibit both SFLLR and thrombin-stimulated platelet Ca2+ mobilization and aggregation, but not the similar responses produced by collagen or TXA2, suggesting some specificity for thrombin receptors (Seiler et al., 1995). However, the potency of C186–65 was relatively low, and partial agonist activity at PAR-1 has been recorded in some cell types. Indeed, recently, this peptide has been found to also have PAR-2 agonist activity in HEK cells (Kawabata et al., 1999b). Nevertheless, using this strategy, a potent antagonist,N-trans-cinnamoyl-p-fluoro-Phe-p-guanidino-Phe-Leu-Arg-NH2(BMS-197525), was synthesized and found to have an IC50 value of approximately 10 nM in radioligand binding assays and 0.2 μM in platelet aggregation studies (Bernatowicz et al., 1996). Furthermore, addition of a single arginine residue at the C terminus further enhanced antagonist potency in functional assays by 5- to 10-fold, whereas further substitution of arginine for ornithine at position 6 generated a peptide suitable for radiolabeling and use in binding studies (Elliott et al., 1999). The relative potencies of these and other analogs were also tested in GTPase assays and Ca2+ mobilization experiments, the results from which agreed well with the initial values obtained in platelets. Similar approaches have generated a number of peptide antagonists with variable potency (Hoekstra et al., 1998; Fujita et al., 1999) (see Table 1).
Despite apparent advances in the development of PAR-1 antagonist peptides, a number of problems remain. Not only do the compounds have only moderate potency for PAR-1, the recent isolation of other PARs has brought into question the relative selectivity of the these compounds and the apparent lack of potency in particular preparations. For example, it has recently been shown that a derivative of BMS-197525 has partial activity at PAR-2, as well as PAR-1 in HEK cells (Kawabata et al., 1999b). Furthermore, although substituted peptide compounds have been shown to inhibit TRAP stimulation in general, they have been shown to be much less effective against thrombin stimulation. Although this may again be due to the use of preparations, where other PARs exist, another likely possibility is the disparity between the conformations achieved by the N-terminal tethered ligand in interaction with the receptor and by receptor-activating peptides in free solution. Considering the spatial constraints of groups in the SFLLRN agonist and the need for a rigid molecular structure, Andrade-Gordon and coworkers (1999) recently synthesized a peptide mimetic PAR-1-selective antagonist RWJ-56110, based on an indole template (Fig.3). This compound demonstrated consistent, relatively potent (approximately 1 to 300 μM), inhibitory actions against both thrombin- and SFLLRN-stimulated responses, including platelet aggregation and smooth muscle Ca2+ mobilization. Other nonpeptide PAR-1 antagonists, including FR171113 (Kato et al., 1999) and SCH 79797, one of a pyrroloquinazoline class of molecules (Ahn et al., 1999, 2000), have recently been identified (Fig. 3). Both compounds strongly inhibited SFLLRN- and thrombin-stimulated platelet aggregation, whereas the latter was also demonstrated to be selective for PAR-1 over PARs 2–4 (see Section VIII.B.). Taken together, these compounds represent good potential lead candidates for the future development of orally active PAR-1 antagonist drugs.
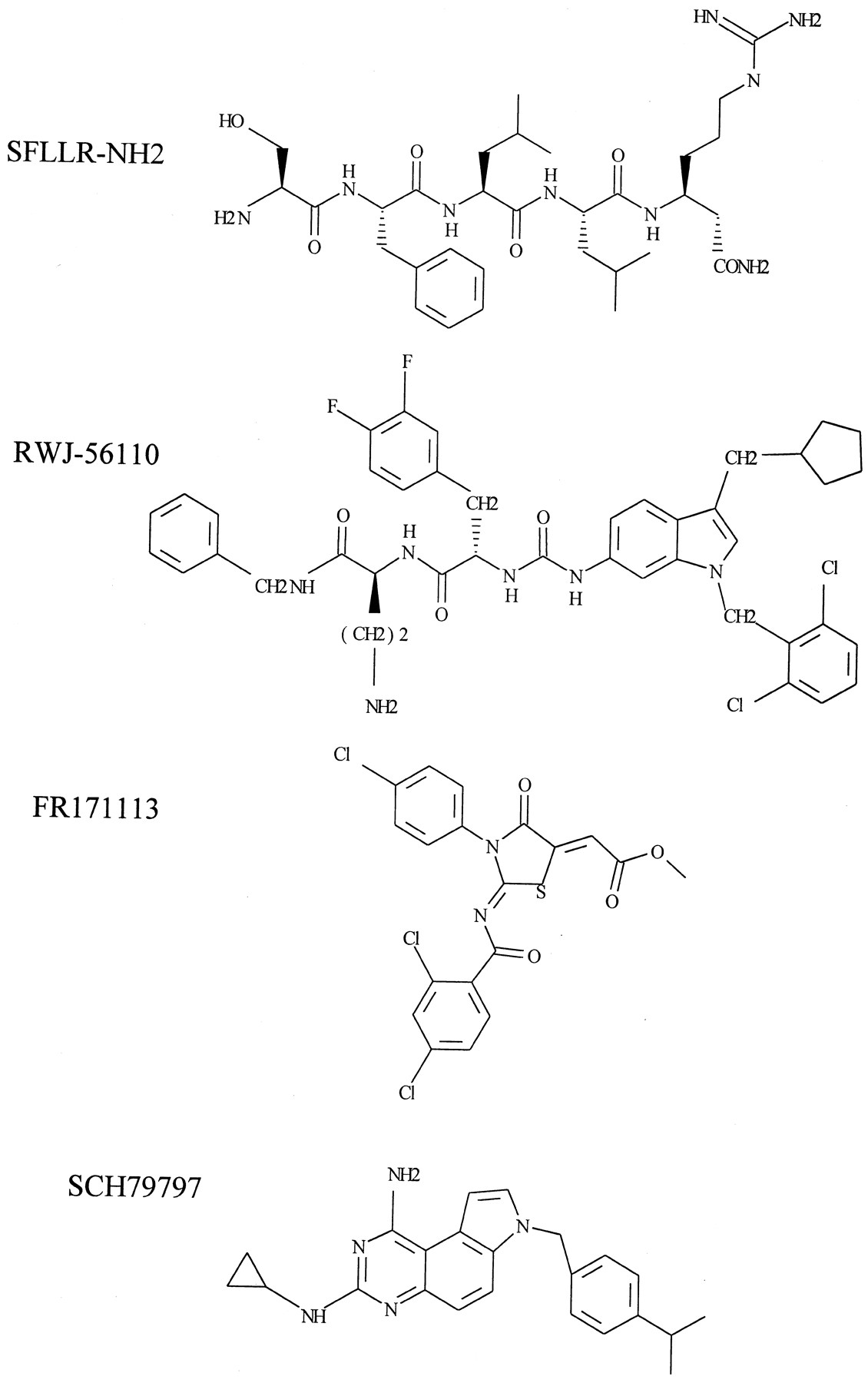
PAR-1 antagonists developed from thrombin receptor-activating peptides. Structural comparison of the thrombin receptor-activating peptide, SFLLR-NH2, and synthetic antagonists. RWJ5610, undeclared; FR171113 = 3-(4-chlorophenyl)-2-(2,4-dichlorobenzoylimino)-5-(methoxycarbonyl methylene)-1,3-thiazolidin-4-one; SCH79797=N-3-cyclopropyl-7-{[4-(1-methyl-ethyl)phenyl]methyl}-7H-pyrrolo[3,2-f]quinazoline-1, 3-diamine.
It should be noted that absolute potency estimations between synthetic agonists generated within different laboratories is difficult due to differences in assay systems used and initial estimates for the EC50 values of TRAPs that vary between studies. This also includes the potential for peptide degradation, since in one study substituting isoserine for Ser1 of SFLLRN was shown to confer resistance to cleavage by aminopeptidase M (Coller et al., 1993). This may generate artifactual differences in potency estimations, depending on the assay system involved. Second, many of these values have been generated using human platelet aggregation and other systems, which may be altered by the presence of PAR-4.
IV. Functional Responses to Proteinase-Activated Receptor-1 Activation
Many of the cellular effects of thrombin are consistent with a primary role in vessel wound healing and revascularization (Carney et al., 1992). This not only includes clot formation, but also effects upon a multitude of cell types known to play a role in the systemic response to vascular damage. Target cells for the effects of thrombin include not only platelets, endothelial, and smooth muscle cells, but also cell types such as neutrophils, leukocytes, neurons, and glial cells. Activation of a wide range of cell types therefore facilitates a co-ordinated response to vessel damage, including platelet aggregation, leukocyte extravasation, angiogenesis, nerve regeneration, and even initiation of a controlled immune response. Since thrombin-generating systems are primarily restricted to blood, few extravascular effects have been reported that cannot be either directly or indirectly extrapolated to vessel damage and repair. However, recent studies have indicated the presence of a thrombin generating system in brain, suggesting potential extravascular sites of thrombin production (Gingrich and Traynelis, 2000). It is therefore clear that thrombin, acting through PAR-1, is capable of affecting a wide range of physiological systems (see Table 2).
Cellular, tissue, and systemic effects of PAR-1 activation
A. Platelet Aggregation
A very large number of studies have now confirmed that thrombin is a major stimulus for platelets, initiating a series of co-ordinated events that result in platelet aggregation in vitro or in vivo (Eidt et al., 1988). Early attempts to characterize the action of thrombin upon platelet aggregation, prior to the cloning of PAR-1, demonstrated that aggregation was not due to generation of an active molecule from the clotting process, but rather involves a direct effect of the enzyme and requires protease activity (Davey and Luscher, 1967; Martin et al., 1975; Tam et al., 1980). Thrombin mediates shape change and stimulates the release of 5-HT (Harmon and Jamieson, 1986b), adenosine triphosphate (Detwiler and Feinman, 1973), thromboxane A2, and other granule contents. It also activates the plasma membrane localization of integrin αIIb/β3, which results in the binding of fibrinogen and von Willebrand factor and platelet aggregation (McGregor et al., 1989; Watts et al., 1989). In addition, thrombin also mediates the translocation of P-selectin and CD40 ligand to the plasma membrane, which facilitate the binding of platelets to endothelial cells (Stenberg et al., 1985; Henn et al., 1998). Other factors such as VEGF may also be released, which may promote endothelial cell growth as an initial step in angiogenesis (Mohle et al., 1997). Numerous studies have confirmed that these responses can be mimicked by PAR-1 activating peptides (Section III.) and involve a number of intracellular signaling events that regulate cytoskeletal reorganisation associated with the aggregation process (Section V.).
As part of the early attempts to characterize the action of thrombin on platelets, several groups demonstrated saturable125I radiolabeled thrombin binding to platelet membranes (Ganguly, 1974; Harmon and Jamieson, 1986b; Greco and Jamieson, 1991). However, extended analysis of radioligand binding data in platelets indicated the presence of three affinity binding states for thrombin with K d values of 0.3, 10, and 3 mM, respectively (Harmon and Jamieson, 1986b; Greco and Jamieson, 1991). Whereas the moderate and low affinity site are related to PAR-1 and possibly PAR-4 interactions, the high affinity thrombin binding site is likely to be associated with an interaction between the anion binding exosite of thrombin with the platelet membrane glycoprotein complexes GP1βα-IX-V complex (Berndt et al., 1986). High affinity thrombin binding is lost in platelets derived from patients with Bernard-Soulier syndrome (Demarco et al., 1991), a condition in which GP1βα is not expressed, or following preincubation with either monoclonal antibodies directed against GP1βα (Greco et al., 1996b) or the metalloprotease Serratia marcesens (Greco et al., 1996a), which removes 70–90% of GP1βα from the platelet surface. In these conditions, thrombin-induced platelet aggregation is either delayed or requires higher concentrations to be maximally effective (Demarco et al., 1991;Greco et al., 1996a,b), suggesting that binding to this site, although nonfunctional, nevertheless enhances thrombin function. Thrombin binding to GP1βα is believed to be within a specific “hirudin-like” extracellular cytoplasmic domain, spanning residues 271–284, within which lies a cluster of negatively charged amino acids that are common to other thrombin binding molecules (Demarco et al., 1994).
Although more recent studies have shown that125I- thrombin binds strongly to GP1βα rich fractions from solubilized platelets (Harmon and Jamieson, 1986a) or to cell lines expressing recombinant GP1βα IX-V functional complexes (Dong et al., 1997), at least one study has identified an additional thrombin binding site on platelets distinct from the GP1βα-IX-V complex. Inhibition of binding to this site by binding of a mutant thrombin, Quick II, enhances rather than reduces thrombin-stimulated platelet activation (Leong et al., 1992), suggesting that occupation of this site results in a negative regulation of platelet responsiveness. Furthermore, an antibody raised against the C terminus of hirudin has recently been shown to bind directly to a site on platelets distinct from GP1βα and PAR-1, despite the presence of a hirudin-like domain within these proteins (Hayes and Tracy, 1999).
Irrespective of the identity of the high-affinity binding site for thrombin in platelets, it is likely to have a functional significance. Clearly, such a site may either positively or negatively regulate the threshold concentration of thrombin required to initiate platelet aggregation. Another possibility may be that the high-affinity site acts to promote chemotaxis, binding low concentrations of thrombin, and targeting platelets to an area of higher thrombin concentration. Such a system would allow platelets to be attracted to specific sites where the formation of a thrombus was necessary, and the higher concentration of thrombin present could cleave PAR-1 and induce platelet aggregation. These possibilities await examination.
B. Endothelial Barrier Dysfunction, Chemotaxis, and Inflammation
A key component of the clotting and wound healing process is the activation of endothelial cells. Thrombin released from platelets stimulates the release of von Willebrand factor, cell surface redistribution of P-selectin, and increased expression of tissue factor and adhesion molecules, ICAM-1, VCAM-1, and E-selectin (Hattori et al., 1989; Bartha et al., 1993; Henn et al., 1998). These actions not only further promote the coagulation process and the binding and aggregation of platelets, but also facilitate the rapid adherence of neutrophils, monocytes, and later lymphocytes to the endothelial cell layer (Malik et al., 1986; Sugama and Malik, 1992; Zimmerman et al., 1994). Thrombin also stimulates endothelial cell contraction and increased permeability (Garcia et al., 1986; Malik et al., 1986; Lum and Malik, 1996) partly through the regulation of cell-to-cell junction organization (Rabiet et al., 1996). These events, along with increased adhesion molecule expression, facilitate rolling and transmigration of neutrophils and other cells to the site of vessel damage.
Concomitant with these effects, thrombin also stimulates aggregation of neutrophils and chemotaxis of neutrophils and monocytes (Bizios et al., 1986). However, several studies have demonstrated that the chemotactic response to thrombin is unrelated to the proteolytic properties of the enzyme, but rather the hirudin binding site appears to be the important feature of the protein (Bizios et al., 1986). The lack of PAR-1 expression on neutrophils (Jenkins et al., 1995), coupled with the noncatalytic nature of the interaction between thrombin and neutrophils, strongly indicates the presence of another binding site for thrombin on these cells. Such a binding site may be similar to that defined in platelets or to the nonproteolytically activated receptor found in other related cell types (Naldini et al., 1998). Further studies are required to confirm the existence and function of such a site on neutrophils; however, it is possible that all cells of megakaryocyte origin may possess a high thrombin affinity site to aid in cell movement.
Although thrombin is unable to induce Ca2+mobilization in neutrophils, consistent with a lack of PAR-1 expression, the PAR-1 agonist peptide SFLLRNPND has been shown to raise intracellular Ca2+ levels (Jenkins et al., 1995). Subsequent studies indicate that PAR-2 is expressed on neutrophils (Howells et al., 1997) and that TRAPs, in addition to activating PAR-1, are capable of activating this receptor (Lerner et al., 1996) (seeSection X.). It is therefore likely that other actions of TRAP on cells unresponsive to thrombin are due to PAR agonist cross-reactivity.
C. Cell Growth and Division
Thrombin, released from platelets, is a potent mitogen for cells of mesenchymal origin. In fibroblasts and vascular smooth muscle and endothelial cells, thrombin stimulates increases in DNA synthesis and promotes cellular proliferation with an efficacy comparable with serum (Chen and Buchanan, 1975; Carney et al., 1978; McNamara et al., 1993). These effects require thrombin serine-protease activity and are mimicked to some extent by SFLLLRNPNDKY-EPF (McNamara et al., 1993;Herbert et al., 1994), consistent with the effect being mediated by PAR-1. At lower concentrations, thrombin can act as a co-mitogen, suggesting roles as both a competence and progression factor. Thrombin is also mitogenic for selected cells of myeloid origin, such as lymphocytes, splenocytes (Chen and Buchanan, 1975), and other cells types, such as oesteoblasts (Abraham and Mackie, 1999). In addition to direct effects upon cell growth, thrombin also facilitates the production and release of promitogenic factors, such as PDGF and ET-1 through induction of PDGF and ET-1 prepro mRNA (Daniel et al., 1986;Yanagisawa et al., 1988; Garcia et al., 1993; Golden et al., 1998) and also regulates the subsequent release of these factors, in particular ET-1 (Kohno et al., 1992). Other similar actions of thrombin include the induction of receptors for VEGF, KDR and Flt (Maragoudakis et al., 2000) and the induction of TGF-β (Bachhuber et al., 1997). These effects provide a basis for synergy between thrombin, or other mitogens and/or the potential for thrombin to mediate mitogenesis indirectly through release of other factors.
Activation of PAR-1 also results in marked effects on the synthesis of extracellular factors that are now known to be important in the normal wound healing process and in the development of vascular disorders (seeSection XV.). Thrombin stimulates procollagen synthesis in smooth muscle cells and lung fibroblasts (Chambers et al., 1998;Dabbagh et al., 1998), and the expression of Cy61 and connective tissue growth factor (Pendurthi et al., 2000). Thrombin also regulates the induction and release of matrix metalloproteinases (MMPs), including progelatinase A (Zucker et al., 1995; Nguyen et al., 1999a) and MMPs 1, 2, and 3. These are key enzymes involved in degradation of the underlying basement membranes which, along with endothelial cell migration and proliferation, is an important first step in the initiation of angiogenesis. Consistent with these findings, thrombin has been shown to stimulate endothelial tube formation in matrigel and to stimulate angiogenesis in the chick chorioallantoic membrane system and in vivo (Tsopanoglou et al., 1993; Haralabopoulos et al., 1997). Thrombin also promotes MMP-2 release in vascular smooth muscle (Fernandez-Patron et al., 1999), suggesting that these events are common to many cells of the vasculature and are likely to participate in a co-ordinated wound healing process.
D. Neuronal Cell Survival
The effects of thrombin upon cell growth and division is not restricted to peripheral tissues. Both PAR-1 and prothrombin mRNA are expressed in a number of regions within the brain, such as the thalamus, hypothalamus, cortex, and cerebellum (Weinstein et al., 1995), indicating the presence of a functional thrombin effector system in the brain. Indeed, in neuronal cells, thrombin or TRAPs mediate neurite retraction and reversal of astrocyte stellation (Gurwitz and Cunningham, 1988; Grand et al., 1989; Cavanaugh et al., 1990; Beecher et al., 1994; Suidan et al., 1996), stimulate astrocyte proliferation (Grabham and Cunningham, 1995), and can protect against neuronal cell death induced by β-amyloid, oxidative stress, or hypoglycemia (Vaughan et al., 1995; Pike et al., 1996). Furthermore, biochemical studies show increased synthesis of nerve growth factor and ET-1 in response to thrombin (Ehrenreich et al., 1993; Neveu et al., 1993), and a decrease in the expression of some subtypes of the metabotropic glutamate receptor (Miller et al., 1996). Taken together, these findings support a role for thrombin in mediating neuronal cell survival at least in response to some environmental insults.
At higher concentrations, thrombin per se causes death of hippocampal neurones (Pike et al., 1996) and in some studies can, at lower concentrations, potentiate β-amyloid-induced cell death (Smith-Swintosky et al., 1995, 1997). These contradictory results suggest that thrombin, as well as aiding neuronal cell survival, may also function as a mediator of some disease states. For example, in Alzheimer's, the levels of an endogenous inhibitor of thrombin, protease nexin-1, have been shown to be reduced (Vaughan et al., 1994,1995). This might lead to neuronal damage due to the presence of higher effective concentrations of thrombin. It has also been postulated that higher levels of systemic thrombin perhaps entering the brain following damage to the blood-brain barrier may act as a neurodegenerative agent.
E. Cardiovascular Responses
Thrombin and TRAPs mediate a substantial endothelial-dependent relaxation of aortic and coronary blood vessels from species such as rat, guinea pig, and dog in vitro (Muramatsu et al., 1992; Tesfamariam, 1994a,b; Zaleski and Ku, 1993; Ku and Dai, 1997). This is likely to be mediated by both release of cyclooxygenase products, including possibly prostaglandin I2 and by nitric oxide (NO), because many of the responses can be reversed by indomethacin andl-NAME or related analogs (Zaleski and Ku, 1993; Ku and Dai, 1997). Following removal of the endothelium thrombin, in some preparations, generates strong contractile responses (Zaleski and Ku, 1993; Ku and Dai, 1997) consistent with expression of PAR-1-linked Ca2+ influx in the underlying smooth muscle (Deblois et al., 1992; Antonaccio et al., 1993; Antonaccio and Normandin, 1994). In other vessels, for example, human umbilical and placental arteries, contractile responses can prevail even in endothelium intact vessels (Tay-Uyboco et al., 1995), indicating differences in the relative expression and function of PAR-1 on endothelial and smooth muscle cells in different vessels. These differences are reflected in whole organ responses to PAR-1 activation: administration of TRAPs causes vasodilation in perfused piglet lung, but vasoconstriction in the guinea pig (Pinheiro et al., 1993; Lum et al., 1994). In coronary vessels in vivo, TRAP generates a transient increase in blood flow followed by a sustained decrease (Damiano et al., 1996a). Indeed, the contractile effects of PAR-1 activation in coronary vessels can also mediate secondary changes in heart function, such as decreases in cardiac output and mean arterial pressure (Damiano et al., 1996a,b), despite the fact that thrombin can directly stimulate both via a positive ionotrophic effect through increased intracellular Ca2+ (Steinberg et al., 1991).
Administration of TRAP to mice in vivo causes a rapid hypotension followed by a sustained moderated hypotension (Darrow et al., 1996;Cheung et al., 1998). However, when NO release is prevented following pretreatment with l-NAME, a rebound hypertension is revealed reflecting the expression of PAR-1 on vascular smooth muscle. Despite these findings, a physiological role for thrombin in the regulation of cardiovascular function is not overwhelming, since in mice deficient in PAR-1, parameters of cardiac function and blood pressure are not different from normal mice (Darrow et al., 1996). It is, therefore, more likely that thrombin plays a role in the control of local blood flow following tissue damage.
V. Proteinase-Activated Receptor-1-Mediated Cellular Signaling
A. Coupling to Heterotrimeric G-Proteins
In common with several other helipthical receptors, PAR-1 has been shown to couple to multiple heterotrimeric G-proteins (Fig.4). In a number of early studies, two main signaling events were characterized that were assumed to involve receptor G-protein coupling. The first event involves the inhibition of cAMP through interactions with inhibitory G-protein of the Gi class (Hung et al., 1992; Kanthou et al., 1996). The second event is stimulation of phospholipase C (PLC)-catalyzed hydrolysis of polyphosphoinositides, resulting in the formation of InsP3, mobilization of intracellular Ca2+, and generation of diacylglycerol, the endogenous activator of protein kinase C (PKC) (Babich et al., 1990;Hung et al., 1992). Thrombin also stimulates the rapid hydrolysis of other phospholipids, implying roles for PLD, PLA2and phosphatidylcholine-specific PLC in the initial generation of lipid activators of protein kinase C isoforms (McNicol and Robson, 1997;Cheng et al., 1999).
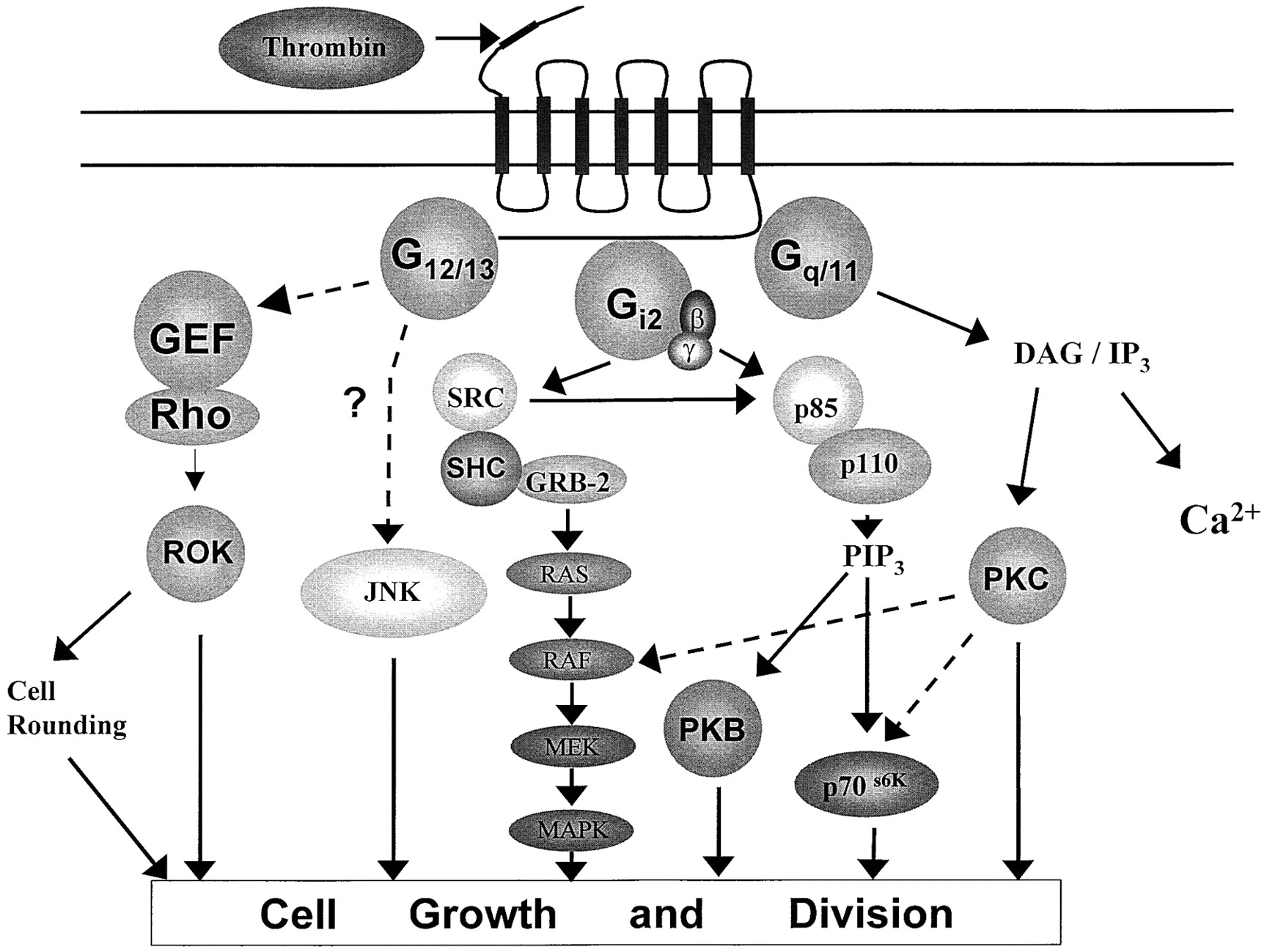
G-protein-dependent signaling pathways regulated through activation of PAR-1. The major signaling events regulated by PAR-1. Dashed lines represent putative pathways where the intermediates have not been identified or fully demonstrated for PAR-1, but are a feature of other G-protein-coupled receptors. Other signaling pathways implicated in PAR-1 activation are outlined in Table 3. Some well identified intermediates and precursors have been omitted for sake of clarity. DAG, diacylglycerol; SRC, pp60src and related kinases; MAPK, p42/44 mitogen-activated protein kinase; PKB, protein kinase B; GER, Rho GTP exchange factor; PIP3, phosphatidylinositol 3,4,5-trisphosphate.
The recent identification of multiple G-protein subunits and their corresponding effector enzymes allowed examination of these transduction mechanisms. Microinjection of antibodies directed against Gq/11 into CCL-39 cells inhibited PAR-1-mediated Ca2+ mobilization (Baffy et al., 1994), whereas the same antibodies abrogated GTPase activity in thrombin-stimulated platelet membranes (Benka et al., 1995). Furthermore, in platelets derived from transgenic mice lacking Gq, thrombin-stimulated phosphoinositide hydrolysis was abrogated (Offermanns et al., 1997).
A direct interaction between PAR-1 and Gq/11 and Gi2 has been recently demonstrated by immunoprecipitation of PAR-1 with Gi2 and Gq/11 in thrombin-stimulated human neuroblastoma SH-EP cells (Ogino et al., 1996), clearly indicating interaction of PAR-1 with these two G-protein subunits. In a number of cell systems, pertussis toxin (PTX)-mediated ADP ribosylation of Gi/Go α-subunits also reduced thrombin-stimulated InsP3 formation and Ca2+, indicating the potential for coupling of PAR-1 to Gi/Go subunits (Babich et al., 1990; Brass et al., 1991). Antibodies to Go also reduced PAR-1-mediated responses (Baffy et al., 1994), suggesting that this subunit contributes to PLC activation. However, Go expression is cell-specific, and it is likely that another pertussis sensitive G-protein, possibly Gi2, may also be involved. At present, it remains unclear for thrombin receptor systems whether βγ subunits derived from Gi2 or Go are able to activate other isoforms of PLC-β, such as PLC-β2 or PLC-β3.
PAR-1 is also linked to other second messenger systems via pertussis-sensitive G-proteins. Thrombin-mediated inhibition of adenylyl cyclase has been demonstrated to involve a direct interaction of the receptor with Gi2 (Hung et al., 1992;Kanthou et al., 1996; Magnaldo et al., 1988; Swift et al., 2000). Stimulation of other phospholipase activity, such as PLD and PLA2 has also been shown to be sensitive to PTX in some cell types (Banga et al., 1988; Suzuki et al., 1996). However, evidence supporting a direct interaction between the receptor and a G-protein α-subunit in a manner analogous to Gq/11/PLC-β1 is minimal. One study has shown that expression of a mutant Gi2 protein can specifically inhibit arachidonic acid release in response to thrombin (Winitz et al., 1994) through a mechanism that does not involve intermediates known to regulate PLA2 activity. In general, regulation of these phospholipases following PAR-1 activation is likely to be downstream of initial activation of PLC-β isoforms and, indeed, in cells where PLC-β1 is poorly expressed, thrombin stimulation of PLD and cPLA2 is diminished (Fee et al., 1994).
Recent studies have also demonstrated that PAR-1 also transduces important cell signals via G12 and G13. In platelet membranes, thrombin stimulates the incorporation of the photoreactive GTP analog [α-32P]GTP azidoanilide into G12 and G13 as assessed by immunoprecipitation studies (Offermanns et al., 1994), suggesting a direct interaction of both G-protein α-subunits with PAR-1. Furthermore, injection of antibodies directed against G12 prevents thrombin-mediated gene transcription and DNA synthesis (see below) strongly implicating a PTX-insensitive, and thus Gi/Go-independent mechanism, as being responsible for many of the cellular effects of PAR-1.
B. Regulation of Kinase Signaling Cascades by Proteinase-Activated Receptor-1
Although thrombin was able to activate PKC isoforms in several cell types, principally by hydrolysis of multiple phospholipids (Baron et al., 1993; Godin et al., 1995), other pathways were implicated in the pro-mitogenic effects of PAR-1 activation. This was based on several key observations. Firstly, thrombin was found to be a potent mitogen relative to other G-protein coupled receptor agonists, despite generating comparable phospholipid and Ca2+signals (Seuwen et al., 1990). Second, PAR-1-activating peptides stimulated inositol phosphate formation to a level comparable with thrombin itself but were unable to stimulate mitogenesis (Vouret-Craviari et al., 1992) and lastly, in a number of cell types thrombin-stimulated mitogenesis was PTX-sensitive while phospholipid hydrolysis was PTX-independent (Babich et al., 1990).
Since the identification of the mitogen-activated protein (MAP) kinases, key signaling events central to the action of thrombin have been identified (see Fig. 4). Multiple signaling paradigms have since been established for PAR-1, including activation of PI-3 kinase (Mitchell et al., 1990; Walker et al., 1998), Src family tyrosine kinases (Cichowski et al., 1992; Rao et al., 1995), stress-activated protein (SAP) kinases (Mitsui et al., 1997, 1998; Malcolm et al., 2000;), Rho kinase (ROK) (Seasholtz et al., 1999; Carbajal et al., 2000), Janus activated kinase-2 (JAK-2) (Rodriguez-Linares and Watson, 1994; Huang et al., 2000), focal adhesion kinase, pp125fak (Negrescu et al., 1995; Choudhury et al., 1996), and proline-rich tyrosine kinase 2 (Pyk-2) (Ohmori et al., 2000) (see Table 3).
PAR-1 regulated kinases
C. Mitogen-Activated Protein Kinase and Phosphatidyl Inositol-3 Kinase Cascades
A paradigm for the activation of p42/44 MAP kinase or extracellular-regulated kinases (ERKs) has now been established for tyrosine kinase-linked receptors (Malarkey et al., 1995). Phosphotyrosine residues within the intracellular domain of an activated receptor interact with the adaptor protein SHC that in turn recruits GRB-mSos resulting in increased rate guanine-nucleotide exchange by the monomeric G-protein p21ras. This initiates binding of Raf-1 isoforms to the plasma membrane for activation by Ras and some other factor, and downstream activation of MEK-1, the direct activator of MAP kinase. Multiple variations of this model can be applied for a number of growth factors and G-protein coupled receptors, and depending on cell type, PAR-1 incorporates many components of such a paradigm (Fig. 4).
Early studies demonstrated that thrombin stimulated p42/44 MAP kinase activation was essential for initiation of DNA synthesis (Pages et al., 1993). However, in contrast with agonists for other G-protein-coupled receptors, thrombin was also found to stimulate a biphasic activation of p42/44 MAP kinase, the sustained phase of which was essentially PTX-sensitive (Kahan et al., 1992). Furthermore, thrombin was found to stimulate GTP/GDP exchange on p21ras indicating the potential for a ‘growth factor-like’ MAP kinase cascade to also be activated via PAR-1 and other G-protein-coupled receptors, such as the receptor(s) for lysophosphatidic acid (Van Corven et al., 1993). In this instance, activation of p21ras was inhibited by both PTX pretreatment and genestein, a nonselective tyrosine kinase inhibitor, suggesting the involvement of both a Gi protein and tyrosine kinase in mediating the activation of p42/44 MAP kinase by PAR-1. Several recent studies have shown for other G-protein-coupled receptors, although not for PAR-1, a role for βγ-subunits in the activation of Src, tyrosine phosphorylation of p52SHC, and formation of SHC-GRB-2 complexes as a mechanism by which Gi-dependent activation of p42/44 MAP kinase could be achieved. Thrombin-mediated stimulation of pp60src and phosphorylation of SHC has been demonstrated in a number of cell types (Chen et al., 1994b, 1996b; Rao et al., 1995), consistent with this model of ERK activation. However, these events are not in all instances PTX-sensitive and indicate the potential for Gi-independent pathways to regulate early events in the MAP kinase signaling cascade (Chen et al., 1996b). Recently in some cell types, G protein-coupled receptor agonists such as lysophosphatidic acid and thrombin have also been found to stimulate the tyrosine phosphorylation of growth factor receptors such as the basic fibroblast growth factor (Weiss and Maduri, 1993) and insulin-like growth factor-1 receptors (Delafontaine et al., 1996) resulting in the recruitment to the receptor of SHC and other intermediate proteins, and the subsequent activation of the MAP kinase signaling cascade, a phenomenon known as transactivation. This is likely to involve pp60src or a similar tyrosine kinase; however, the events that regulate these events have not been elucidated.
A similar mode of activation of other signaling pathways may also be a feature of PAR-1. PI-3 kinase plays important roles in thrombin-mediated regulation of cytoskeletal structure, cell motility, cell survival, and mitogenesis and, also in some cell types, functions as an intermediate in activation of ERKs (Malarkey et al., 1995;Touhara et al., 1995). Thrombin stimulates the accumulation of PtdIns(3,4,5)P3 in platelets, neutrophils, human and bovine airway smooth muscle cells, and others through activation of multiple PI-3 kinase isoforms, including a novel 110-kDa isoform that can be directly activated by G-protein βγ-subunits, rather than through binding of the tyrosine kinase receptor-associated protein p85 PI-3 kinase. In platelets, thrombin-stimulated PI-3 kinase activity involves the small molecular weight G-protein Rho (Zhang et al., 1995). In addition, activity can also be regulated by sequestration of G-protein βγ-subunits, consistent with a role for γ-p110 and thus a Gi/Go-dependent pathway. This latter model of activation is likely to be restricted to certain cell types, where PAR-1 mediated second messenger formation is largely PTX-sensitive, although it is likely that even in a single cell type multiple pathways for activating PI-3 kinase isoforms exists. In platelets, the activity of other small molecular weight G-proteins, such as Rac and Cdc42, may also be regulated through PAR-1 activation, although their inter-relationship with PI-3 kinase signaling and aggregation remains unclear (Azim et al., 2000).
In both human and bovine airway smooth muscle cells and in pulmonary artery fibroblasts, PI-3 kinase is implicated in PAR-1-mediated activation of p70s6k (Belham et al., 1997; Walker et al., 1998; Johanson et al., 1999; Krymskaya et al., 1999), and protein kinase B (Walker et al., 1998), two important regulators of cell survival and mitogenesis. Although thrombin-stimulated p70s6k is partially PTX-sensitive in pulmonary artery fibroblasts, suggestive of the involvement of a βγ-regulated form of PI-3 kinase, in human and bovine airway smooth muscle cells, thrombin has been shown to stimulate the tyrosine phosphorylation of the classical growth factor receptor-associated p85/110 isoform (Walker et al., 1998; Krymskaya et al., 1999). Thus, as with activation of p42/44 MAP kinase cascade, intermediate stimulation of pp60src and/or transactivation of a growth factor receptor is also likely to be involved in the activation of this pathway.
D. G12-Dependent Proteinase-Activated Receptor-1 Signaling
In some cell types where PTX-independent cellular responses to PAR-1 have been recorded, G12/G13 signaling pathways have been implicated. Injection of antibodies directed against G12 abolished thrombin-stimulated DNA synthesis in 1321N1 astrocytoma cells (Aragay et al., 1995), whereas reconstitution of PAR-1 with G12 in COS-7 cells gives rise to substantial AP-1-mediated gene expression in response to thrombin (Post et al., 1996). This is in turn likely to be mediated via Ras- or Rac-dependent activation of JNK, as expression of a dominant negative mutant of MEKK-1, an upstream regulator of JNK, or mutant forms of Ras and Rac inhibit thrombin-stimulated AP-1 gene expression in NIH3T3 cells (Collins et al., 1996). Recent evidence also suggests that G12 is essential for thrombin-stimulated tyrosine phosphorylation of SHC and AP-1 reporter activity (Collins et al., 1997), whereas Src has been implicated in JNK activation mediated by G12 (Nagao et al., 1998). Thus, it is possible that PAR-1 may utilize G12 to activate Src, resulting in the phosphorylation of SHC, activation of JNK and regulation of AP-1 activity. Further studies are required, however, to confirm whether this pathway mediates JNK activation in other PAR-1 expressing cell systems. Thrombin has also been shown to activate both JNK and p38 MAP kinase in other cell types, and both PTX-sensitive G-proteins and PKC have been implicated, suggesting additional roles for Gi- and Gq-dependent pathways (Mitsui et al., 1997, 1998; Malcolm et al., 2000).
In other cell types however, G12 has also been implicated in the regulation of Rho-dependent events initiated via PAR-1. As well as being implicated in the regulation of PI-3 kinase, JNK and others, Rho plays an intimate role in the regulation of cellular responses to thrombin through activation of a number of target kinases, including in particular ROK. PAR-1-mediated responses in which Rho or ROK have been implicated include: the activation of cell rounding and apoptosis in cultured neurones and astrocytes (Donovan et al., 1997; Majumdar et al., 1998), stimulation of smooth muscle DNA synthesis and cell migration (Seasholtz et al., 1999), stress fiber formation (Crouch, 1997), platelet aggregation (Zhang et al., 1995), endothelial cell and smooth muscle contraction (Essler et al., 1998), and endothelial cell barrier dysfunction (Vouret-Craviari et al., 1998;Carbajal et al., 2000). Many of these events are also activated by G12- or G13-dependent mechanisms, and recent studies have provided evidence for the direct coupling of G12 to Rho via a group of Rho-specific guanine nucleotide exchange factors (Majumdar et al., 1998; Fukuhara et al., 1999). Taken together, these studies suggest that the G12/Rho/Rho kinase axis may represent a new and important pathway in mediating PAR-1 response in a variety of cell types.
VI. Desensitization of Proteinase-Activated Receptor-1
The intramolecular basis for PAR-1 activation through the generation of a tethered N terminus ligand has important implications for the magnitude and kinetics of thrombin responses. Firstly, a single thrombin molecule may proteolytically cleave multiple thrombin receptors and secondly, cleavage could result in sustained activation of each receptor. This does not seem to be the case, however, because the extent of phosphoinositide hydrolysis is directly proportional to the concentration of thrombin (Ishii et al., 1993). This implies the generation of a fixed “quanta” of second messenger followed by a rapid termination mechanism. Thus, PAR-1 desensitization has been examined in considerable detail and closely compared with that observed in other G-protein-coupled receptors activated through a normal ligand/receptor mechanism (Hein et al., 1994; Vouret-Craviari et al., 1995). For such receptors, desensitization essentially entails uncoupling of the receptor from the G-protein, followed by subsequent internalization (Bohm et al., 1997) (Fig.5). Desensitization also includes the potential for the long-term down-regulation of mRNA expression (Weinstein et al., 1998); however, relatively few studies of this type have been performed regarding PAR-1.
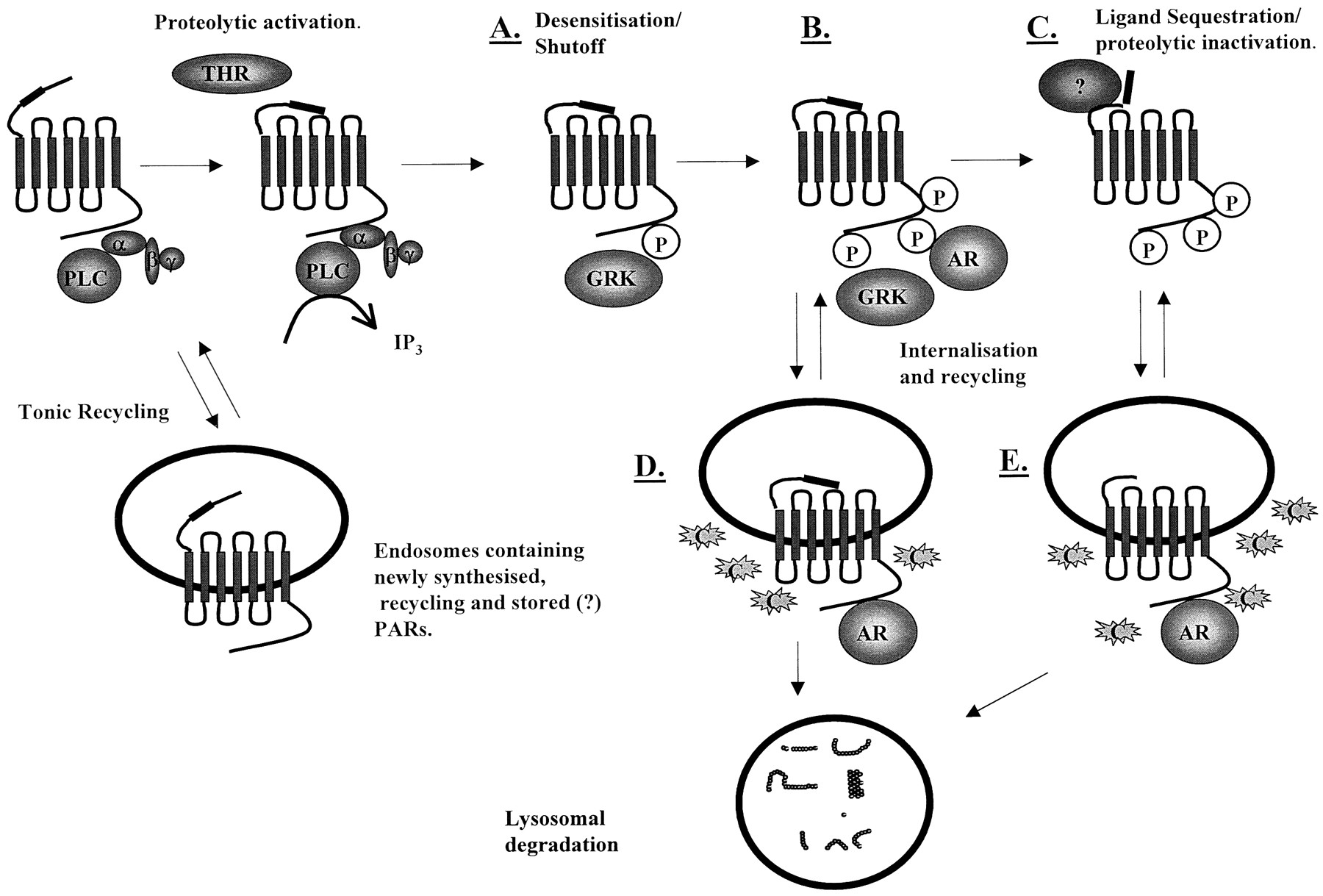
Modes of PAR-1 desensitization. C-terminal phosphorylation at distinct residues can mediate rapid receptor shutoff (A), followed by further phosphorylation and receptor internalization (B). Additional modes of receptor desensitization may involve either sequestration and or proteolytic degradation of the tethered ligand (C). It is unclear whether receptor internalization predominantly occurs immediately following phosphorylation (D), and a portion of this population recycled to the plasma membrane before sequestration, or whether the ligand sequestration event occurs prior to any internalization (E). Indeed, if the latter scenario is the case, then this pool of internalized receptors must itself recycle to some degree and presumably remain sensitive to soluble agonist. AR, β-arrestin; C, clathrin; α, β, and γ, G-protein subunits; THR, thrombin.
A. Phosphorylation and Internalization
In Rat1a fibroblasts transfected with PAR-1, thrombin stimulates a rapid, PKC-independent phosphorylation of the receptor (Ishii et al., 1994). This finding and the presence of consensus GRK phosphorylation sites in PAR-1 (Ser-391 and Ser-395) strongly suggest a principal role for G-protein receptor kinases in agonist-induced receptor phosphorylation. Indeed, injection of GRK-3 into oocytes substantially reduces thrombin-stimulated Ca2+ signaling (Ishii et al., 1994). Furthermore, it has been shown that GRK-3 is more potent in producing this effect than GRK-2, demonstrating receptor specificity in the GRK involved. In transgenic mice overexpressing GRK-3, thrombin-stimulated MAP kinase signaling is selectively inhibited, whereas AII receptor-mediated events remain unaffected (Iaccarino et al., 1998). Cell-type specificity is likely to be a feature of GRK-mediated phosphorylation of PAR-1, however, since in endothelial cells GRK-5 rather than GRK-3, is involved (Tiruppathi et al., 2000). GRK-mediated desensitization is dependent upon receptor occupancy and, at high concentrations of thrombin, other kinases may be activated that regulate phosphorylation. This may involve protein kinase C isoforms, since the PKC activator phorbol 12-myristate 13-acetate has previously been shown to promote PAR-1 phosphorylation (Ishii et al., 1994), and PKC-β has been demonstrated to be involved in heterologous desensitization of thrombin receptor in endothelial cells (Yan et al., 1998). Recently, Ido et al. (1996) isolated a novel 33-kDa kinase from platelets, which is able to phosphorylate a GST-fusion protein of the PAR-1 cytoplasmic tail (Ido et al., 2000), although it is unclear if it functions similarly in vivo.
Structure/function studies have also confirmed that the main site for phosphorylation dependence is within the C-terminal tail. The 5-HT2 receptor is characterized by a slow and very limited form of desensitization that does not involve phosphorylation (Vouret-Craviari et al., 1995). However, replacement of the C-terminal tail of the 5-HT2 receptor with that of PAR-1 confers a rapid and substantial desensitization in response to 5-HT, with similar kinetics to desensitization of thrombin-stimulated PAR-1, accompanied by marked phosphorylation of the receptor. Modification of the Ser/Thr phosphorylation sites within the C terminus to Ala also renders PAR-1 insensitive to GRK-3 and also potentiates thrombin-stimulated Ca2+signaling (Ishii et al., 1994).
At present, these studies have not identified specific AA residues within the C terminus that are critical for desensitization, although a recent study has demonstrated that phosphorylation sites within the C terminus region, between Ser391 and Ser406, reduce receptor inactivation time (“shutoff”) (Hammes et al., 1999). However, these residues appeared to have no effect on internalization, suggesting that there are two separate components of the desensitization process. It is also unclear whether sites within other intracellular regions of the receptor can also contribute to desensitization, such as the third intracellular loop, as with the α2A-adrenergic receptor (Jewell-Motz et al., 2000), or if only the C terminus defines the characteristics of PAR-1 desensitization.
In addition to intracellular phosphorylation events, other recent studies have also provided evidence in favor of additional extracellular proteolytic events that mediate the inactivation of tethered ligand. Initially, it was found that aminopeptidase M, a plasma protease, was able to inhibit PAR-1-induced platelet aggregation in response to TRAPs, but not thrombin (Coller et al., 1992) through cleavage of the peptide's N-terminal serine residue. However, a later study noted that whereas responses to SFLLRN could be reversed by treatment with aminopeptidase M or thermolysin (Chen et al., 1996a), only thermolysin reversed the response to thrombin. Since these thermolysin-desensitized receptors remained responsive to SFLLRN, this suggests a specific protease-mediated destruction of the N terminus tethered ligand. In support of this idea, plasmin has also been demonstrated to desensitize thrombin-dependent Ca2+ signaling through cleavage at sites distal to Arg41 (Kuliopulos et al., 1999).
Interestingly, since thrombin-desensitized receptors can be activated by soluble ligand peptide (Hoxie et al., 1993; Hammes and Coughlin, 1999) and yet peptide stimulation itself leads to rapid receptor phosphorylation (Hammes et al., 1999), there may be some additional mechanism of receptor shutoff involving removal of the N-terminal ligand from within the binding pocket of ECL-2, a process known as ligand sequestration (see Fig. 5). This is supported by the observation that a mutant thrombin receptor possessing an SFFLRN-trypsin cleavage site C-terminal to the thrombin cleavage site can be activated by trypsin after thrombin desensitization (Hammes and Coughlin, 1999). Although both proteolytic degradation and sequestration of the N-terminal ligand are of interest, it is unclear whether these processes are physiologically relevant or apply to more than a small proportion of the total PAR-1 receptor population. Clearly, further studies are required to separate closely interlinked events in the overall process of desensitization and their relative importance.
B. Protein-Activated Receptor-1 Endocytosis and Trafficking
Differences in endocytosis have been observed between PAR-1 and other G-protein-coupled recycling have receptors. Initially as with other receptors, PAR-1 is sequestered from the cell surface into coated pits and then into endosomes within the first 60 s of activation (Hoxie et al., 1993). Interestingly, cleavage of PAR-1 is not required to promote internalization because the peptide-simulated receptor also follows this route, suggesting the unique activation mechanism does not define the mode of internalization. However, whereas PAR-1 is internalized into the early endosomes, unlike several other receptors, a large proportion of PAR-1 then moves to the lysosomes for degradation. The C terminus of PAR-1 is crucial in directing lysosomal sorting, as a PAR-1 mutant bearing the cytoplasmic tail of the substance P receptor is able to immediately recycle to the plasma membrane (Trejo et al., 1998). A substance P receptor with a PAR-1 cytoplasmic tail is, however, directed to lysosomes (Trejo and Coughlin, 1999). Recycled PAR-1 with a substance P receptor C terminus seems to be constitutively active, a condition that may not reflect the fate of endogenous PAR-1, since a proportion of PAR-1 that escapes lysosomal sorting and returns to the surface cannot normally be reactivated by thrombin (Hoxie et al., 1993). This confirms that phosphorylation within specific regions of the C terminus may cause dissociation of the tethered ligand from the receptor activation site and receptor shutoff per se. This also provides further evidence that receptor inactivation and internalization may be distinct processes.
The resensitization of PAR-1 responses also involves a number of distinct mechanisms. In a number of cell types, PAR-1 resides both on the cell surface and in a substantial intracellular pool. Naive receptors cycle tonically between the cell surface and this pool by an undefined mechanism that is physically distinct from agonist-triggered trafficking and is independent of C-terminal S/T phosphorylation (Shapiro et al., 1996). Studies using a series of C-terminal mutants showed that tonic cycling required a domain between Lys397 and Tyr407 within the cytoplasmic tail, a region also involved to some extent in agonist-induced internalization (Shapiro et al., 1996; Shapiro and Coughlin, 1998). Thus, phosphorylation within this domain and others may therefore distinguish agonist-induced trafficking and tonic cycling of PAR-1. The tonic cycling of nonactivated receptors is not surprising because it provides a rapid source of free receptor for reactivation without recourse to new receptor synthesis.
This potential intracellular pool of PAR-1 is not, however, likely to be involved in PAR-1 resensitisation in every cell type. The intracellular pool of PAR-1 is limited to membranes of the surface connecting system in platelets, limiting the capacity of the cells to regain thrombin responsiveness (Molino et al., 1997a). In other megakaryoblastic cell lines, PAR-1 recovery is also slow and likely to involve new protein synthesis because there appears to be no intracellular pool of receptors (Hoxie et al., 1993; Brass et al., 1994). In contrast, cells of endothelial origin tend to possess substantial intracellular pools probably associated with the Golgi apparatus (Storck et al., 1997), which can lead to partial recovery of thrombin responsiveness within 90 min (Storck and Zimmermann, 1996;Ellis et al., 1999). Studies in human umbilical vein endothelial cells have demonstrated that cleaved receptors are internalized in two distinct steps, with 60% being internalized rapidly and the rest requiring several hours, with no recycling of cleaved receptors (Woolkalis et al., 1995). In megakaryoblastic cells, however, more than 90% of receptors are internalized rapidly, with up to 40% of cleaved receptors being recycled. Thus, resensitization is likely to be cell type-specific, dependent upon the initial mechanism of desensitization, the availability of intracellular receptor pools and other mechanisms. Other studies indicate differences in resensitization profiles in cells at different stages of confluency (Woolkalis et al., 1995) and in cultured cells relative to cells studies in situ (Mizuno et al., 2000), suggesting the involvement of other mechanisms currently undefined.
VII. Cloning of Proteinase-Activated Receptor-2
Although the cloning of PAR-1 was a major advance in the understanding of the physiological actions of thrombin, the possibility of other serine-protease-activated receptors was likely. It had been noted that the effects of thrombin on cells could not entirely be reproduced by addition of activating peptide (Vouret-Craviari et al., 1992; Kinlough-Rathbone et al., 1993). Hence, the presence of a second thrombin receptor in platelets was postulated. However, Southern blotting experiments with genomic DNA failed to identify a candidate until a unique DNA sequence encoding a G-protein-coupled receptor was isolated from a mouse genomic library (Nystedt et al., 1994).
Moderate stringency hybridization with a mixture of two oligonucleotide primers corresponding to regions of the bovine substance K receptor was used to probe the mouse library. A cosmid clone containing a 3.7-kb Pst-1 fragment with an open reading frame encoding a putative 395 amino acid protein similar to that of the human thrombin receptor. Hydropathy analysis revealed seven putative transmembrane-spanning helices and an amino terminal sequence probably corresponding to a signal peptide. The amino acid sequence was found to be most closely related to the human thrombin receptor, with 30% identity and shared 28% identity with the mouse isoform. Significant heterogeneity was observed in the extramembranous domains, including the C-terminal tail and the N terminus that is 29 amino acids shorter than in the thrombin receptor and lacks a hirudin-like thrombin-binding domain.
However, when the putative receptor was expressed in Xenopusoocytes, thrombin was unable to stimulate calcium release. Low concentrations of trypsin had also been demonstrated to activate the thrombin receptor, and this protease was now found to strongly activate calcium release from oocytes containing the receptor, now designated PAR-2. Half-maximal response to trypsin was found to be about 1 nM, several hundred-fold lower than displayed by oocytes expressing human thrombin receptor.
Analysis of the PAR-2 N-terminal amino acid sequence revealed a possible trypsin cleavage site at Arg34 (Fig. 2). The peptide, SLIGRL, derived from the receptor sequence corresponding to the probable tethered ligand, was able to elicit calcium release from PAR-2 expressing oocytes with an approximate EC50 of 5 μM. Mutation of receptor Ser35 to a trypsin-resistant Pro, yielded a receptor that could not be activated by trypsin, whereas activation by SLIGRL remained unaffected. Furthermore, Northern blot analysis revealed PAR-2 transcripts in tissues, such as the kidney, small intestine, stomach, and eye—a distribution markedly different from that observed for PAR-1.
Despite the initial cloning of the new receptor, it was possible that the PAR-2 sequence isolated by this strategy did not represent the entire protein. Since the PAR-2 construct had been cloned from genomic DNA, it was possible that RNA splicing could produce a transcript that encoded a different receptor. Indeed, use of an exon trap vector strategy (Buckler et al., 1991) allowed the isolation of a PAR-2 PCR fragment containing a splice acceptor site (Nystedt et al., 1995b). Hybridization of a mouse stomach cDNA library using a probe derived from the genomic PAR-2 sequence identified a clone that contained an open reading frame of 1197 nucleotides. This cDNA was identical to the genomic sequence, except for the 5′ sequence up to codon 30. This resulted in the alteration of five amino acids in the mature PAR-2 from that previously described, but with no alteration in the proposed trypsin cleavage site.
The gene encoding human PAR-2 was then isolated from a human genomic DNA library, using hybridization to a probe derived from the 3′ exon of the mouse PAR-2 gene (Nystedt et al., 1995a) and subsequently cloned from human kidney cDNA (Nystedt et al., 1995a; Bohm et al., 1996b). Consistent with PAR-1, the human PAR-2 gene was also found to consist of two exons and was localized to chromosome 5q13, separated from PAR-1 by only 90 kb of DNA.
However, whereas human and mouse PAR-2 isoforms were shown to share 83% overall identity, trypsin -mediated cleavage at Arg36 and Ser37 in hPAR-2 generated a distinct N-terminal tethered ligand sequence, SLIGKV. Chinese Hamster Ovary cells transfected with human PAR-2 were found to respond to trypsin, both human (SLIGKV) and mouse (SLIGRL)-activating peptides and in addition hTRAP (SFLLRNP) (Nystedt et al., 1995a). Cells derived from tissues shown to be rich in PAR-2 mRNA, kidney, pancreas, small intestine, colon, and skin were also found to respond to these agents, additionally confirming the presence of a functionally physiologically relevant receptor (see below).
VIII. Functional Responses to Proteinase-Activated Receptor-2 Activation
Since the cloning of PAR-2 and its identification within a number of tissues, numerous studies, particularly in isolated vessels or cell preparations, have elucidated functional responses in vascular, airways, and intestinal smooth muscle, neuronal tissue, leukocytes, osteoblasts, and other lymphoid tissues (see Table4). Although many of these studies show a range of responses comparable with PAR-1 activation, the distinct distribution of PAR-2 implicates potentially unique roles in airway relaxation, intestinal function, and skin development.
Cellular, tissue, and systemic effects of PAR-2 activation
A. Cardiovascular Responses
Expression of PAR-2 in vascular tissue and highly vascularized organs has been widely documented in humans and other species (Nystedt et al., 1994, 1995a; Bohm et al., 1996; D'Andrea et al., 1998). These studies, coupled with those discussed above, indicated a potential role for PAR-2 in the regulation of vascular tone. Numerous studies have now shown that trypsin and PAR-2APs cause an endothelium-dependent relaxation of isolated preparations from rat (Al-Ani et al., 1995) and rabbit aorta (Roy et al., 1998), porcine coronary (Hwa et al., 1996;Hamilton et al., 1998), and basilar arteries (Sobey and Cocks, 1998;Sobey et al., 1999). Inhibitors of nitric oxide reverse the PAR-2-mediated relaxation in the large majority of these preparations consistent with a role for NO as the intermediate in this response. Evidence suggests that this is likely to be as a direct result of PAR-2 induced Ca2+ mobilization and subsequent activation of endothelial NO synthase. However, one study has shown that, in rat aorta, SLIGRL-induced NO release is inhibited by the endothelin receptor B receptor antagonist BQ-788, suggesting that ET-1 functions as an intermediate in this response (Magazine et al., 1996). In other preparations, such as the GP and mouse trachea, prostacyclin rather than NO is implicated as the relaxant effects of trypsin, or PAR-2 peptides can be abolished by indomethacin pretreatment (Lan et al., 2000; Ricciardolo et al., 2000).
In vascular preparations, vasoconstriction has been observed following endothelium denudation in some preparations, such as rabbit aorta (Komuro et al., 1997), and this correlates with expression of PAR-2 in the smooth muscle layers of these species. However, recent studies have also shown that trypsin and high concentrations of PAR-2APs can also initiate endothelium-dependent contraction in both rat pulmonary artery (Roy et al., 1998) and human umbilical vein (Saifeddine et al., 1998), possibly through the release of a unidentified contractile factor from human endothelial cells. This response is likely to be mediated via a novel PAR-2 receptor subtype (see Section X.).
The coupling of PAR-2 to vessel relaxation via the NO pathway is reflected in the hemodynamic responses observed in response to PAR-2 activation. Intravenous infusion of SLIGRLETQPPI or SLIGKV was found to cause a transient decrease in mean arterial pressure in anesthetized rats (Emilsson et al., 1997; Cicala et al., 1999) and additionally in mice a sustained moderate hypotension (Cheung et al., 1998). The effect of PAR-2 activation in these models was again shown to be at least partially dependent upon NO release, because the hypotensive response was inhibited by prior infusion of nitric oxide inhibitors. Trypsin has also been shown to stimulate a similar hypotensive response that was sensitive to the trypsin inhibitor SGKR-chloromethylketone, further confirming the involvement of a proteinase-activated receptor (Cicala et al., 1999). Significantly, and in contrast to PAR-1, no rebound hypertension was observed either in control conditions or following infusion of NO inhibitor (Cheung et al., 1998), indicating a lack of PAR-2 function in vascular smooth muscle of the mouse.
Despite these findings, the physiological and pathophysiological role of PAR-2 in regulating cardiovascular responses remains unclear. In mice deficient in PAR-2 (Damiano et al., 1999a), SLIGRL-mediated hypotension was abolished; however, basal mean arterial pressure was not altered. Furthermore, the vasodilatory responses to PAR-1 activation were enhanced, indicating a functional interaction between the two receptors (see Section XIV.), which may result in a compensatory mechanism operating when PAR-2 is nonfunctional, and suggesting the potential of receptor redundancy.
Recent data tends to support PAR-2 involvement in disease conditions, although it is unclear if activation of the receptor mediates a disease condition or is activated to protect against it. NO-mediated vasodilatation in response to SLIGKV is enhanced in cerebral arteries of SHR rats relative to WKY controls (Sobey et al., 1999), whereas in the isolated rat heart PAR-2 activation protects against ischemia-reperfusion injury (Napoli et al., 2000). In contrast SLIGRL-induced hypotension was enhanced following LPS pretreatment, suggesting that PAR-2 is a mediator of some of the deleterious cardiovascular effects of endotoxin infection (Cicala et al., 1999). Clearly, future studies are required, including further utilization of the PAR-2 knockout mice to clarify the acute function of PAR-2 under different physiological and pathophysiological conditions.
It should be noted that an additional feature of the acute cardiovascular effects of PAR-2 possibly involves an increase in receptor expression. Previously, it has been shown that, in endothelial cells, following either LPS or tumor necrosis factor-α pretreatment, PAR-2 receptor expression was significantly enhanced (Nystedt et al., 1996). Increased PAR-2 expression was also observed in two of the studies outlined above (Cicala et al., 1999; Napoli et al., 2000) and also during restenosis following balloon angioplasty (Damiano et al., 1999b), indicating that enhanced receptor expression may be an important feature in the cardiovascular actions of PAR-2.
In addition to acute effects upon vascular tone, PAR-2 as with PAR-1, is strongly implicated in chronic responses associated with vessel inflammation and wound healing. Trypsin and PAR-2 APs stimulate the activation of T-cells and neutrophils, and promote leukocyte rolling and adhesion (Vergnolle, 1999; Vergnolle et al., 1999). PAR-2 also promotes leukocyte extravasation in vivo, which is facilitated by increased capillary permeability and enhanced production of cytokines. PAR-2 is also linked to enhanced production of von Willebrand factor, smooth muscle, and endothelial cell growth and increases in tissue factor mRNA and activity (Mirza et al., 1996; Storck et al., 1996;Langer et al., 1999), responses all relevant to the aspects of wound healing. Since several cardiovascular responses, including wound healing, involve inflammation, it is still unclear if PAR-2 activation of these responses is part of normal vessel physiology or associated with the development of certain cardiovascular diseases. Further studies are required to clarify these points.
B. Proteinase-Activated Receptor-2 Involvement in Gastrointestinal Function
PAR-2 is expressed in numerous cell types within the GI tract, suggesting both multiple functions and numerous modes of receptor activation. Once again, it is unclear if PAR-2 activation predominates in normal physiological conditions or is activated preferentially following inflammation of the GI tract.
PAR-2 is strongly expressed in enterocytes in both basolateral and apical membranes (D'Andrea et al., 1998). Application of PAR-2AP or trypsin to the serosal side of rat jejunal strips mounted in Ussing chambers stimulates a short circuit current, which was mediated by active Cl− ion transport (Vergnolle et al., 1998). This response is in turn likely to be Ca2+-dependent since a number of intestinal cell lines, in particular hBRIE, have previously been shown to respond to PAR-2 activation with an increase in intracellular Ca2+ (Kong et al., 1997). However, the effect of PAR-2 activation is likely to involve the prior release of prostaglandins, possibly PGE2, since the effect upon short circuit current was abolished by indomethacin (Vergnolle et al., 1998). Structure/function studies suggest the involvement of a novel PAR-2 in the mediation of this response (see Section X.). PAR-2 activation at the apical site by PAR-2-activating peptides and trypsin also strongly stimulates intracellular Ca2+ and the secretion of PGE2 and PGF1α (Kong et al., 1997); however, to date, no studies have indicated that this is directly linked to Cl− ion transport.
Although these studies suggest that PAR-2 regulates ion channels at both poles of the enterocyte, several key differences may be observed. Apical PAR-2 is likely to be directly activated by trypsin and at the serosal surface, in the human system, by α-tryptase released from mast cell degranulation (see Section IX.). Second, PAR-2-mediated activation at the serosal side may be indirect, possibly reliant upon the release of mediators, such as histamine, substance P prostaglandins and opiates from mast cells, fibroblasts, and PAR-2 responsive enteric neurones. Indeed, recent studies have identified PAR-2 receptors on 50 to 60% of enteric neurones (Corvera et al., 1999), and in the porcine ileum, prior addition of δ-opiate agonist inhibited ion transport stimulated by trypsin (Green et al., 2000). Interestingly, a similar system has also been described for trypsin-induced contraction of the guinea pig bronchus, with release of neurokinins from sensory nerves being responsible for the final contractile response (Carr et al., 2000).
PAR-2 is also strongly expressed in a number of smooth muscle preparations, including mouse gastric fundus, guinea pig Taenia coli, rat duodenum, and colon (Corvera et al., 1997; Cocks et al., 1999b; Kawabata et al., 1999a). In mouse gastric fundus, previous studies have implicated a PAR-2-mediated contractile response; however, a recent study has shown that this may mask an initial relaxation. In rat colon, spontaneous motility is also abolished, whereas in GPTaenia coli and rat duodenum no relaxation and only a minor contraction were observed, suggesting species and tissue selectivity in the response of intestinal smooth muscle to PAR-2 activation. Relaxations in these and in related smooth muscle preparations, such as the mouse ureter (Moffatt and Cocks, 1999), do not seem to involve intermediate transmitter nerves innervating the smooth muscle, and are independent of either NO or prostaglandin synthesis (Corvera et al., 1997). Rather, a recent study has implicated a role for activation of Ca2+-dependent K+ channels and subsequent closure of L-type voltage-operated calcium currents as a mechanism for PAR-2-mediated relaxation, because responses to trypsin or SLIGRL were inhibited by apamin and ryanodine (Cocks et al., 1999b).
PAR-2 has also been detected in other digestive tract organs, most notably the pancreas where the receptor has been identified in acinar cells and linked to the release of amylase (Bohm et al., 1996b). Furthermore, activation of PAR-2 led to increased Ca2+-activated Cl− and K+ conductances in pancreatic duct epithelial cells, further linking activation of the receptor with pancreatic secretory function (Nguyen et al., 1999b). Indeed PAR-2 has also been linked to increased exocrine secretion from salivary, parotid, and sublingual glands (Kawabata et al., 2000c,d), suggesting a common secretory role throughout tissues of the intestinal tract. The mechanism involved in this process is unclear, but may involve a tyrosine kinase cascade since secretion was found to be inhibited by genestein, a general tyrosine kinase inhibitor (Kawabata et al., 2000c) (see Section XI.). Activation PAR-2 in renal collecting duct epithelial cells has also been identified as activating the secretion of Cl− ions (Bertog et al., 1999).
C. Proteinase-Activated Receptor-2 Regulation of Skin Function
One of the most active areas of study on PAR-2 has been in the epidermis, where PAR-2 is expressed at moderate to high levels within the layers of epidermal keratinocytes that constitute the majority cell type in this tissue (D'Andrea et al., 1998; Steinhoff et al., 1999).
These particular studies have revealed differential expression of the receptor between layers of the epidermis, such that PAR-2 expression was found to be higher in the more differentiated granular layer of keratinocytes than in the suprabasal spinous layer or the proliferative basal layer. The exact reasons for these differing levels of PAR-2 remains to be elucidated, although a role in the control of the latter stages of keratinocyte terminal differentiation may be one explanation for the pattern of expression.
The original studies into the role that PAR-2 may play in epidermal keratinocytes noted that, in addition to increasing intracellular levels of calcium (Santulli et al., 1995), activation of the receptor inhibited cellular proliferation and differentiation (Derian et al., 1997). The latter of these studies notes that the effect may have been due to PAR-2-mediated cytokine production and subsequent release, rather than a PAR-2 specific event. In particular, a role for TGF-β was suggested, because this was the only other agent affecting keratinocytes that had similar effects on both proliferation and differentiation (Choi and Fuchs, 1990). The effect of PAR-2 agonists on the production of TGF-β as an autocrine agent has not been assessed; however, PAR-1 has been shown to increase the synthesis and release of this cytokine (Yamabe et al., 1997), and therefore a similar response to PAR-2 activation is possible.
One laboratory has carried out several studies assessing the effect of trypsin on different epidermal parameters. These studies have implicated trypsin as an agent capable of decreasing hair growth and increasing epidermal thickness (Seiberg et al., 1997b), improving skin elasticity, decreasing both follicular papillae apoptosis and urticuli size (Seiberg et al., 1997a), and altering skin pigmentation (Seiberg et al., 1997b, 2000a). In addition to these studies, this group has also established a link between PAR-2 and both melanosome uptake (Seiberg et al., 2000b) and general phagocytosis in keratinocytes (Sharlow et al., 2000). The involvement of PAR-2 was not indicated in either of the two earlier studies; however, in the latter paper, a strong link was made between the effect of trypsin on melanogenesis and the activation of PAR-2 by the protease (Seiberg et al., 2000a).
IX. Endogenous Activators of Proteinase-Activated Receptor-2
Following the initial characterization of PAR-2 as a trypsin-sensitive receptor (Nystedt et al., 1994), the potential for trypsin itself to be the preferred endogenous activator of PAR-2 in all tissues remains controversial. The high level of expression of PAR-2 in the small intestine and colon and lower levels of expression in the stomach (Bohm et al., 1996; Corvera et al., 1997; Kong et al., 1997;D'Andrea et al., 1998) suggest the potential for direct activation of PAR-2 to occur by trypsin released from its zymogen precursor trypsinogen by enteropeptidases within the duodenum. The concentration of trypsin generated in the intestine (Green and Nasset, 1980) is within the range required to activate both PGE2and PGF1α formation and stimulate Cl− secretion in vitro, consistent with its role as an activator of PAR-2. In other parts of the gastrointestinal tract, such as the pancreas, it is unlikely that sufficient trypsin is generated to directly activate PAR-2 despite high levels of receptor expression. However, during pancreatitis, trypsin prematurely produced within the acini and secreted is likely to result in the activation of PAR-2 in both acinar cells, by an autocrine mechanism, and duct cells.
Although this mode of activation restricts the role of trypsin as a PAR-2 activator to the GI tract, except during pancreatitis where trypsin can be released into the bloodstream, recent studies have shown that several cell types also express trypsinogen (Koshikawa et al., 1997; Koshikawa et al., 1998; Miyata et al., 1999). These studies indicate that autocrine activation of PAR-2 by trypsin may be more widespread than previously thought, a case that is supported by a recent study indicating that extrapancreatic trypsin-2 can activate PAR-2 in a model system (Alm et al., 2000). Since trypsinogen expression has been demonstrated in areas removed from the major site of trypsin production, such as the stomach, colon, airway epithelium, skin, neuronal and vascular endothelial cells, trypsin may well prove to be the main activator of PAR-2 in many systems. However, activation of PAR-2 under these conditions would also require processing of trypsinogen either extra- or intracellularly by specific enteropeptidases. To date, few examples of such enzymes have been demonstrated in human tissue without the GI tract, although expression of enteropeptidase has been reported in the stomach, colon, and brain of the rat (Yahagi et al., 1996).
Several other serine proteases have been assessed for the ability to activate PAR-2. One of the main candidates is tryptase, a chymotrypsin-like protease, which is abundant in mast cells, particularly of the MCTC subtype found in the skin, intestine, and lung tissue (Schwartz, 1994). Tryptase has been shown to mimic the actions of trypsin in cells transfected with PAR-2 (Mirza et al., 1997; Molino et al., 1997b) and in numerous other cell types expressing the receptor endogenously (Corvera et al., 1997, 1999;Compton et al., 1998; Schechter et al., 1998; Steinhoff et al., 1999;Akers et al., 2000), strongly suggesting it to be an endogenous PAR-2 activator. However, although tryptase has been shown to directly cause proteolytic cleavage of PAR-2 (Fox et al., 1997; Molino et al., 1997b), it is less potent than trypsin itself. This would be consistent with high local concentrations of tryptase that can occur following migration of mast cells to local sites of inflammation and subsequent degranulation (Santos et al., 1998). At higher concentrations, tryptase is also able to activate PAR-1, albeit in transfected COS-7 cells and not in cells expressing an endogenous level of PAR-1 (Molino et al., 1997b), and to cleave PAR-2 at sites other than Ser36. The potential functional relevance of these observations is unclear, although they may represent some form of down-regulation mechanism.
A number of other tissue-specific enzymes that can activate PAR-2 have been identified. Using a peptidyl chloromethane inhibitor (biotinyl-Ser-Lys-Gly-Arg-CH2Cl) based on the cleavage site of PAR-2, Fox and co-workers (1997) showed the sperm enzyme acrosin more rapidly initiated receptor cleavage than either trypsin or tryptase. This finding is consistent with the presence of PAR-2 in oocytes and epithelium of the seminiferous tubules, and a recent study that indicates acrosin activation of native PAR-2 on oocytes (Smith et al., 2000). Other tissue-specific proteases that have been shown to activate PAR-2 include a proteolytic fragment of the neuronal protein B-50/GAP-43 (SFRB60) (Hollenberg et al., 2000), tissue factor Xa, (Camerer et al., 2000), a brain-derived trypsin-like serine protease (P22) (Sawada et al., 2000), and gingipain-R, a serine protease released from Porphyromonas gingivalis and implicated in adult periodontitis (Lourbakos et al., 1998). Taken together, these studies strongly implicate the presence of tissue-type specific activators of PAR-2.
Although such tissue-type specific activators of PAR-2 are likely, it is possible that these activators may act in tandem with trypsin, tryptase, or indeed other proteases to fully stimulate the receptor. A recent study has shown that the coagulation factors VIIa and Xa are able to activate PAR-2 in endothelial cells. Factor VIIa was seen to act both indirectly by generating factor Xa and directly following binding to tissue factor expressed on the cell surface, presumably near the PAR-2 receptor N terminus (Camerer et al., 2000) (Fig.6C). This implies the potential for PAR-2 to be stimulated by single or multiple endogenous activators, and also to be dependent upon membrane proteins that present the relevant serine protease to PAR-2. However, a subsequent independent report has indicated that the intracellular effects of factor VIIa may not be PAR-2-dependent (Petersen et al., 2000), and further studies are required to resolve this question fully. Other membrane proteins could also play a role, such as membrane-type serine protease 1 that has been shown to activate PAR-2 (Takeuchi et al., 2000) (Fig. 6D), or the recently identified transmembrane serine protease 3 (Wallrapp et al., 2000).

Models of PAR activation involving coreceptor activation tethered proteins and multiple serine proteinases. A, PAR-3 functions to provide thrombin as a ligand for PAR-4. B, tethered ligand from PAR-1, exposed by thrombin-mediated proteolysis, interacts with PAR-2 to activate PAR-2 signaling. C, tissue factor (TF) binds factor VIIa (FVIIa), which produces active factor X (FXa) from its zymogen Fx. Both FXa and TF-tethered FVIIa are then capable of proteolytic activation of PAR-2. D, membrane-type serine protease-1 (MT-SP1) functions to activate PAR-2 independently of circulating trypsin or a tryptase.
From these studies it seems clear that several endogenous activators of PAR-2 can exist, however, which activator is predominant within each cell system has not been properly defined. Such a definition is likely to be dependent upon the concentration of the activator in the extracellular fluid, the presence of tethering proteins/enteropeptidases, the relative binding affinities of each activator for the PAR-2 N terminus and their relative enzymatic activities. Certainly under different physiological and pathophysiological conditions, it is possible that different endogenous activators may activate PAR-2 expressed on the same cell type.
X. Pharmacology of Proteinase-Activated Receptor-2
Very few studies have tackled the structure/function relationship between peptide agonist and PAR-2. Investigations initially focused on the hexapeptide sequences SLIGRL, the murine tethered ligand sequence, and SLIGKV the human variant. SLIGRL was found to activate murine PAR-2 expressed in Xenopus laevis oocytes with an EC50 of 5 μM (Nystedt et al., 1994) and was also active at this receptor expressed in Chinese hamster ovary cells (Nystedt et al., 1995a). In later studies on the human receptor, SLIGKV-NH2 was found to have an EC50 for mobilization of intracellular calcium on A549 cells of 4.6 μM, comparing well with the earlier murine studies. It should be noted that, in all models tested, the murine peptide has been consistently shown to be more potent than the human peptide agonist in the same experiments (Blackhart et al., 1996).
An important series of classical pharmacological experiments, using organ bath techniques, investigated how changes in agonist peptide structure affected PAR-2 activation in rat aortic ring and gastric longitudinal muscle preparations. The “classical” agonist peptide, SLIGRL, and its amino variant, SLIGRL-NH2, were tested and found to have similar EC50 values in both tissue types (Al-Ani et al., 1995; Saifeddine et al., 1996), although the peptides were 10-fold less active in the contractile model, compared with the vascular relaxation system.
Reduction of the peptide chain length to an amino-pentapeptide, SLIGR-NH2, slightly reduced agonist potency in the aortic ring preparation, but lead to a substantial loss of potency in the gastric longitudinal muscle preparation (Saifeddine et al., 1996). This apparent discrepancy may indicate different receptor subtypes in the two tissues studied. Interestingly, the pentapeptide SLIGR had lower potency as a vasodilator in the aortic ring than both the hexapeptide derivatives and the amino-pentapeptide variant (Hollenberg et al., 1996). The potency of the amino-pentapeptide was further reduced by the removal of Ser1, giving rise to the peptide LIGR-NH2, indicating the importance of this amino acid in agonist function (Hollenberg et al., 1996). The same study also revealed that alanine substitution of the Leu2 and Arg5 of the hexapeptide, respectively abolished or substantially reduced aortic vasodilation. These studies expanded work on a series of alanine-substituted analogs of SLIGRL, in which substitution of any of the agonist peptide residues gave rise to marked decreases in agonist effectiveness (Blackhart et al., 1996).
Later studies further demonstrated the effects on PAR-2 activation of mutation and chemical alteration (Hollenberg et al., 1997). Alanine substitution of Ile3 from murine PAR-2 agonist peptide, giving SLAGRL-NH2, caused a great decrease in the peptides' ability to relax the rat aortic preparation. This may be related to a change in the shape of the peptide molecule, because it involved the substitution of alanine, an amino acid with a methyl hydrocarbon chain, for the bulkier Ile. Ile is also an aliphatic amino acid, as is alanine, but has a three-carbon chain, methylated at position 1. This substitution may, therefore, have altered the overall conformation of the peptide. Changes in the stability of the molecule in aqueous solution, due to alteration of hydrophobic interactions resulting from the change in hydrocarbon chain length, may also have played a part in the observed decrease in activity. Replacement of Gly4 in the hexapeptide by Ala only slightly reduced the potency of the agonist, in agreement with earlier work (Blackhart et al., 1996). A final substitution of Ser1 with a Thr residue greatly decreased agonist potency in both experimental models (Hollenberg et al., 1997), as with previous work in the oocyte expression system (Blackhart et al., 1996), therefore further stressing the critical nature of this position in receptor activation. The N-terminal N-acetylation of SLIGRL produced a completely inactive peptide derivative in the rat aortic model of vascular relaxation (Hollenberg et al., 1997).
Two later studies have assessed the agonist potential of another chemically modified peptide, designed to be specific for PAR-2. Thetrans-cinnamoyl-LIGRLO-NH2(tc-LIGRLO-NH2) was found to be less active than SLIGRL in active ion transport assay, but was found to have similar or identical EC50 values to SLIGRL in Ca2+ mobilization and vascular relaxation experiments, respectively (Roy et al., 1998; Vergnolle et al., 1998). The latter study also demonstrated the tc-LIGRLO-NH2 derivative to be inactive in endothelium-denuded artery preparations, whereas SLIGRL-NH2 was fully active.
PAR-1 agonist peptide SFLLRN has also been shown to activate PAR-2. Such an interaction had initially been suggested in theXenopus expression system (Blackhart et al., 1996; Lerner et al., 1996). These studies indicated SFLLRN, or SFLLRNP-NH2 had activated expressed PAR-2. Hollenbergs' group reported that substitution of the N-terminal Ser residue with Thr in SFLLRN produced an agonist with less potency in the aortic relaxation model, but greatly increased the selectivity of the peptide in the gastric contraction system, compared with the PAR-2 agonists (Hollenberg et al., 1997).
In contrast to the numerous studies using mutated PAR-1 to assess the characteristics of the tethered ligand interaction, few studies have utilized PAR-2 chimeric proteins in the same way. One previous study confirmed the ECL-2 as critical in receptor activation (Lerner et al., 1996). Recent work by this group has also indicated the importance of ECL-2, in particular the sequence PEE, in defining the respective interactions of PAR-2 peptides and the tethered ligand with the receptor (Al-Ani et al., 1999), although clearly additional studies are require in this area.
XI. Proteinase-Activated Receptor-2-Mediated Intracellular Signaling
In contrast to the large number of studies examining PAR-1-mediated intracellular signaling, there have been relatively few studies examining PAR-2 systems. The reason for this is unclear, but may relate to the relatively low level of endogenous expression of PAR-2, relative to PAR-1, in cells such as fibroblasts, where intracellular signaling pathways are easily manipulated. However, it is known that both trypsin and PAR-2 AP stimulate [3H]IP formation and Ca2+in numerous cell types, consistent with coupling to the heterotrimeric G-proteins Gq/G11 and PLC isoforms. Furthermore, PTX-sensitive Ca2+signaling has been demonstrated in Xenopus oocytes in response to trypsin (Schultheiss et al., 1997), further indicating the potential involvement of Go/Gi-dependent transduction mechanism in a manner similar to PAR-1. However, to date, no direct studies have been performed regarding coupling of PAR-2 to these G-proteins or to others such as G12 or G13.
In HEK-293 cells transfected with PAR-2, SLIGKV has been shown to increase c-fos promoter activity in a PTX-sensitive manner (Yu et al., 1997), a response associated with tyrosine phosphorylation of SHP-2, a tyrosine phosphatase previously observed to play a role in PAR-1 mitogenic signaling. Other aspects of tyrosine kinase signaling directly associated with PAR-1, such as tyrosine phosphorylation of SHC and growth factor receptor transactivation, have not been demonstrated for PAR-2 activation. This is despite several recent studies showing that trypsin can activate elements further downstream in the cascade, including Raf-1, MEK, and both p42/44 isoforms of ERK (Belham et al., 1996; Dery et al., 1998; Sabri et al., 2000). In other cell systems, such as cardiac myocytes and transfected skin epithelial cells, PAR-2 has been shown to be linked to the SAP kinases, JNK, and p38 MAP kinase (Kanke et al., 2000 3;Sabri et al., 2000). Coupling to this pathway would be consistent with linkage to a number of proinflammatory responses in target cell types. Trypsin and peptide stimulation of the nuclear factor-κB signaling pathway has also recently been demonstrated in a transfected keratinocyte cell line and in coronary smooth muscle cells (Bretschneider et al., 1999; Macfarlane et al., 2000), further supporting the potential of direct coupling of proinflammatory signaling pathways to PAR-2.
XII. Proteinase-Activated Receptor-2 Desensitization
The termination of PAR-2 responses represents an important consideration for the physiological actions of PAR-2 activation, similar to the case for PAR-1. Early studies utilizing isolated blood vessel preparations showed that responses to trypsin and SLIGKV were rapidly desensitized, with recovery indicative of an internalization and trafficking system similar to that for PAR-1 (Bohm et al., 1996a). However, far fewer studies have examined the mechanisms involved. Bunnet and co-workers (Dery et al., 1999) have shown that PAR-2 is internalized probably through clathrin-coated pits to the early endosomes followed by redistribution to the lysosomes. Resensitization is inhibited by both brefeldin and cyclohexamide (Bohm et al., 1996a), indicating the presence of trafficking of PAR-2 and mechanisms for resynthesis of the receptor. Similar results were obtained using whole vessels, suggesting that the process is universal (Cocks and Sobey, 1998; Hamilton et al., 1999). PAR-2 endocytosis has been found to be mediated by β-arrestin (DeFea et al., 2000b), consistent with several recent studies that have examined the role of this protein in the internalization of G-protein-coupled receptor (Ferguson et al., 1996;Goodman et al., 1996). However, in contrast to PAR-1, endocytosis may not represent the major mechanism for signal termination since prevention of internalization using PAO has little effect upon the desensitization of trypsin-mediated Ca2+signaling (Bohm et al., 1996a). Indeed, endocytosis may be required for efficient activation and intracellular targeting of p42/44 MAP kinase or other signaling intermediates (DeFea et al., 2000a). In this context, it is clear that further studies are required to clarify the role, if any, of C-terminal phosphorylation in the regulation of internalization and “shutoff”. Although a number of putative phosphorylation sites for GRK and other kinases are present within the C-terminal tail, at present only indirect studies using inhibitors have been used to implicate a role for PKC in the termination of PAR-2 signaling.
XIII. Identification and Function of Proteinase-Activated Receptor-3 and Proteinase-Activated Receptor-4
A. Proteinase-Activated Receptor-3
As noted previously, pharmacological studies had indicated anomalies between the effects of thrombin and PAR-1APs in several systems (Kinlough-Rathbone et al., 1993), suggesting the possible existence of a separate thrombin-sensitive receptor. Later studies indicated the presence of the second receptor by targeted disruption of the thrombin gene (Connolly et al., 1996). Platelets derived from surviving mice still responded strongly to thrombin, whereas fibroblasts were found to be insensitive to both thrombin and SFLLRN. An initial candidate for this receptor was cloned from rat platelet mRNA (which is more abundant than mouse or human mRNA) by polymerase chain reaction using a selection of degenerate primers corresponding to conserved regions of PARs 1 and 2, and also peptide glycoprotein mRNA (Ishihara et al., 1997). Primers based on the subsequent clone were then used to isolate the human and murine forms of the receptor (Ishihara et al., 1997). The human isoform of the protein was found to retain 27% amino acid sequence homology with hPAR-1 and 28% with hPAR-2. A serine protease cleavage site within the human PAR-3 N-terminal sequence at Lys38/Thr39 was also identified, as was a hirudin-like binding domain, FEEFP, C-terminal to the cleavage site (Fig. 2). Genomic analysis located the PAR-3 gene to the same 100-kb spanning gene cluster as both PAR-1 and PAR-2, and the gene was also found to have a similar two exon structure to the other family members (Kahn et al., 1998; Schmidt et al., 1998).
The molecular identification of human PAR-3 was complemented by cellular studies, indicating that phosphoinositide hydrolysis in response to thrombin via the receptor, expressed in COS-7 cells, was dependent upon the presence of the Lys38/Thr39 cleavage site. Synthetic peptides that mimic the putative tethered ligand of PAR-3 were, interestingly, found to be inactive (Ishihara et al., 1997). This suggests that the tethered ligand conformational specificity may be more rigorous at PAR-3 than is the case with other PAR family members. Alternatively, the cleavage of the receptor may simply switch the receptor on, as suggested by the authors (Ishihara et al., 1997). The overall consequence of the lack of a specific activator has been that PAR-3 function has remained poorly defined in the majority of human cell types where it has been identified (Schmidt et al., 1998). At a molecular level, the presence of a very short C-terminal tail region in PAR-3 (see Fig. 2) also suggests potential differences in the way in which this receptor signals and is desensitized relative to other PARs, although no studies to date have examined these phenomena.
Although human PAR-3 was found to be expressed in a variety of tissues, including heart, small intestine, bone marrow, airway smooth muscle, vascular endothelium, and astrocytes (Ishihara et al., 1997; Hauck et al., 1999; Bartha et al., 2000), no expression of the receptor was detected in platelets. This is in contrast to the murine form of the receptor that was found to be strongly expressed in mouse megakaryocytes, but at low levels in other mouse tissues, e.g., brain and lung, indicating possible species-specific differences in the function of PAR-3 (Ishihara et al., 1997). Indeed, although studies using both PAR-3-deficient mice (Kahn et al., 1998) and PAR-3-specific antibodies (Ishihara et al., 1998) strongly suggest that PAR-3 is important in achieving full thrombin-mediated platelet activation in mice, murine PAR-3 itself may not be a fully functional receptor, but rather may play a role as a tethering protein for thrombin (Section XIV.). This idea is further supported by the differences in signaling measured from human and murine PAR-3 in response to thrombin. As noted previously, the human form of PAR-3 activated phosphoinositide signaling in response to thrombin when overexpressed in COS-7 cells (Ishihara et al., 1997). However, subsequent experiments indicated that the murine form of the receptor did not signal upon exposure to the protease (Nakanishi-Matsui et al., 2000). These observations, therefore, provide evidence that the human and murine forms of PAR-3 may indeed have different, species-specific, physiological functions, and augments the idea of murine PAR-3 acting as a thrombin binding site rather than a fully active receptor.
B. Proteinase-Activated Receptor-4
The cloning of a fourth proteinase-activated receptor was carried out simultaneously by two laboratories after they identified PAR-like sequences following searches of Expressed Sequence Tag (EST) libraries (Kahn et al., 1998; Xu et al., 1998). Xu et al. discovered a sequence with 34% identity within the fourth transmembrane region of PAR-2. A full-length clone (4.9 kb) was then obtained, from a lymphoma Daudi cell line library, using a 600 bp DNA probe from the EST sequence. Kahn et al. (1998) likewise found an 11 amino acid sequence that was 73% identical to PAR-2. They employed 5′ Rapid Amplification of cDNA Ends to isolate a full-length clone from mouse embryo DNA and subsequently cloned the human PAR-4, using RT-PCR with K562 mRNA, with primers based on the mouse sequence.
The human receptor protein was found to be 385 amino acids in length, and possessed both a signal peptide and a putative serine protease cleavage site at Arg47/Gly48 in the N-terminal sequence. This new PAR family member was shown to share 33% amino acid sequence homology with the other three human PARs; however, both the N and C termini of PAR-4 were noted to be markedly different from the other receptors, as was the cleavage site. In addition to these differences, no hirudin-like thrombin binding sequence was identified in PAR-4. This was reflected in the ability of both α- and γ-thrombin to activate PAR-4-dependent phosphoinositide hydrolysis in COS-7 cells with equal efficacy (Xu et al., 1998). Activation of PAR-4 by either thrombin or trypsin was prevented by mutation of Arg47 to Ala, whereas the response to the synthetic peptide, GYPGQV, which corresponds to the PAR-4 tethered ligand, was unaffected by the mutant, confirming the protease mediated mechanism of activation.
The tissue distribution of PAR-4 was found to be distinct from the other PAR family members, with the highest levels of receptor mRNA detected in lung, pancreas, thyroid, testis, and small intestine, although lower levels were detected in most tissues tested. It was also noted that, although the PAR-4 gene shared a similar two exon structure to PARs 1–3, the gene was located on a different chromosome, at position 19p12 (Xu et al., 1998).
PAR-4 is relatively insensitive to thrombin, with an EC50 for the protease approximately 50-fold higher than the corresponding figure for PAR-1 (Kahn et al., 1998; Xu et al., 1998). This suggests that PAR-4 may function as a low affinity thrombin receptor that is activated in conditions where high concentrations of thrombin are achieved. The finding that PAR-4 antibodies only block Ca2+ signaling and platelet activation at high concentrations of thrombin gives strong support to this hypothesis (Kahn et al., 1999). A temporal aspect to PAR-4 activation in platelets is also apparent, as a recent study has shown (Kahn et al., 1999). Biphasic Ca2+ signaling by thrombin was resolved into a rapid PAR-1-mediated signal and a slower sustained PAR-4-mediated response, the latter of which is associated with the late phase of the platelet aggregation process. Thus, although PAR-1 seems to be the predominant receptor involved in both platelet aggregation and the clotting process itself (Andersen et al., 1999;Kahn et al., 1999), PAR-4 may help to sustain aggregation in response to thrombin during a period when PAR-1 becomes rapidly inactivated. This idea is consistent with the finding that PAR-4 is not rapidly phosphorylated following thrombin treatment and is slowly desensitized relative to PAR-1 (Shapiro et al., 2000), and indicates a lack of consensus sequences in the C-terminal tail necessary for GRK-mediated phosphorylation (see Fig. 2).
Trypsin was also identified as a PAR-4 agonist, with an EC50 of 5 nM, indicating that both trypsin and thrombin were equipotent at the new receptor (Xu et al., 1998), and that the receptor may represent a more general serine protease receptor than other PAR family members. This possibility is further supported by the finding that PAR-4 is also strongly activated by the neutrophil granule protease cathepsin G (Sambrano et al., 2000). In addition to the protease agonists for this receptor, the development of a PAR-4-specific peptide AYPGKF has been reported (Faruqi et al., 2000). The development of this peptide provides the opportunity for future studies to investigate PAR-4-mediated effects in systems where PAR-4 is expressed in conjunction with other PAR family members.
It remains to be determined if, as in platelets, PAR-4 functions as a low affinity receptor whose cellular effects are delayed. However, it is clear that, in those cellular systems where biphasic PAR-1 signaling has been identified, reappraisal of the receptors involved may be required. In two cases where the roles of PAR-1 and PAR-4 have been assessed in the relation to a dual effect of thrombin, duodenal motility was shown to be PAR-1-mediated (Kawabata et al., 1999a), whereas in esophageal tissue roles for both PARs1 and 4 were identified (Kawabata et al., 2000a). The demonstration of functional coupling between PAR-4 and Ca2+ signaling in human astrocytoma cells (Kaufmann et al., 2000), and functional responses in mouse airway endothelium and rat longitudinal smooth muscle (Hollenberg et al., 1999; Lan et al., 2000) have, however, not elucidated features of PAR-4 function that distinguish it from other PARs.
Thus, it is likely that, in different cell types, PAR-4 and also PAR-3 may function as a primary receptor or as an adjunct to other PARs, particularly PAR-1. Furthermore, given that thrombin, trypsin, and cathepsin G are all activators of PAR-4, then a different array of PAR-4-generated responses may be expected in different pathological conditions.
XIV. Functional and Molecular Interactions Between Proteinase-Activated Receptors
The expression of several PARs within one cell type has lead to studies assessing their functional interactions (Hwa et al., 1996;Molino et al., 1997c; Hollenberg et al., 1999; Lan et al., 2000;Vergnolle, 2000). Interactions between PARs expressed in platelets have been most closely studied, since the original identification of multiple binding sites for thrombin in platelet membranes (Ishihara et al., 1997; Kahn et al., 1998; Kahn et al., 1999; Nakanishi-Matsui et al., 2000) (Fig. 6).
In mouse platelets, the expression of PAR-3 was found to be necessary for full activation by thrombin, since in PAR-3-deficient mouse platelets expressing only PAR-4, the response to thrombin is delayed and less sensitive than in platelets derived from normal mice (Kahn et al., 1998). As noted previously, stimulation of mPAR-3 with thrombin does not result in intracellular signaling or functional activation despite evidence to support ongoing cleavage of the receptor. Recently, Coughlin and colleagues (Nakanishi-Matsui et al., 2000) showed that PAR-3, although not linked to a cellular response per se, facilitated thrombin stimulation of PAR-4 by functioning as a tethering protein for the protease (Fig. 6A). This would perhaps allow high affinity binding to PAR-4 in the absence of a hirudin-like binding domain. This model is unlikely to apply in human platelets, where, although PAR-4 is expressed, only very low levels of PAR-3 are detectable (Schmidt et al., 1998). Indeed, no compelling evidence has been presented, except in transfected cell systems, to suggest that the two receptors actually interact in this manner. However, in human platelets, it is possible that GP1βα (discussed in Section IV.A.), or another protein may also function to present thrombin to either PAR-1 or PAR-4, circumventing a role for PAR-3 in this context.
Potential interactions between other PAR family members have also been recently documented. As outlined above (Section VIII.A.), mice lacking the PAR-2 gene have enhanced responses to PAR-1 (Damiano et al., 1999a), suggesting the possibility of a direct functional interaction between the two receptors. Recently, it has been shown that in cells coexpressing PAR-2 and a PAR-1 mutant capable of being cleaved but not able to signal, thrombin was still able to stimulate accumulation of [3H]inositol phosphates indicative of ongoing PLC activity (O'Brien et al., 2000). This suggests potential transactivation of PAR-2 by the tethered ligand of PAR-1, which is consistent with the ability of the free PAR-1 ligand SFLLRN to activate PAR-2 in different cell types (Blackhart et al., 1996) (Fig. 6B). It is again unclear whether this phenomenon has any physiological relevance, since these studies were performed in transfected cell systems. However, such interactions could represent a mechanism that provides a dual receptor-activating system for thrombin in cell types where PAR-4 is not strongly expressed. Further identification of PAR-2 activators, such as serine protease 1, which is a membrane bound protein itself, indicates the potential for such interactions (Fig. 6D).
These studies are very significant given the recent findings indicating that some G-protein-coupled receptors dimerize as part of the activation process (Overton and Blumer, 2000; Salahpour et al., 2000;Zeng and Wess, 2000). The presentation of thrombin by mPAR-3 to PAR-4 (Nakanishi-Matsui et al., 2000) and transactivation of PAR-2 by PAR-1 (O'Brien et al., 2000) clearly indicated the potential for PAR-PAR activation to occur, although no direct evidence for dimerization has been presented to date. Furthermore, the species differences in the apparent function of PAR-3 may indicate evolutionary changes in PAR function from roles as tethering proteins to fully functional receptors. This may also be reflected in the relatively poor functional activation of PAR-4 by thrombin.
Indeed, there appears to be a variety of mechanisms besides simple protease binding by which PARs can be activated, either through nonenzymatic transactivation by a second PAR, cleavage by a protease bound to another PAR, or cleavage by a membrane-bound protease (Fig.6). Clearly, further studies will reveal more novel aspects of the interaction between members of the PAR family.
XV. Proteinase-Activated Receptors as Therapeutic Targets in Disease States
A. Proteinase-Activated Receptors in Genetic Disorders
As outlined previously, the human PAR-1 gene is localized to band q13 of chromosome 5, a site now known to be contiguous to the common breakpoint found in the majority of patients with 5q syndrome (Bahou and Demetrick, 1997). This disease is associated with refractory anemia and dysmegakaryocytopoiesis (Van Den Berghe et al., 1974). In addition to its effects upon platelets, thrombin has been shown to inhibit growth of human megakaryocytes in vitro (Vittet et al., 1992; Plantier et al., 1994), and hence a role for PAR-1 disruption has been postulated in this form of disorder. However, although studies with patients presenting this disease have confirmed that the PAR-1 gene is indeed centromeric to the common breakpoint (Demetrick et al., 1996), no PAR-1 gene deletions or rearrangements have been recorded to date (Bahou and Demetrick, 1997).
Despite the fact that PAR-2 has the same genetic localization as PAR-1, no association with a genetic abnormality has been identified. Nevertheless, a PAR-2 polymorphism has been discovered that has a pharmacology distinct from PAR-2 (Compton et al., 2000). Although the investigators identified the polymorph in the human population, any association with a disease state remains uncertain. It should be noted that the polymorphism represents a Phe240 to Ser240 substitution in ECL-2. Since this sequence of PAR-2 is thought to be involved in agonist recognition (seeSection X.), any PAR-2-specific drug may interact differently with this polymorphic receptor.
B. Proteinase-Activated Receptor-1-Mediated Thrombosis and Vascular Remodeling
The vital role of thrombin in the regulation of platelet aggregation and clot formation points to this process as potentially the most important therapeutic target for PAR-1 receptor inhibition. Initial research has focused on the development of thrombin inhibitors, rather than receptor antagonists, as antithrombotic agents. These compounds are based either on inhibition of the active site of thrombin, the exo-anion site or both (Maraganore et al., 1990; Hauptmann and Markwardt, 1992; Stubbs and Bode, 1993; Feng et al., 1997). However, although effective in vivo, complete enzymatic inhibition of thrombin may also result in prolonged bleeding. Inhibitors of other enzymes in the coagulation cascade, such as factor Xa, that do not have this additional effect may turn out to be more clinically applicable (Sinha et al., 2000)
Given the limitations of thrombin inhibitors, the development of a selective PAR-1 antagonist represents a potentially useful adjunct or alternative to current antithrombotic therapies. Studies in monkeys have shown that an antibody directed against the exosite binding region (51–64) of PAR-1 can reduce platelet-dependent cyclic flow, and abolish ex vivo platelet aggregation (Cook et al., 1995). Although this study provides indirect evidence that blockade of the receptor may be of therapeutic value, clinical studies using highly selective and orally available PAR-1 antagonists have not been presented to date. Furthermore, the discovery of PAR-4 on human platelets has required a reappraisal of the potential of PAR-1 antagonists as therapeutic agents. Preferential blockade of this receptor may successfully limit platelet activation in response to excess thrombin, since PAR-4 is only activated by high concentrations of thrombin, and thus may be a more effective and suitable antithrombotic target than PAR-1.
Thrombin and PAR-1 activation have been implicated in several other cardiovascular diseases. Balloon catheter injury causes an increase in PAR-1 mRNA within 6 h with concomitant changes in receptor expression, and coupled with the well known mitogenic effects on vascular smooth muscle cells (see Section V.B.), suggests a role for PAR-1 in remodeling (Wilcox et al., 1994). Indeed, in normal human arteries PAR-1 is expressed in the endothelial cell layer, whereas in human atheroma, PAR-1 is widely expressed in regions rich in vascular smooth muscle cells and macrophages (Nelken et al., 1992). Studies in PAR-1-deficient mice have shown that, following vascular injury, the absence of PAR-1 results in reduced neoitima, increased cell density in the vessels, and impaired remodeling, possibly as a result of decreased matrix deposition (Cheung et al., 1999). Indeed, desulfatohirudin has been shown to be useful in limiting luminal narrowing following balloon angioplasty without inhibiting cell proliferation (Ragosta et al., 1996). It is through effects upon both matrix deposition and proliferation that thrombin receptor inhibition may be beneficial. Other disease states, such as pulmonary fibrosis (Hernandez-Rodriguez et al., 1995), and acute lung injury (Hoffmann et al., 1990) also implicate PAR-1 activation and suggest that blockade of the receptor may be a potential site of therapeutic intervention.
As well as involving effects upon cell proliferation and remodeling, many of the disease conditions outlined clearly involve an inflammatory component. Constitutive PAR-1 activation is also associated with crescentic glomerulonephritis, a renal inflammatory condition associated with glomerular inflammatory cell infiltration and increased fibrin deposition (Xu et al., 1995). The clinical features of the disease can be reduced in normal mice using hirudin, whereas in PAR-1-deficient mice reduced crescent formation and inflammatory cell infiltration is observed (Cunningham et al., 2000).
C. Cancer
The recognition that thrombin plays an important role in angiogenesis has also implicated a role for PAR-1 in tumor formation and metastasis. Thrombin can be synthesized by some tumor cells, whereas PAR-1 is highly expressed in tumor cells, invasive cell lines, and in breast carcinoma biopsy specimens (Wojtukiewicz et al., 1995;Zacharski et al., 1995; Even-Ram et al., 1998; Henrikson et al., 1999). Thrombin promotes tumor cell adhesion to endothelial cells, subendothelial matrix, fibronectin, and Von Willebrand factor under static conditions, and platelet-dependent adhesion to endothelial cells under flow conditions (Klepfish et al., 1993; Wojtukiewicz et al., 1993; Nierodzik et al., 1995; Wojtukiewicz et al., 1995; Dardik et al., 1998). This process is mediated by enhanced expression of α-IIbβ-3 and other cell surface molecules such, as P-selectin. Thrombin also promotes the invasion of aggressive breast tumor cells (Henrikson et al., 1999) and is implicated in the development of experimental pulmonary metastasis (Nierodzik et al., 1995, 1998). Although no clinical studies have been published to date using PAR-1 antagonists, it has been shown that antisense cDNA directed against PAR-1 is able to inhibit breast carcinoma invasion in a model system (Even-Ram et al., 1998), suggesting a possible therapeutic use for PAR-1 blockade in some forms of cancer. These studies indicate that thrombin antagonists, such as RWJ-56110, may prove useful in the treatment of cancers.
Although PAR-1 has been more extensively studied, PAR-2 activation may also play a role in regulation of some forms of cancer. Several cancer cell lines, including those from the stomach and colon, show high expression of trypsinogen 1 and 2 and secretion of active trypsin (Bernard-Perrone et al., 1998; Miyata et al., 1999). In another gastric carcinoma cell line, MKN-1, trypsin stimulates an integrin α5β1-dependent adhesion to fibronectin and proliferation through PAR-2 (Miyata et al., 2000). Taken together, these suggest the potential for chronic activation of PAR-2 during intestinal cancer. However, a recent study in another pancreatic cell line showed a PAR-2-dependent decrease in DNA synthesis (Kaufmann et al., 1998). Further studies are required to clarify the reasons for these differences. A possible scenario is that PAR-2 may regulate normal cell growth and differentiation at physiological levels of expression and activation; but, when pathophysiological levels of PAR-2 activators are produced, sustained proliferative responses may be observed. This model may be extended to cells of the skin, lung, and other tissues that have recently been shown to express novel serine proteases. However, such a model will clearly require full examination following the synthesis of PAR-2 selective antagonist drugs.
D. Proteinase-Activated Receptors and Neurological Disorders
As outlined previously, cellular studies in neurones have indicated a possible role for PAR-1 in neurological disorders (seeSection IV.D.). Disruption of thrombin synthesis and its inhibition by the endogenous proteins PN1 (Choi et al., 1990) and neuroserpin (Hastings et al., 1997) are likely to profoundly affect brain function and are therefore of considerable clinical importance. This may occur following a severe brain insult that results in perturbation of the blood-brain barrier leading to a dramatic rise in thrombin levels in the central nervous system. In conditions such as head wounds, hemorrhagic shock, subdural hematomas, and even following surgical procedures, thrombin is able to enter the interstitial fluid and initate neuronal cell cytotoxicity either alone or in synergy withN-methyl-d-aspartate (Gingrich et al., 2000). Thrombin may also profoundly affect glial function stimulating astrogliosis, promoting infiltration of inflammatory cells and induction of angiogenesis (Nishino et al., 1993). This results in scarring, which forms a physical barrier for regenerating axons, therefore impeding neural repair. Thrombin is also implicated in ischemic cell death during brain insult and posttraumatic hyperexcitibility and seizure (Striggow et al., 2000; Willmore, 1990). Several models of brain trauma in which PAR-1 activation has been enhanced or prevented, including the use of tissue plasminogen activator deletion mice (Wang et al., 1998), indicate that a PAR-1 antagonist may be of clinical use.
High levels of thrombin are associated with PAR-4 activation (seeSection XIII.B.), and this receptor has been identified in human astrocytoma cells (Kaufmann et al., 2000). However, to date, no studies have fully mapped PAR-3 or PAR-4 expression in the brain. If these subtypes are not expressed at high levels, then PAR-1 antagonists alone may be potentially useful, allowing effective therapies to be developed for neurological disorders.
Although PAR-2 has been found to be expressed in hippocampal neurons and to be associated with cell death in these cells (Smith-Swintosky et al., 1997), its functional role in the brain has not been elucidated. However, candidate PAR-2-activating proteinases have recently been demonstrated in brain tissue (Hollenberg et al., 2000; Sawada et al., 2000). It is likely that, during brain inflammation, PAR-2 activation will occur and subsequently mediate diverse neuronal responses, depending upon the severity of the inflammatory challenge. In addition, a recent study has demonstrated functional PAR-2 in human primary meningiomas, suggesting a link to brain tumor formation (Kaufmann et al., 1999).
E. Proteinase-Activated Receptor-2 and Inflammatory Diseases
At present there is not enough data available to determine in which inflammatory disease states activation or blockade of PAR-2 may be clinically useful. It seems likely that acute activation of PAR-2 functions to aid normal physiological processes, whereas in conditions of hyperactivation associated with inflammation, PAR-2 is deleterious. Overall, the relative cellular specificity of PAR-2 expression and the potential modes of activation do make antagonism of this receptor potentially important for therapies aimed at reducing inflammation.
In skin, PAR-2 is expressed in the major epidermal cell types and may be linked to certain disease conditions (Steinhoff et al., 1999), although no currently available data directly support this contention. However, since levels of tryptase in the skin are associated with the severity of psoriasis (Toruniowa and Jablonska, 1988; Harvima et al., 1993), blockade of PAR-2 activation may be desirable. PAR-2 has also been associated with pigmentation in the epidermis (Seiberg et al., 2000a,b) and thus PAR-2 antagonists may be effective in combating diseases associated with disrupted pigmentation (Hermanns et al., 2000).
The potential for regulation of PAR-2 activation has already been identified for inflammatory pain, intestinal inflammation, and asthma (Cocks and Moffatt, 2000; Vergnolle, 2000; Vergnolle et al., 2001). The role of PAR-2 in nociception has begun to receive more attention following the identification of the receptor on sensory afferent nerves, where it was found to mobilize intracellular Ca2+ in these neurones (Steinhoff et al., 2000). Subsequently, the receptor has been found to mediate a long-lasting thermal hyperalgesia (Vergnolle et al., 2000) in addition to inducing hyperalgesia in the intestine of conscious rats (reviewed in Vergnolle et al., 2001).
Therefore, the potential involvement of PAR-2 in the pain associated with conditions, such as inflammatory bowel disease and Crohns disease, where inflammatory processes may lead to the activation of PAR-2 on the sensory nerves, may prove to be an important target for therapuetic intervention. Additionally, PAR-2 activation is likely to play some part in pancreatitis, and therefore blockade of the receptor may provide part of a strategy of treatment for the condition.
In contrast with these other studies, PAR-2 has been associated with a protective, relaxant effect of the airways, implicating PAR-2 activation rather than inhibition as a possible therapeutic strategy for asthma (Cocks et al., 1999). A key consideration in this regard is the relative contribution of PAR-2 expressed on the epithelium and smooth muscle airways to bronchial/tracheal tone (Chow et al., 2000; Cicala et al., 1999; Lan et al., 2000; Ricciardolo et al., 2000). In the case of PAR-1, any relaxant effect of thrombin mediated through the epithelium is usually counterbalanced by a direct bronchoconstrictor effect on the airway smooth muscle itself. However, it has been shown that disruption of the epithelial layer of isolated murine trachea by mechanical rubbing or introduction of influenza virus does not inhibit the relaxation induced by PAR-2 activation (Lan et al., 2000). These findings therefore suggest that the underlying smooth muscle may also be involved in mediating the relaxant effects of PAR-2. If this were true in human airways, then it would represent an additional action for PAR-2-activating drugs, particularly if an additional PAR-2 subtype were found to be involved. However, the potential for PAR-2 activation to result in the production of inflammatory cytokines in the epithelium of the lung is likely although, as yet, it has not been fully investigated. If PAR-2 activation results in the release of cytokines, such as interleukin-6 and granulocyte-macrophage colony-stimulating factor, as with other cell types, the usefulness of PAR-2 agonists may be limited in more severe forms of asthma. Moreover, the situation is made more complex by the presence of other PAR family members, including PAR-4. Although PAR-4 is also coupled to airway relaxation in mice and rats, the effect of PAR-4 activation upon human airway vessel tone or smooth muscle cell proliferation has not been elucidated. Thus, further studies on human tissues defining the functions of PARs 2–4 are required.
The role of PAR-2 in airway function is unlikely to be restricted to asthma. Since other lung cell types express PAR-2 (Akers et al., 2000), and findings indicate the presence of not only tryptase, but also other trypsin-like proteases (Yamaoka et al., 1998); the likelihood is that the receptor will prove to be integral to a wide range of pulmonary functions under both physiological and disease states. One condition in which the antagonism of PAR-2 may be of use is α1-antitrypsin deficiency, a condition resulting in obstructive lung disease (Blank and Brantly, 1994; Coakley et al., 2001), as well as affecting other systems (Fortin et al., 1991;Davis et al., 1992). Although this condition is most strongly linked with neutrophil enzyme-mediated tissue damage, it is likely that the lack of this important plasma proteinase inhibitor will also affect PAR-2 agonist serine proteinases such as trypsin and tryptase, potentially leading to hyperactivation of PAR-2 in the airways and other pulmonary cells. PAR-2 therefore has a clear potential as a useful therapeutic target for a range of diseases associated with pulmonary function.
To define the role of PAR-2 in inflammation adequately and clearly, extended studies are required utilizing PAR-2-deficient mice and disease models coupled with the development of potent and selective PAR-2 agonists and antagonists. One such study using PAR-2-deficient mice has noted that lack of the receptor affects the onset of inflammatory responses (Linder et al., 2000), indicating that blockade of PAR-2 may be of some use in the therapeutic control of inflammation. Such approaches should allow further advances in the utilization of PAR-2 as a therapeutic target.
XVI. Future Perspectives
In the last 10 years, major developments have been recorded in the understanding of the novel family of receptors known as PARs. Working models regarding modes of activation, intracellular signaling, desensitization, and functional responses have been established for PAR-1 and, to a lesser extent, PAR-2. However, many outstanding questions remain to be addressed. The cellular functions of PAR-2 and, in particular, PAR-3 and PAR-4, remain to be fully elucidated. For PAR-2, this includes a more detailed investigation, in a manner similar to PAR-1, of the intracellular signaling pathways involved in the cellular effects of PAR-2 stimulation. At a functional level, fuller exploitation of the available PAR-2 knockout mice is required, particularly in models of inflammation and other diseases. Matching a therapeutic target with the development of selective PAR-2 agonist and antagonist drugs will clearly be a major thrust of several pharmaceutical companies in the next decade.
For both PAR-3 and PAR-4, initial studies investigating functional responses in relation to tissue distribution need to be expanded. Again, this needs to be complemented by investigation of the intracellular signaling pathways coupled to these receptors and the molecular basis for their desensitization and resensitization. These studies will also benefit from the future development of PAR-4 knockout mice to assess roles in inflammation, immune function, and coagulation.
It is likely that other PARs exist that remain unidentified, allowing further development of selective drugs with therapeutic potential. The presence of multiple PARs accords with the increasing number of serine proteases that have now been identified, have cell type-specific expression, and the potential to cleave PARs. In this context, the recent demonstration of receptor coactivation and the roles for other tethering proteins in PAR activation are important extensions of the normal receptor-ligand paradigm. The potential for other undiscovered PARs to have similar modes of activation is a fascinating prospect. This, and the potential for multiple serine proteases to active the same PAR, makes understanding the mechanisms of endogenous PAR activation a challenging area of future research.
Acknowledgments
The authors thank Drs. Andrew Paul, Brian Furman, and C. M. Belham for reading the manuscript and for constructive comments, and to Callum Sloss for drawing the diagrams. We also wish to acknowledge that limited space necessitates omission of a number of important studies in this field. Work in the author's laboratory is funded by Kowa Co. Ltd., Japan.
Footnotes
-
↵1 Address for correspondence: Robin Plevin, Department of Physiology and Pharmacology, University of Strathclyde, Strathclyde Institute for Biomedical Sciences, 27 Taylor St., Glasgow G4ONR, UK. E-mail: r.plevin{at}strath.ac.uk
-
↵3 T. Kanke, S. Macfarlane, M. J. Seatter, E. Davenport, A. Paul, and R. Plevin, manuscript submitted.
Abbreviations
- PAR
- proteinase-activated receptor
- AP
- agonist peptide
- AP-1
- activating protein-1
- ECL
- extracellular loop
- ERK
- extracellular regulated kinase
- EST
- expressed sequence tag
- ET-1
- endothelin-1
- BMS-197525
- N-trans-cinnamoyl-p-fluoro-Phe-p-guanidino-Phe-Leu-Arg-NH2
- GI
- gastrointestinal
- GP
- guinea pig
- GP1βα
- glycoprotein-1βα
- GRB-2
- growth factor receptor binding protein-2
- GRK
- G-protein-coupled receptor kinase
- GST
- glutathionine S-transferase
- 5-HT
- 5-hydroxytryptamine
- ICAM-1
- intracellular adhesion molecule-1
- IP
- inositol phosphate
- InsP3
- inositol 1,4,5-trisphosphate
- kb
- kilobase(s)
- JNK
- c-jun N-terminal kinase
- l-NAME
- Nω-nitro-l-arginine methyl ester
- LPS
- lipopolysaccharide
- MAP
- mitogen-activated protein
- MEK
- mitogen-activated protein kinase kinase
- MMP
- matrix metalloproteinase
- NO
- nitric oxide
- PAO
- phenylarsine oxide
- PDGF
- platelet-derived growth factor
- PGF1α
- prostaglandin-F1α
- PGE2
- prostaglandin E-2
- PI-3 kinase
- phosphatidylinositol 3-kinase
- PKC
- protein kinase C
- PLA2
- phospholipase A2
- PLC
- phospholipase C
- PLD
- phospholipase D
- PTX
- pertussis toxin
- ROK
- Rho-dependent kinase
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- SAP
- stress-activated protein
- SHC
- Src homology collagen
- SHP-2
- Src homology phosphatase-2
- TRAP
- thrombin receptor-activating peptide
- TGF-β
- transforming growth factor-β
- TXA2
- thromboxane
- VCAM-1
- vascular cell adhesion molecule-1
- VEGF
- vascular endothelial growth factor
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





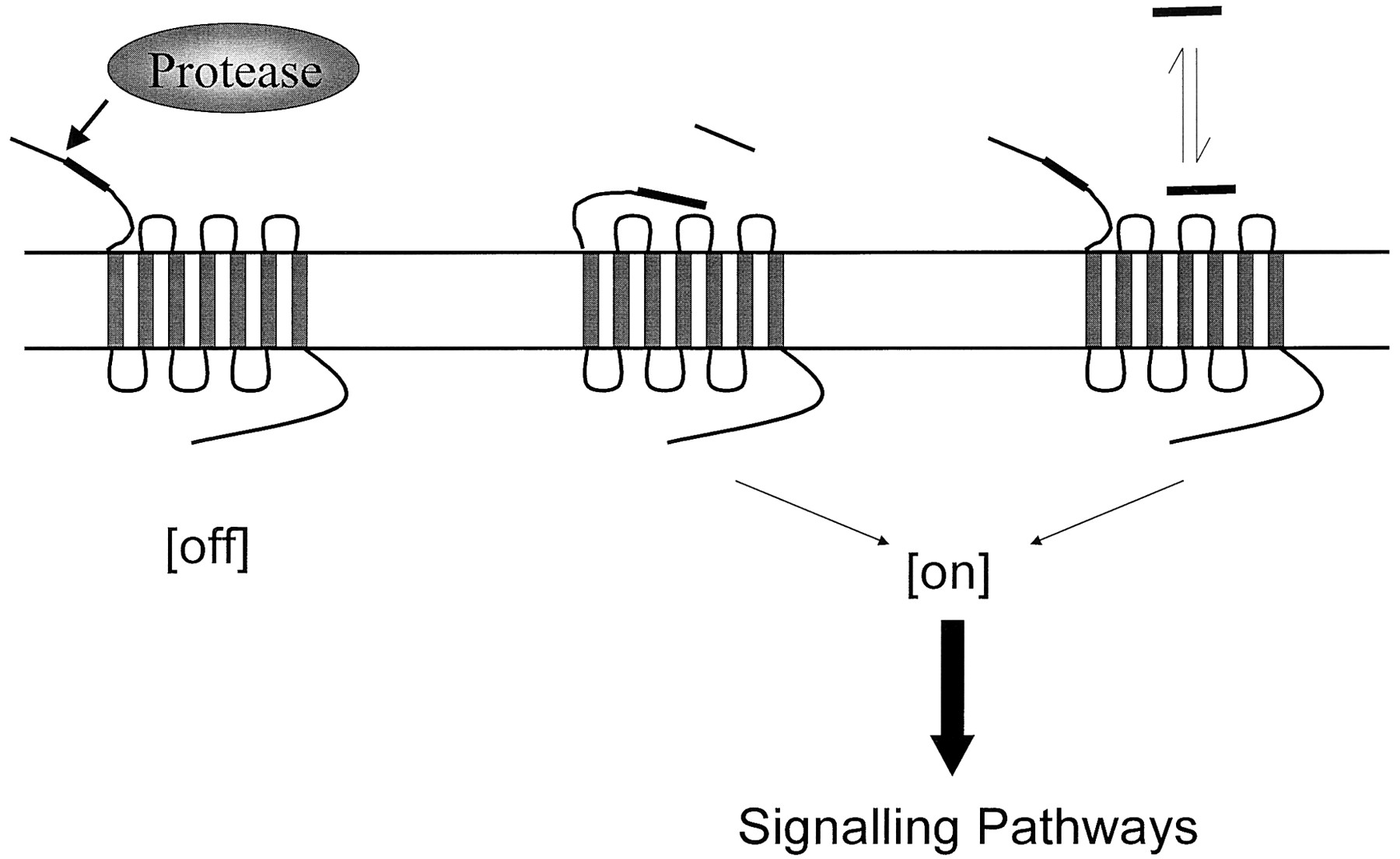{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Historical Perspectives—Cellular Effects of Thrombin and the Cloning of the Thrombin Receptor, Proteinase-Activated Receptor-1
- III. Pharmacology of Proteinase-Activated Receptor-1
- IV. Functional Responses to Proteinase-Activated Receptor-1 Activation
- V. Proteinase-Activated Receptor-1-Mediated Cellular Signaling
- VI. Desensitization of Proteinase-Activated Receptor-1
- VII. Cloning of Proteinase-Activated Receptor-2
- VIII. Functional Responses to Proteinase-Activated Receptor-2 Activation
- IX. Endogenous Activators of Proteinase-Activated Receptor-2
- X. Pharmacology of Proteinase-Activated Receptor-2
- XI. Proteinase-Activated Receptor-2-Mediated Intracellular Signaling
- XII. Proteinase-Activated Receptor-2 Desensitization
- XIII. Identification and Function of Proteinase-Activated Receptor-3 and Proteinase-Activated Receptor-4
- XIV. Functional and Molecular Interactions Between Proteinase-Activated Receptors
- XV. Proteinase-Activated Receptors as Therapeutic Targets in Disease States
- XVI. Future Perspectives
- Acknowledgments
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters