


I. Introduction
Adenosine is an endogenous nucleoside that modulates many physiological processes. Its actions are mediated by interaction with specific cell membrane receptors. Four subtypes of adenosine receptors have been cloned: A1, A2A, A2B, and A3. Significant advancement has been made in the understanding of the molecular pharmacology and physiological relevance of adenosine receptors, but our knowledge of A2B receptors lags behind that of other receptor subtypes. The lack of selective pharmacological probes has hindered research in this area. Perhaps because of their lower affinity for adenosine compared with other receptors, it is often assumed that A2B receptors are a low-affinity version of the A2A receptor and are of lesser physiological relevance. It has been only recently that potentially important functions have been discovered for the A2B receptor, prompting a renewed interest in this receptor type. It is also recently recognized that A2B receptors are coupled to intracellular pathways different from those of A2A receptors, a finding that may provide the basis for their distinct physiological role. A2B receptors have been implicated in mast cell activation and asthma, vasodilation, regulation of cell growth, intestinal function, and modulation of neurosecretion. We try to review the recent advances made in the study of A2B receptors and underscore areas in which more progress is needed. We discuss some of the characteristics of A1, A2A, and A3 receptors only to highlight their similarities and differences with A2B receptors. Recent reviews on specific adenosine receptor subtypes can be found elsewhere (Linden, 1991, 1994; Dalziel and Westfall, 1994; Fredholm, 1995; Palmer and Stiles, 1995; Sebastião and Ribeiro, 1996; Daval et al., 1996; Ongini and Fredholm, 1996).
II. Classification of Adenosine Receptors
The properties of extracellular adenosine as a protective autacoid have been known since the study of its cardiovascular effects conducted in 1929 by Drury and Szent-Györgyi (1929). The purinergic receptors that mediate the effects of adenosine were classified as P1 receptors, whereas the receptors activated by nucleotides like adenosine 5c-triphosphate (ATP) were classified as P2 receptors (Burnstock, 1978). Adenosine receptors were found to modulate intracellular levels of adenosine 3c,5c-cyclic monophosphate (cAMP) and were initially subdivided into A1 and A2 subtypes based on their ability to inhibit or stimulate adenyl cyclase, respectively (van Calker et al., 1979; Londos et al., 1980). The alternative classification of adenosine receptors as Ri and Ra (Londos et al., 1980) was replaced by the A1 and A2 terms (van Calker et al., 1979). The further division of A2receptors into two subtypes was proposed originally by Daly et al. (1983) based on the finding of high-affinity A2receptors in rat striatum and low-affinity A2receptors throughout the brain, both of which activated adenyl cyclase. The existence of subtypes of A2 receptors was also suggested by the finding, independently reported by Elfman et al. (1984), of high-affinity A2 receptors in cultured neuroblastoma cells and low-affinity A2 receptors in glioma cells. These high- and low-affinity receptor subtypes were later designated as A2A and A2B, respectively (Bruns et al., 1986). The classification of P1 receptors has been validated by the recent success in molecular cloning and expression of all three anticipated A1, A2A, and A2B adenosine receptors and the previously unrecognized A3 receptor (Maenhaut et al., 1990;Libert et al., 1991; Zhou et al., 1992; Rivkees and Reppert, 1992;Pierce et al., 1992). This classification has been endorsed by IUPHAR Committee on Receptor Nomenclature and Drug Classification (Fredholm et al., 1994, 1996b, 1997).
III. Molecular Characterization of A2B Receptors
Adenosine A2B receptors were cloned from rat hypothalamus (Rivkees and Reppert, 1992), human hippocampus (Pierce et al., 1992), and mouse mast cells (Marquardt et al., 1994), employing standard polymerase chain reaction techniques with degenerate oligonucleotide primers designed to recognize conserved regions of most G protein-coupled receptors. The human A2Breceptor shares 86 to 87% amino acid sequence homology with the rat and mouse A2B receptors (Rivkees and Reppert, 1992; Pierce et al., 1992; Marquardt et al., 1994) and 45% amino acid sequence homology with human A1 and A2A receptors (fig.1). As expected for closely related species, the rat and mouse A2B receptors share 96% amino acid sequence homology. By comparison, the overall amino acid identity between A1 receptors from various species is 87% (Palmer and Stiles, 1995). A2Areceptors share 90% of homology between species (Ongini and Fredholm, 1996), with most differences occurring in the 2ndextracellular loop and the long C-terminal domain (Palmer and Stiles, 1995). The lowest (72%) degree of identity between species is observed for A3 receptor sequences (Palmer and Stiles, 1995). Differences in amino acid sequence of adenosine receptors between species may result in distinct pharmacological characteristics. For example, the rat A1 receptor has a rank order of potency (R)-N6-phenylisopropyladenosine (R-PIA) > 5′-N-ethylcarboxamidoadenosine (NECA) > (S)-N6-phenylisopropylaolenesine (S-PIA), and the bovine A1 receptor has a potency order R-PIA > S-PIA > NECA (Klotz et al., 1991), whereas the canine A1 receptor binds NECA with a higher affinity than that of R-PIA (Tucker and Linden, 1993). The differences between amino acid sequences of A3 receptors are reflected in the insensitivity of the rat A3receptor to antagonism by methylxanthines (Zhou et al., 1992), a phenomenon that is not observed in the human or sheep A3 receptor (Linden et al., 1993; Salvatore et al., 1993). These interspecies differences between adenosine receptors explain why the adenosine agonist xanthine amine congener (XAC) is a selective A1 agonist in the rat, but not in the human or rabbit (Jacobson et al., 1992; Jacobson and Suzuki, 1996). Few comparisons have been made between A2B receptors from different species. No differences in pharmacological profiles were found between A2B receptors from fibroblasts of murine and human origin (Bruns, 1981; Brackett and Daly, 1994) or between human A2B receptor expressed in Chinese hamster ovary (CHO) cells and guinea pig brain A2B receptors (Alexander et al., 1996).
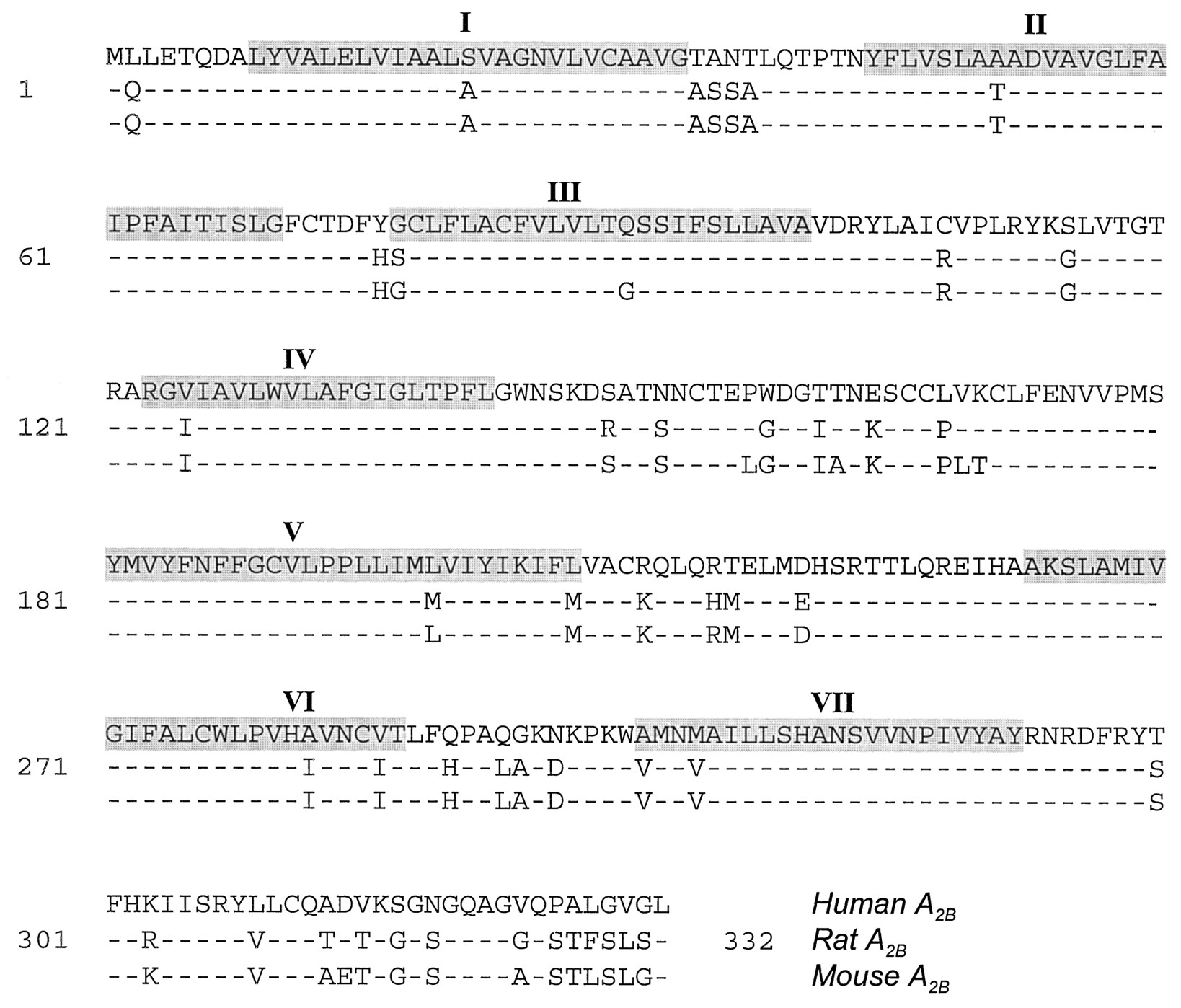
Comparison of amino acid sequences of human (M97759) (Pierce et al., 1992), rat (M91466) (Stehle et al., 1992), and mouse (U05673) (Marquardt et al., 1994) A2B receptors. Dashed lines indicate amino acid identity. Predicted transmembrane spanning domains are highlighted and indicated by roman numerals.
The proposed membrane structure of A2Breceptors is typical of G protein–coupled receptors, with seven transmembrane domains connected by three extracellular and three intracellular loops, and flanked by an extracellular N-terminus and an intracellular C-terminus (Rivkees and Reppert, 1992; Pierce et al., 1992; Marquardt et al., 1994; fig. 2). The highest degree of identity in amino acid sequences between A2B receptors of different species is found in the transmembrane domains (fig. 1). The 2ndextracellular loop of the human, mouse, and rat A2B receptors contains two potential N-glycosylation sites (Rivkees and Reppert, 1992; Pierce et al., 1992;Marquardt et al., 1994). It should be noted that enzymatic treatment failed to demonstrate N-glycosylation of A2Breceptors in T84 epithelial cells (Puffinbarger et al., 1995). However, it is not clear whether A2B receptors are glycosylated in other cells or glycosylation can alter A2B function. A2A receptors were found to be glycosylated in canine striatum and liver membranes (Palmer et al., 1992), but the binding characteristics of A2A receptors for 4-[(N-ethyl-5′-carbamoyladenos-2-yl)-aminoethyl]-phenylpropionic acid (CGS 21680) appear to be the same in both glycosylated or unglycosylated forms of the receptor expressed in COS M6 cells (Piersen et al., 1994).

Amino acid sequence of the human A2Breceptor. The receptor is drawn according to the seven-membrane spanning motif common to the G protein-coupled receptor superfamily. Possible sites of N-linked glycosylation on the 2ndextracellular loop are highlighted.
The predicted molecular mass of A2B receptors is similar to that of A1 and A3 receptors (36–37 kDa), whereas A2A receptors have a larger predicted size (45 kDa). The greater molecular mass of the A2Areceptor is explained by the presence of a longer intracellular C-terminus. Together with the 3rd intracellular loop, the intracellular C-terminus is thought to be involved in the coupling of A2A receptors to G proteins (Palmer and Stiles, 1995). To date, no mutational analysis of A2B receptor-G protein coupling has been reported. However, some parallels could be drawn from studies using chimeric A1/A2A adenosine receptors (Tucker et al., 1996; Olah, 1997). Using this approach, it has been shown that the amino terminal portion of the 3rd intracellular loop of the A2A receptor determines its selective coupling with Gs (Olah, 1997). This 15-mer portion of the A2A receptor shares 57% amino acid sequence homology with the A2B receptor, both of which are coupled to Gs, and only 27% with the A1 receptor, which is not coupled to Gs (fig. 3). In addition, the nature of the amino acids in the 2nd intracellular loop may indirectly modulate A2A receptor coupling. In particular, lysine and glutamic acid residues in that portion of the molecule were found to be necessary for efficient A2A adenosine receptor-Gs coupling (Olah, 1997). These amino acid residues are also present in the A2Breceptor. The long intracellular C-terminal tail of the A2A receptor, which represents a major structural difference with the A2B, does not appear to be involved in the determination of receptor coupling to Gs protein. The removal of the C-terminal tail of the A2A receptor, or its replacement with a cytoplasmic tail of the A1 receptor, does not impair stimulation of adenyl cyclase when these truncated or chimeric receptors are expressed in CHO cells (Tucker et al., 1996; Palmer and Stiles, 1997; Olah, 1997). The data generated from these studies, however, leave the possibility that this region can still play a role in the modulation of the coupling of A2Areceptors to G proteins. For example, it was suggested that the C-terminal tail confers the A2A receptors’ ability to couple tightly to Gs, a feature considered to be unique for this receptor subtype (Nanoff et al., 1991).

Comparison of amino acid sequences of 2nd and 3rd intracellular loops and C-terminus of human A2A receptors, with corresponding regions of human A1 and A2B receptors. Dashed lines indicate amino acid identity. Asterisks represent gaps in the sequence introduced to highlight amino acid homologies. The amino acid residues discussed in the text are highlighted.
Mutational studies of A2A receptors revealed that a threonine residue (Thr298) of the C-terminal tail of the A2A receptor, located in proximity to the seventh transmembrane span (fig. 3), is essential for the development of rapid agonist-mediated desensitization (Palmer and Stiles, 1997). This amino acid residue is also present in the human A2B receptor (Thr300), but its role in receptor desensitization has not been explored. Although the mechanisms of desensitization are not completely identified, it is of interest that rapid desensitization of A2A as well as A2B receptors can be mediated by G protein-coupled receptor kinase 2 (Mundell et al., 1997). It should be noted that A2B receptors can be coupled to other intracellular signaling pathways in addition to Gs and adenyl cyclase. The similarities and differences in A2B and A2Areceptor coupling to G proteins warrant studies involving mutational analysis of A2B receptors, and possibly chimeric A2A/A2B receptors, to better understand determinants of A2B-G protein coupling.
The human A2B receptor gene was mapped to chromosome 17p11.2-p12 (Jacobson et al., 1995; Townsend-Nicholson et al., 1995). A single intron interrupts the coding sequence of the human A2B receptor gene in a region corresponding to the 2nd intracellular loop between Leu111and Arg112 (Jacobson et al., 1995). In this respect, the human A2Breceptor gene is similar to the other human adenosine receptor genes in that it also contains a single intron in its coding sequence (Ren and Stiles, 1994; Peterfreund et al., 1994; Murrison et al., 1996). Some G protein-coupled receptors are known to have multiple introns in the coding sequences of their corresponding genes. Alternative splicing of their primary transcripts results in heterogeneity in protein sequences, as observed with EP3 prostanoid receptors (Neglishi et al., 1995), D2 dopamine receptors (Giros et al., 1989), lutropin/choriogonadotropin receptors (Aatsinki et al., 1992), and fibroblast growth factor receptors 2 (Dell and Williams, 1992). The presence of only one intron within the coding region of the human A2B receptor gene precludes structural variations of A2B receptors by alternative splicing.
In addition to the human A2B receptor gene, an A2B pseudogene with 79% identity with the A2B receptor complementary deoxyribonucleic acid (cDNA), has been localized to chromosome 1q32 (Jacobson et al., 1995;Townsend-Nicholson et al., 1995). When compared with the coding sequence of the A2B receptor, the pseudogene contained multiple deletions, point mutations, and frame shifts and two in-frame stops (Jacobson et al., 1995). It is doubtful that with all these changes the pseudogene would encode a functional adenosine receptor. However, further studies are needed to determine whether the A2B pseudogene is transcriptionally competent. For example, dopamine D5 pseudogene transcripts can be detected in human brain tissues (Nguyen et al., 1991). The same possibility should always be considered in Northern blot analysis or in situ hybridization of A2B receptor in various tissues, because the use of sequences common between the functional A2B cDNA and the A2Bpseudogene as probes could potentially lead to misinterpretation of results.
IV. Pharmacology of A2B Receptors
Highly selective and potent agonists have been designed for A1, A2A, and A3 receptors. These compounds have been important tools in the characterization of adenosine receptors and the determination of their functions. All four subtypes, including A2B receptors, have a typical order of potency for agonists (table 1; fig.4). However, no selective agonist for A2B receptors has been found so far. The adenosine analog NECA remains the most potent A2Bagonist (Bruns, 1981; Feoktistov and Biaggioni, 1993, 1997; Brackett and Daly, 1994), with a concentration producing a half-maximal effect (EC50) for stimulation of adenyl cyclase of approximately 2 μm. It is, however, nonselective and activates other adenosine receptors with even greater affinity, with an EC50 in the low nanomolar (A1 and A2A) or high nanomolar (A3) range (table 1; fig. 4). The characterization of A2B receptors, therefore, often relies on the lack of effectiveness of compounds that are potent and selective agonists of other receptor types. A2B receptors have been characterized by a method of exclusion, i.e., by the lack of efficacy of agonists that are specific for other receptors. The A2A selective agonist CGS 21680 (Webb et al., 1992), for example, has been useful in differentiating between A2A and A2B adenosine receptors (Hide et al., 1992; Chern et al., 1993; Feoktistov and Biaggioni, 1995; van der Ploeg et al., 1996). Both receptors are positively coupled to adenyl cyclase and are activated by the nonselective agonist NECA. CGS 21680 is virtually ineffective on A2B receptors but is as potent as NECA in activating A2A receptors, with an EC50 in the low nanomolar range for both agonists (Jarvis et al., 1989; Nakane and Chiba, 1990; Webb et al., 1992; Hide et al., 1992; Feoktistov and Biaggioni, 1993; Alexander et al., 1996). A2B receptors have also a very low affinity for the A1 selective agonist R-PIA (Feoktistov and Biaggioni, 1993; Brackett and Daly, 1994) as well as for the A3 selective agonist N6-(3-iodobenzyl)-N-methyl-5′-carbamoyladenosine (IB-MECA) (Feoktistov and Biaggioni, 1997). The agonist profile NECA > R-PIA = IB-MECA > CGS 21680 was determined in human erythroleukemia (HEL) cells for A2B-mediated cAMP accumulation. The difference between EC50 for NECA and the rest of the agonists is approximately 2 orders of magnitude. Therefore, responses elicited by NECA at concentrations in the low micromolar range (1–10 μm), but not by R-PIA, IB-MECA or CGS 21680, are characteristic of A2B receptors.
Pharmacological characteristics of adenosine receptor subtypes

Chemical structure and radioligand binding data on the affinity of adenosine agonists. Ki values (nm) for rat A1/A2A/A2B/A3 receptors are shown, except as indicated (h, human). Numbers in brackets represent EC50 of adenosine agonists for cAMP accumulation in HEL cells. Data compiled from Feoktistov and Biaggioni (1993), van Galen et al. (1994), Jacobson and Suzuki (1996), and Feoktistov and Biaggioni (1997).
Pharmacological characterization of receptors based on apparent agonist potencies, however, is far from ideal, because it depends not only on agonist binding to the receptor but also on multiple processes involved in signal transduction. Selective antagonists are preferable for receptor subtype identification (Kenakin et al., 1992). Highly selective and potent A2B antagonists are not yet available, but, whereas A2B receptors have a lower affinity for agonists compared with other receptor subtypes, this is not true for antagonists. The structure-activity relationship of A2B receptors for adenosine antagonists has not been completely characterized, but at least some xanthines are as potent antagonists at A2B receptors as at other adenosine receptors (Feoktistov and Biaggioni, 1993; Brackett and Daly, 1994).
The antiasthmatic drug enprofylline (3-n-propylxanthine), is the most selective A2B antagonist known to date. In early studies, enprofylline was found to be about 20 times more potent in blocking hippocampal A2 receptors compared with rat fat cell A1 receptors (Fredholm and Persson, 1982). It was initially proposed, therefore, that enprofylline can selectively block a subtype of A2 receptors in the hippocampus (Fredholm and Persson, 1982). However, enprofylline was then found to be a poor antagonist of A2receptors in thymocytes (Fredholm and Sandberg, 1983) and platelets (Ukena et al., 1985). More recently, enprofylline has also been found to have a low affinity for A3 receptors (Linden et al., 1993). These findings led to the conclusion that enprofylline was not an adenosine receptor antagonist. These original studies need to be reinterpreted in the light of our current knowledge of adenosine receptor subtypes. It is now known that accumulation of cAMP in hippocampal slices, which was shown to be blocked by enprofylline, is mediated by A2B receptors (Lupica et al., 1990), and that platelets, found to be insensitive to enprofylline, express mainly A2A receptors (Feoktistov and Biaggioni, 1993; Dionisotti et al., 1996; Ledent et al., 1997). Therefore, previous contradictory results can now be explained by a selective antagonism of A2B receptors by enprofylline. Indeed, it was recently demonstrated that enprofylline is equipotent to theophylline as an A2B receptor antagonist in HEL cells, with a dissociation constant of antagonist-receptor complex (KB) of 7 μm (Feoktistov and Biaggioni, 1995). An analysis of the original results in the hippocampus (Fredholm and Persson, 1982) reveals an approximate KB of 6 μm. An identical Ki for enprofylline (7 μm) was found in CHO cells stably transfected with A2Busing radioligand binding with [3H]1,3 diethyl-8-phenylxanthine (Robeva et al., 1996). This value also correlated well with the KB estimated from inhibition of NECA-induced cAMP generation in a similar cell model (23 μm) (Alexander et al., 1996). Enprofylline is also an effective antagonist of A2B receptors in human HMC-1 mast cells (Feoktistov and Biaggioni, 1995) and canine BR mastocytoma cells (Auchampach et al., 1996). In comparative radioligand binding studies on all four human adenosine receptors permanently expressed in CHO cells, enprofylline has been shown to be 22-fold selective for A2B versus A1, five-fold versus A2A, and six-fold versus A3 (Robeva et al., 1996). Enprofylline, therefore, can be considered a relatively selective, though not potent A2B antagonist.
More potent but nonselective A2B receptor antagonists have been also characterized. These compounds include 1,3-dipropyl-8-(p-sulfophenyl)xanthine (DPSPX), 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), and XAC (Feoktistov and Biaggioni, 1993; Brackett and Daly, 1994). The xanthine antagonist DPSPX is 20-fold more potent at A2B receptors in HEL cells (KB = 141 nm) compared with platelet A2A receptors (Feoktistov and Biaggioni, 1993). However, the affinity of A2B receptors for DPSPX (Feoktistov and Biaggioni, 1993) is similar to those of sheep A3 (Linden et al., 1993) and rat A1 (Ukena et al., 1986) receptors. Among nonxanthine compounds, 2,4-dioxobenzo[g]pteridine (alloxazine) was reported to be nine-fold more potent as an antagonist of A2B receptors in VA13 and NIH 3T3 cells compared with A2A receptors in PC12 cells (Brackett and Daly, 1994; fig. 5).

Chemical structure and radioligand binding data on the affinity of adenosine antagonists. Ki values (nm) for rat A1/A2A/A2B/A3 receptors are shown, except as indicated (h, human; gp, guinea pig; s, sheep; m, mice). Numbers in brackets represent KB of adenosine antagonists. Data compiled from Feoktistov and Biaggioni (1993),Brackett and Daly (1994), van Galen et al. (1994), Jacobson et al. (1996), Robeva et al. (1996), Jiang et al. (1996), van Rhee et al. (1996), Zocchi et al. (1996a), and Jacobson and Suzuki (1996). *, selective for rat, but not for human or rabbit A1 receptors (Jacobson and Suzuki, 1996).
A2B receptors are frequently found with other adenosine receptor subtypes in the same tissue, and are even coexpressed in the same cells. Recent advances in the development of selective A1, A2A, and A3 antagonists (table 1; fig. 5) provide a new approach to the study of A2B receptors; the nonselective agonist NECA can be used in conjunction with highly selective antagonists of other adenosine receptor subtypes to selectively stimulate A2B receptors. The ability to selectively block other adenosine receptors is particularly useful in situations in which they are present with A2Breceptors.
The first selective A1 antagonist DPCPX was discovered by two independent groups of investigators (Martinson et al., 1987; Bruns et al., 1987) and has become the reference A1 receptor antagonist. It is highly selective for A1 versus A2A (80- to 500- fold across species) (Jacobson et al., 1992; Robeva et al., 1996). In recent radioligand binding studies involving all four human recombinant adenosine receptors, DPCPX has been confirmed to be 20-fold selective for A1 versus A2B(Robeva et al., 1996). Selective blockade of A1receptors with DPCPX was successfully used to reveal functional A2B receptors in tissues coexpressing both A1 and A2B receptors (Mogul et al., 1993; Murthy et al., 1995; Nicholls et al., 1996). Other compounds have been identified with even greater selectivity for the A1 receptor; C8-(N-methylisopropyl)-amino-N6-(5′-endohydroxy)-endonorbornan-2-yl-9-methyladenin (WRC-0571) binds to human A1 receptors with a Ki of 3 nm and to human A2B receptors with a Ki of 19 μm. This compound, therefore, is approximately 6300-fold selective for A1 versus A2B (Robeva et al., 1996).
Among the new generation of A2A antagonists, 4-(2-[7-amino-2-)2-furyl(triazolo {2,3-a}-[1,3,5]triazin-5-ylamino]ethyl)phenol (ZM 241385) was reported to be 30- to 80-fold selective for A2Aversus A2B (Poucher et al., 1995). Another antagonist, 5-amino-7-(phenylethyl)-2-(1-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (SCH 58261), has a high affinity (Ki = 0.7–2.2 nm) for A2A receptors (Belardinelli et al., 1996; Lindström et al., 1996; Dionisotti et al., 1996;Zocchi et al., 1996a,b; Ongini et al., 1996; Ongini and Fredholm, 1996) but was found not to block NECA-induced vasorelaxation of guinea pig aorta, a process thought to be mediated by A2Breceptors (Zocchi et al., 1996a). The selectivity of SCH 58261 for A2A versus A2B has been also confirmed in a cellular system; this compound was ineffective on HEL cell A2B receptors up to a concentration of 100 nm, whereas it inhibited the CGS 21680-induced cAMP accumulation in HMC-1 cell (A2A receptor) with a KB of 0.1 nm (Feoktistov and Biaggioni, 1997). SCH 58261, therefore, can be useful in the discrimination of A2B function in cells also coexpressing A2A receptors. This approach was applied to the study of adenosine receptors in the human mast cells HMC-1 (fig. 6). The concentration-response relationship of the nonselective adenosine agonist NECA for cAMP accumulation in these cells follows a curve with a Hill slope of 0.64 ± 0.07 best fitted to a two-site model with an apparent pD2 of 7.69 ± 0.42 and 5.92 ± 0.21 for the high- and low-affinity sites, respectively. Upon complete blockade of A2A receptors with 100 nm SCH 58261, the concentration-response curve of NECA was transformed into a typical sigmoid curve with a Hill slope of 0.93 ± 0.06 and a pD2 of 5.68 ± 0.03, consistent with activation of A2B receptors. Blockade of A2A receptors in the same cells with SCH 58261 did not affect NECA-induced calcium mobilization, confirming that this process is mediated solely via A2Breceptors (Feoktistov and Biaggioni, 1997), as it has been previously suggested on the basis of the lack of CGS 21680 effectiveness (Feoktistov and Biaggioni, 1995).

Selective activation of A2B receptors with NECA in HMC-1 human mast cells coexpressing A2A and A2B receptors, made possible by blockade of A2Areceptors with the selective antagonist SCH 58261 (Feoktistov and Biaggioni, 1997). See text for details.
Recently, several antagonists with A3 selectivity versus A1 and A2A receptors have been introduced. These compounds include the flavonoid derivative 3,6-dichloro-2′-isopropyloxy-4′-methylflavone (MRS 1067) and the dihydropyridine derivatives 3-ethyl 5-benzyl 2-methyl-phenylethynyl-6-phenyl-1,4(±)dihydropyridine-3,5-dicarboxylate (MRS 1191), 3,5-diethyl 2-methyl-4[2-(4-nitrophenyl)-(E)-vinyl-6-phenyl-1,4-(±)-dihydro-pyridine-3,5-dicarboxylate (MRS 1222), and 3,5-diethyl 2-methyl, 6-phenyl-4-[2-[phenyl-(trans)-vinyl]-1,4(±)dihydropyiridine-3,5-dicarboxylate (MRS 1097) (fig. 5), which are selective for the human A3 receptor by a factor of 45- to 1700-fold, versus rat A1 and A2Areceptors, as determined from radioligand binding studies (Jiang et al., 1996; Karton et al., 1996; van Rhee et al., 1996). It should be noted, however, that the highest degree of selectivity for these compounds is observed when their effects on human A3 receptors are compared with their effects on rat A1 and A2A receptors. For example, MRS 1191 was selective for the human A3 receptor by factor of 1300-fold, whereas for the rat A3 receptor, the selectivity was only 11-fold versus the rat A1 receptor (Jiang et al., 1996). Among other compounds, the triazolonaphthyridine derivative 6-carboxymethyl-5,9-dihydro-9-methyl-2-phenyl-[1,2,4]-triazolo-{5,1-a}-[2,7]-naphthyridine (L-249313) (fig. 5) is highly potent on human A3 receptors (Ki =13 nm), but not on rat A3 receptors (Ki = 58 μm). The thiazolopyrimidine derivative 3-(4-methoxyphenyl)-5-amino-7-oxo-thiazolo-[3,2]-pyrimidine (L-268605) was also shown to be a potent antagonist on human A3 receptor (Ki = 18 nm). Both compounds are highly selective for the human A3 receptor versus the human A1 (>300-fold) and A2A(>1400-fold) receptors (Jacobson et al., 1996). Unfortunately, A2B receptors have not been included when characterizing the selectivity of A3 antagonists. Additional studies of A3 antagonists with respect to A2B receptors are required to verify whether they can be useful to discriminate between A3 and A2B-mediated effects.
In summary, potent and selective agonists and antagonists are available for all adenosine receptors except for the A2Bsubtype. The characterization of A2B receptors has been based on apparent potencies of agonists selective to other adenosine receptor subtypes. The development of selective A1, A2A, and A3 antagonists provides a new approach when used in conjunction with the nonselective agonist NECA to selectively stimulate A2B receptors. This approach is particularly useful in tissues or cells expressing more than one adenosine receptor. However, much progress in this field could be achieved by the development of selective A2Breceptor antagonists. Because of the low affinity of this receptor for agonists, the design of selective and potent A2Bantagonists seems to be more promising than the development of selective agonists.
V. Distribution of A2B Receptors
The generation of cDNA for A2B receptors has made possible the identification of the tissue distribution of this receptor subtype. A2B receptor messenger ribonucleic acid (mRNA) was originally detected in a limited number of rat tissues by Northern blot analysis, with the highest levels found in cecum, bowel, and bladder, followed by brain, spinal cord, lung, epididymis, vas deferens, and pituitary (Stehle et al., 1992). The use of more sensitive reverse transcriptase-polymerase chain reaction techniques revealed a ubiquitous distribution of A2B receptors. mRNA encoding A2B receptors was detected at various levels in all rat tissues studied, with the highest levels in the proximal colon and lowest in the liver (Dixon et al., 1996). In situ hybridization of A2B receptors showed widespread and uniform distribution of A2B mRNA throughout the brain (Stehle et al., 1992; Dixon et al., 1996). The expression of A2B receptors in a variety of human and murine tissues has been confirmed by Western blotting and by immunostaining with an anti-A2B receptor antibody (Puffinbarger et al., 1995).
Pharmacological identification of A2B receptors, based on their low affinity and characteristic order of potency for agonists, also indicates a widespread distribution of A2B receptors. In brain, functional A2B receptors are found in neurons (Mogul et al., 1993; Okada et al., 1996; Kessey et al., 1997) and glial cells (van Calker et al., 1979; Elfman et al., 1984; Hösli and Hösli, 1988; Altiok et al., 1992; Peakman and Hill, 1994, 1996; Fiebich et al., 1996a). Although there is no evidence that A2B receptor are present in microglia (Fiebich et al., 1996b), there is ample data that show that they are expressed in astrocytes and in different glioma cell lines (Elfman et al., 1984;Hösli and Hösli, 1988; Altiok et al., 1992; Peakman and Hill, 1994, 1996; Fiebich et al., 1996a). The expression of A2B receptors in glial cells, which represent a majority of the brain cell population, can explain the original observation that slices from all brain areas examined showed an adenosine-stimulated cAMP response (Sattin and Rall, 1970; Daly, 1976).
Functional A2B receptors have been found in fibroblasts (Brackett and Daly, 1994) and various vascular beds (Martin, 1992; Martin et al., 1993; Chiang et al., 1994; Martin and Potts, 1994; Haynes et al., 1995; Rubino et al., 1995; Prentice and Hourani, 1996; Dubey et al., 1996b). Contamination with these cells may also contribute to the widespread pattern of A2Breceptor distribution in all organs. This possibility should always be considered, especially when data from crude tissue preparations are analyzed. The presence of functional A2Breceptors also has been demonstrated in hematopoietic cells (Feoktistov and Biaggioni, 1993; Porzig et al., 1995), mast cells (Marquardt et al., 1994; Feoktistov and Biaggioni, 1995), myocardial cells (Liang and Haltiwanger, 1995), intestinal epithelial (Strohmeier et al., 1995) and muscle cells (Murthy et al., 1995; Nicholls et al., 1996), retinal pigment epithelium (Blazynski, 1993; Gregory et al., 1994), endothelium (Iwamoto et al., 1994), and neurosecretory cells (Casado et al., 1992;Gharib et al., 1992; Mateo et al., 1995).
Coexpression of A2B receptors together with other adenosine receptors has been reported in various cell preparations and cell lines. Functionally coupled A2B and A2A receptors are coexpressed in rat pheochromocytoma PC12 cells (Hide et al., 1992; Chern et al., 1993; van der Ploeg et al., 1996), T-cell leukemia Jurkat cells (van der Ploeg et al., 1996), mouse bone marrow-derived mast cells (Marquardt et al., 1994), human mast HMC-1 cells (Feoktistov and Biaggioni, 1995), human aortic endothelial cells (Iwamoto et al., 1994), human umbilical vein endothelial cells (Feoktistov and Biaggioni, unpublished observations), and human neutrophil leukocytes (Fredholm et al., 1996c). mRNA encoding A2A, A2B, and A3, but not A1 receptors, have been found in rat RBL 2H3 mast cells (Ramkumar et al., 1993;Marquardt et al., 1994).
Functional A1 receptors can also be coexpressed with A2A and/or A2Breceptors. In most cases, a selective blockade of A1 receptors is required to unmask functional A2B receptors. This approach was successfully used in dispersed guinea pig small intestinal muscle cells (Murthy et al., 1995), in rat duodenum longitudinal muscle muscularis mucosae cells (Nicholls et al., 1996), and in guinea pig pyramidal neurons from the hippocampal CA3 region (Mogul et al., 1993). Similarly, uncoupling of A1 receptor using pertussis toxin unmasks the presence of A2A and A2Breceptors in ventricular myocytes (Liang and Haltiwanger, 1995). By contrast, it was not necessary to block A1receptors in various glial cells to observe either A2A or A2Breceptor-mediated stimulation of adenyl cyclase (Elfman et al., 1984;Altiok et al., 1992; Peakman and Hill, 1994, 1996; Fiebich et al., 1996a). Also, the balance between A1- and A2-mediated responses can be modulated. For example, corticosteroid treatment of DDT1 MF2 smooth muscle cells increased A1 receptor number and signaling and decreased A2 receptor signaling (Gerwins and Fredholm, 1991). A similar decrease in the A2B signaling upon dexamethasone treatment was also reported in Jurkat cells (Svenningsson and Fredholm, 1997).
Coexistence of different adenosine receptor types in cells obtained from primary tissue cultures (Iwamoto et al., 1994; Peakman and Hill, 1994, 1996) may be attributed to the presence of different subpopulations of cells, each one expressing a single type of adenosine receptor. However, studies on established cell lines (Hide et al., 1992; Feoktistov and Biaggioni, 1995; van der Ploeg et al., 1996) have confirmed the coexpression of adenosine receptors in a single target cell. Moreover, studies performed on single cells have also demonstrated the presence of more than one adenosine receptor subtype (Liang and Morley, 1996; Strickler et al., 1996), including A2B receptors (Liang and Haltiwanger, 1995).
Coexpression of A2B and A2Areceptors has been demonstrated even in clonal cell lines originally used to describe prototypic A2A (PC12 cells) and A2B receptors (Jurkat cells) (van der Ploeg et al., 1996). These cells predominantly express A2Aand A2B receptors, respectively, and the presence of the other receptor type was recognized only after carefully conducted studies using differential responses to a series of 2-substituted adenosine analogs (Hide et al., 1992; van der Ploeg et al., 1996). It is entirely possible, therefore, that more examples of cells coexpressing adenosine receptors may become apparent after selective adenosine antagonists are applied in the characterization of these cells.
The functional meaning of this simultaneous expression of multiple adenosine receptor subtypes in a single target cell is not known. Because A1 and A2Areceptors have a higher affinity for adenosine, in many cellular systems, these receptors need to be blocked before A2B-mediated effects are apparent (Mogul et al., 1993; Liang and Haltiwanger, 1995; Murthy et al., 1995; Nicholls et al., 1996; Kessey et al., 1997; Feokstistov and Biaggioni, 1997). This, however, is not always the case. Both A1 and A2B receptors are present in glial cells of rat astrocytes, and stimulation of A2B receptors with the nonselective agonist NECA induces cAMP accumulation that is evident even in the presence of A1 receptors (Elfman et al., 1984; Altiok et al., 1992; Peakman and Hill, 1994, 1996; Fiebich et al., 1996a). It is possible that the relative importance of A2B receptors is greater in situations in which high interstitial levels of adenosine are reached, e.g., in tissues in which metabolic demands are increased or oxygen supply is decreased, whereas the high affinity A1 and A2A receptors may modulate cellular functions in response to lower concentrations of this autacoid. The recent recognition that in cells coexpressing other adenosine receptors, A2B receptors can be coupled to distinct intracellular pathways (Feoktistov and Biaggioni, 1995), may also provide the basis for a differential physiological role.
VI. Intracellular Pathways Regulated by A2B Receptors
It is generally accepted that A2A and A2B receptors are coupled to Gs proteins, because both activate adenyl cyclase in virtually every cell in which they are expressed. Although activation of adenyl cyclase is arguably an important signaling mechanism for A2A receptors, this is not necessarily the case for A2B receptors, as other intracellular signaling pathways have been found to be functionally coupled to A2B receptors in addition to adenyl cyclase (fig. 7).

Schematic representation of intracellular pathways coupled to adenosine A2B receptors in various cells. A2B receptors are coupled to adenyl cyclase (AC) in all cells shown. Activation of this pathway results in accumulation of cAMP and stimulation of protein kinase A (PKA). (A) A2Breceptors are coupled to phosphatidylinositol-specific phospholipase C (PI-PLC) via a G protein of the Gq family [G(q)] in mast cells (Marquardt et al., 1994; Feoktistov and Biaggioni, 1995; Auchampach et al., 1996). Activation of this pathway results in increase in diacylglycerol (DAG) and inositol trisphosphate (IP3). Diacylglycerol stimulates protein kinase C (PKC). Inositol trisphosphate activates mobilization of calcium from intracellular stores. (B) A2B receptors potentiate calcium influx directly by coupling with Gsprotein in HEL cells (Feoktistov et al., 1994). (C) In contrast, A2B receptors potentiate calcium influx via cAMP and activation of protein kinase A in pyramidal neurons from the CA3 region of guinea pig hippocampus (Mogul et al., 1993).
Recombinant rat A2B receptors expressed inXenopus oocytes activate calcium-dependent chloride conductance presumably by stimulation of phospholipase C (Yakel et al., 1993). Likewise, it has been proposed that A2Breceptors stimulate phospholipase C in mouse bone marrow-derived mast cells (Marquardt et al., 1994). Regulatory proteins of the Gq family are thought to play a role in the coupling of A2B receptors to β-phospholipase C in human mast HMC-1 cells (Feoktistov and Biaggioni, 1995) and canine BR mastocytoma cells (Auchampach et al., 1996), because this process is unaffected by treatment with pertussis or cholera toxins. A2B receptor-mediated stimulation of β-phospholipase C results in mobilization of intracellular calcium in HMC-1 cells and eventually promotes synthesis of interleukin-8 (IL-8) (Feoktistov and Biaggioni, 1995; fig. 7a). In contrast to A2B receptors, there is no evidence that A2A receptors can stimulate phospholipase C under physiological conditions, even though cotransfection of human A2A receptors with murine Gα15 and human Gα16, but not with Gαq, Gα11or Gα14, results in A2A-mediated stimulation of phospholipase C in COS-7 cells (Offermanns and Simon, 1995). However, promiscuous coupling of Gα15 and Gα16 has been observed when these G proteins are coexpressed with receptors which are otherwise not normally coupled to phospholipase C (Milligan et al., 1996). Also, expression of Gα15 and Gα16 is limited only to a subset of hematopoietic cells (Amatruda et al., 1991; Wilkie et al., 1991).
Stimulation of A2B receptors also increases intracellular calcium in HEL cells but not through a mechanism involving phospholipase C activation (fig. 7b). In contrast to the cholera toxin- and pertussis toxin-insensitive mobilization of intracellular calcium observed in HMC-1 mast cells, A2B receptors facilitate calcium influx through a cholera toxin-sensitive mechanism in HEL cells. This effect was observed only when intracellular calcium levels were elevated, either by receptor-dependent (e.g., by thrombin) or -independent (e.g., thapsigargin) mechanisms. Even though this process is coupled to Gs-proteins, it is cAMP-independent. It has been suggested that αGs, coupled to A2B receptors, can directly stimulate a putative calcium channel (Feoktistov et al., 1994), as proposed for other Gs-coupled receptors (Imoto et al., 1988; Scamps et al., 1992).
Of interest, a similar mechanism has been suggested for A2A receptors in fetal chicken ventricular myocardium cells. These cells coexpress A2A and A2B receptors, and both are positively coupled to stimulation of adenyl cyclase and myocyte contractility (Liang and Haltiwanger, 1995). Selective activation of A2Areceptors with CGS 21680 results in cAMP-independent calcium entry in pertussis toxin-treated cells. This effect does not involve simulation of phospholipase C and was blocked by the selective A2A antagonist 8-(3-chlorostyryl)caffeine (Liang and Morley, 1996). This study did not explore the possibility that A2B receptors share a common mechanism of Gs-mediated stimulation of calcium entry with A2A receptors. This could be tested by using the nonspecific A2 agonist NECA in the presence of a selective A2A antagonist such as SCH 58261.
In another example of positive modulation of intracellular calcium, it has been reported that activation of A2Breceptors results in significant potentiation of P-type, but not N-type, calcium currents in pyramidal neurons from the CA3 region of guinea pig hippocampus. This mechanism was thought to be mediated by adenyl cyclase, because this potentiation could be inhibited by blocking the cAMP-dependent protein kinase (Mogul et al., 1993; fig.7c).
It has recently been recognized that intracellular signaling of A2B receptors can be modulated by interaction with other receptor systems (Fredholm, 1995; Fredholm et al., 1996a; fig. 8). For example, agents that increase intracellular calcium or activate protein kinase C significantly potentiate A2B-mediated cAMP production in various cells (Hollingsworth et al., 1985; Norstedt and Fredholm, 1987; Fredholm et al., 1987; Norstedt et al., 1989; Kvanta et al., 1989, 1990; Altiok et al., 1992; fig. 8a). On the other hand, bradykinin-stimulated calcium entry caused inhibition of A2B receptor-stimulated adenyl cyclase in astrocytoma D384 cells (fig. 8b), but direct stimulation of protein kinase C enhanced the A2B response (Altiok et al., 1992; Altiok and Fredholm, 1993). The exact mechanism of the interaction between protein kinase C and A2B-mediated pathways is not known, but it cannot be considered a unique feature of A2B receptors. For instance, activation of thrombin-induced phospholipase C pathways potentiate cAMP accumulation stimulated by IP prostanoid receptors in HEL cells (Turner et al., 1992; Feoktistov et al., 1997). It has been suggested that protein kinase C does not exert its effect at the level of the receptor but rather affects the coupling of the stimulated Gs protein with adenyl cyclase (Fredholm, 1995). The synergistic interaction between the A2Breceptors and the calcium/protein kinase C pathway can occur further down in the signaling cascade. Thus, A2Breceptors greatly potentiate the phorbol 12-myristate 13-acetate-induced synthesis of IL-8 in human mast cells (Feoktistov and Biaggioni, 1995). In T-lymphocytes, the T-receptor is known to activate immediate early gene transcription, leading to the activation of the AP-1 transcription factor (Kvanta et al., 1992). A2B receptors significantly potentiate this response, implying that cAMP, and calcium/protein kinase C pathways, may act in concert in the regulation of gene transcription (Kontny et al., 1992; Kvanta and Fredholm, 1994).

Modulation of A2B receptor signaling. (A) In Jurkat cells, stimulation of calcium entry and of protein kinase C (PKC) by T-cell receptor activation increases the magnitude of the cAMP response to stimulation of A2B receptors (Kvanta et al., 1989; Fredholm et al., 1996a). (B) In human astrocytoma cells, D384 activation of protein kinase C is also able to enhance A2B receptor-mediated cAMP accumulation, but the stimulation of calcium entry via the bradykinin B2 receptor leads to inhibition of A2B receptor-mediated cAMP accumulation (Altiok and Fredholm, 1993; Fredholm et al., 1996a).
In summary, A2B receptors are coupled to adenyl cyclase through Gs proteins in every cell studied. Current evidence suggests that the actions of A2B receptors can be mediated not only by cAMP, but also by other intracellular pathways that may vary between cells. A2B receptors can couple to calcium channels through Gs, but additional studies are needed to determine the type of channel involved. Similarly, it remains to be determined which member of the Gq family is responsible for A2B receptor coupling to phospholipase C. It is of interest that, as far as intracellular pathways are concerned, A2B receptors have as much in common with A1 or A3 receptors (activation of phospholipase C), as with A2A receptors (activation of adenyl cyclase). It would be important to determine which domain of the A2B receptor defines the differences in G protein coupling between A2A and A2B receptors.
VII. Physiological Functions of A2B Receptors
A. Control of Vascular Tone
Adenosine-induced vasodilation has been traditionally attributed to activation of A2A receptors. However, the recent finding of the presence of A2B receptors in some vascular beds raised the possibility that they participate in the regulation of vascular tone. Indeed, there are vascular beds in which the nonselective agonist NECA produces profound vasodilation, but the selective A2A agonist CGS 21680 has little effect, suggesting that adenosine-induced vasodilation is mediated via A2B receptors (for review, see Webb et al., 1992). This phenomenon is observed in guinea pig aorta and dog saphenous vein (Hargreaves et al., 1991), and in dog coronary arteries (Balwierczak et al., 1991). This effect is not due to species differences, because both A2A and A2B receptors may mediate vasodilation in the same species. In guinea pig, for instance, A2Areceptors mediate relaxation of coronary vessels, whereas A2B receptors produce vasodilation of the aorta (Martin, 1992; Martin et al., 1993). Likewise, the A2A agonist CGS 21680 lowers blood pressure in the intact dog (Levens et al., 1991), presumably by inducing vasodilation, despite its lack of efficacy in the coronary arteries of this species.
The vasodilatory effects of adenosine can be accounted for by a direct relaxing action on vascular smooth muscle cells. However, recent studies have suggested that the endothelium contributes to, or is even essential for, the vasodilatory effects of intravascular adenosine. It has been shown that most of the labeled adenosine administered intra-arterially is contained within endothelial cells, and very little escapes this endothelium trap to reach the underlying vascular smooth muscle (Nees et al., 1985). Similarly, intravascular administration of adenosine linked to macromolecules, and therefore less likely to cross the endothelium, is still able to produce vasodilation (Olsson et al., 1977).
In vitro studies, however, have yielded conflicting results as to whether the vasodilatory actions of adenosine are different in vascular preparation with intact or denuded endothelium (Rubanyi and Vanhoutte, 1985; Yen et al., 1988; Falcone et al., 1993; Maekawa et al., 1994). Evaluation of a putative endothelium-dependent vasodilation by adenosine is challenging in ring preparation, because adenosine will produce vasodilation in preparations with or without endothelium. This is particularly true when stable agonists are used, because they are not trapped by the endothelium in the way adenosine is and have more ready access to the underlying vascular smooth muscle. Other endothelium-dependent vasodilators will constrict vascular smooth muscle in the absence of endothelium (Furchgott, 1984), making their distinction easier. Conversely, adenosine-induced vasodilation could conceivably produce flow-related release of nitric oxide (NO), giving the appearance of NO-mediated vasodilation (Olsson, 1996).
Methodological difficulties notwithstanding, more fundamental differences may explain the apparent discrepancies regarding the role of the endothelium on adenosine-induced vasodilation. Given the diversity of endothelial cell types, it is possible that endothelial vasodilatory responses to adenosine vary between species, and within the same species depending on the vascular bed being studied. It is unclear to what degree endothelial A2A or A2B receptors may contribute to these differences. This issue will be resolved only if studies that examine the endothelium-dependency of adenosine-induced vasodilation also define the adenosine receptor subtype involved.
A2B receptors have been shown in endothelial cells. Both A2B and A2Areceptors regulate cAMP production in human aortic (Iwamoto et al., 1994) and human umbilical vein (Feoktistov and Biaggioni, unpublished observations) endothelial cells, and A2B receptor mRNA has been detected in human aortic endothelial cells (Iwamoto et al., 1994). Few studies have directly examined the possible interaction between A2B receptors and endothelium-derived vasodilation, and results vary depending on the vascular bed studied. A2B receptors mediate vasodilation in the rat mesenteric arterial bed (Rubino et al., 1995) and in the isolated blood-perfused rat lung preparation (Haynes et al., 1995). In both cases, A2B-mediated vasodilation seems to be independent of NO generation, because they were not reversed by inhibition of NO synthase by NG-nitro-l-arginine methyl ester (l-NAME) (Haynes et al., 1995; Rubino et al., 1995). On the contrary, isolated rat renal artery rings contain A2B receptors that are located exclusively on the endothelium and cause NO release and vasodilation, because this vasodilation can be blocked with l-NAME and prevented by removal of the endothelium (Martin and Potts, 1994). Similarly, A2B receptors also appear to vasodilate the rabbit corpus cavernosum, and this effect is reduced by removal of the endothelium (Chiang et al., 1994).
In summary, both A2A and A2B receptors mediate vasodilation. The relative contribution of A2B receptors to adenosine-induced vasodilation is not defined. There are also conflicting results as to the importance of NO generation in adenosine-induced and A2B-induced vasodilation, but there are some examples in which the endothelium contributes to A2B-mediated vasodilation. To complicate things further, in some vascular beds, adenosine-induced vasodilation is endothelium-dependent but does not appear to be mediated by NO because it is not blocked by inhibition of NO synthase, raising the possibility that other endothelial factors, such as endothelium-dependent hyperpolarizing factor, may be involved (Headrick and Berne, 1990). The precise nature of the interaction between A2Breceptors and endothelial cells and their role in the regulation of vascular tone are areas where more research is needed.
B. Cardiac Myocyte Contractility
Adenosine has important protective effects against ischemia in the myocardium, but these effects are largely attributed to A1 receptors (for review, see Olsson and Pearson, 1990). It has been reported recently that myocytes isolated from fetal chick ventricles, but not from the atria, possess functional A2B and A2A receptors. Both receptors are capable of augmenting myocardial contractility in this model. These adenosine effects, however, become evident only after inhibitory A1 receptor pathways are inactivated with pertussis toxin (Liang and Haltiwanger, 1995). Presence of A2 receptors, capable of stimulating cAMP accumulation, was demonstrated in cultured adult rodent myocardial cells after A1 receptor blockade (Romano et al., 1989; Stein et al., 1993; Xu et al., 1996). These results could be explained by a possible contamination of myocardial preparations with fibroblasts and endothelial cells expressing A2Breceptors. However, studies performed on single cells argue against this possibility. A positive inotropic response mediated via A2 receptors was demonstrated in cultured rat and guinea pig ventricular myocytes (Stein et al., 1993; Xu et al., 1996;Dobson and Fenton, 1997). The role of myocardial A2 receptors in mediating a positive inotropic effect remains a controversial issue (Olsson, 1996), and their physiological significance is unclear, given that their effects become evident only under blockade of A1 receptors.
C. Modulation of Neurosecretion and Neurotransmission
Adenosine is in general considered to be a depressor of neurons, inhibiting neurotransmitter release and other neuronal functions (Phillis et al., 1993a) and acts as a neuroprotective against ischemia (Dragunow and Faull, 1988). Many of these inhibitory actions are mediated by A1 receptors (Dunwiddie and Fredholm, 1989). A2 receptors, on the other hand, have been shown to mediate excitatory actions on the nervous system (Sebastião and Ribeiro, 1996). Earlier studies did not use specific agonists or antagonists to allow a precise identification of the A2 receptor subtype involved, and relatively little information is available for the A2Breceptor. More recently, several excitatory actions have been linked to the A2A receptor, including enhancement of the release of several neurotransmitters, including acetylcholine, the excitatory amino acids glutamate and aspartate, dopamine, and norepinephrine (for review, see Sebastião and Ribeiro, 1996). However, gene knockout mice lacking A2A receptors exhibit aggressive behavior and lack the stimulant effect of caffeine (Ledent et al., 1997), suggesting that A2Areceptors normally exert a tonic central depressant action. This is in agreement with several observations indicating a depressant effect of A2A agonists on locomotor activity (Sebastião and Ribeiro, 1996). It should be noted, however, that comparisons between molecular mechanisms of excitation and integrated physiological responses need to be done with care. For example, adenosine depresses sympathetic nerve activity and blood pressure when injected into the nucleus tractus solitarii (Tseng et al., 1988) via activation of A2A receptors (Barraco et al., 1991). This apparent depressant action, however, is mediated by local stimulation of the release of the excitatory amino acid glutamate (Mosqueda-Garcia et al., 1989, 1991), mediated by A2A receptors (Castillo-Melendez et al., 1994).
A2B receptors are widespread in the brain, but little is known about their function. There are, however, several examples of neuroexcitatory actions. Adenosine agonists increase the release of the excitatory amino acid aspartate in rat cerebral cortex cup superfusates in vivo while depressing the release of the inhibitory amino acid GABA (Phillis et al., 1993b). The agonist profile was suggestive of an A2B receptor, in that it was produced by a high concentration of N6-cyclopentyladenosine (CPA), but not by the A2A agonist CGS 21680. If confirmed, these results would suggest that A2B receptors would lead to greater tissue injury if activated during ischemia, an action that is in sharp contrast to the postulated protective effects of A1 receptors. In this same model, A2B receptors also enhance basal release of acetylcholine (Phillis et al., 1993a).
Acutely isolated pyramidal neurons from the CA3 region of guinea pig hippocampus contain A1 receptors that inhibit N-type Ca2+ currents. After blockade of A1 receptors, adenosine agonists potentiate a P-type Ca2+ current with a pharmacological profile consistent with A2B receptors, inasmuch as the A2A agonist CGS 21680 had no effect (Mogul et al., 1993). Likewise, after A1 receptor blockade, A2B receptors induce long-term potentiation in the CA1 region of rat hippocampus (Kessey et al., 1997). Although these studies were not designed to explore the site of action of A2B receptors, the results were consistent with a postsynaptic site of action. It should be noted that the selective A2A agonist CGS 21680 was also found to facilitate long-term potentiation in the hippocampal CA1 area (de Mendonca and Ribeiro, 1994), whereas A1receptors inhibit long-term potentiation in this area of the brain (Arai and Lynch, 1992). The relative importance of these contrasting pathways coupled to adenosine receptor subtypes remains to be defined under physiological and pathological conditions. Similarly, A1 receptors inhibit dopamine release in rat striatum, but selective blockade of A1 receptors reveals a stimulatory effect of A2B receptors on dopamine release (Okada et al., 1996).
Adenosine also inhibits norepinephrine release from peripheral noradrenergic nerve terminals (Wakade and Wakade, 1978). This effect is thought to be mediated by putative presynaptic A1receptors (Paton, 1981), but the inhibition of norepinephrine release in isolated canine pulmonary arteries was better explained by A2B receptors, based on rank order of potencies of agonists (Tamaoki et al., 1997). Similarly, the pharmacological profile for adenosine-induced inhibition of neurotransmission in rabbit corpus cavernosum is consistent with that of an A2B receptor (Chiang et al., 1994). On the other hand, the nonselective A2 agonist NECA potentiated acetylcholine release evoked by electrical stimulation of the rat bronchial smooth muscle and was 100-fold more potent than the A2A agonist CGS 21680 (Walday and Aas, 1991). Furthermore, this process was blocked by 10 μmenprofylline. The effect of exogenous acetylcholine was not affected by NECA. Taken together, these results provide evidence for an A2B presynaptic receptor that enhances neurally mediated release of acetylcholine and thereby induces contraction of bronchial smooth muscle (Walday and Aas, 1991).
Adenosine also modulates release of catecholamines from chromaffin cells. These adrenal medullary cells are under neural control through cholinergic nicotinic receptors. No specific binding was found using selective ligands for the A1 or A2A receptors in chromaffin cells from bovine adrenal medulla, but specific binding was obtained using [3H]NECA. These results were interpreted as evidence that only A2B receptors are expressed in these cells (Casado et al., 1992). At high (20 μM) concentrations, the nonselective agonist NECA inhibits catecholamine release induced by nicotinic stimulation, presumably by activation of A2B receptors (Mateo et al., 1995). This effect has an unusually slow time course and is seen only after 20 to 30 minutes of preincubation with NECA, and its physiological relevance is not clear.
D. Cell Growth and Gene Expression
Whereas most studies of the cardiovascular effects of adenosine have focused on its acute actions on vascular tone and adrenergic neurotransmission, recent evidence suggests that adenosine may also play a long-term modulatory role in smooth muscle growth. Exogenous adenosine was shown to inhibit rat aortic smooth muscle cell growth induced by fetal calf serum, as assessed by a decrease in thymidine incorporation and in cell number (Dubey et al., 1996b). These inhibitory effects were reversed by the A2receptor antagonist KF 17837, but not by the A1antagonist DPCPX, and were not mimicked by the A2A agonist CGS 21680, suggesting that this effect is mediated by A2B receptors (Dubey et al., 1996b). Activation of adenyl cyclase is postulated as the signaling pathway involved, because this effect is mimicked by 8-bromo-cAMP. These investigators later showed that stimulation of these vascular smooth muscle cells by fetal calf serum can trigger release of endogenous adenosine, which then acts in an autocrine fashion to inhibit growth. The importance of endogenous adenosine is less certain, because inhibition of vascular smooth muscle growth by endogenous adenosine is evidenced only if adenosine deaminase is inhibited (Dubey et al., 1996a). It is postulated that this reflects a limitation of the experimental model, due to the presence of adenosine deaminase in the fetal calf serum used to stimulate vascular smooth muscle growth. If a role for endogenous adenosine is confirmed, this finding would establish a novel cardioprotective effect of adenosine, with relevance to vascular remodeling process observed in hypertension and atherosclerosis.
A2B receptors can also modulate gene expression, in some cases leading to inhibition of protein synthesis. For example, stimulation of A2B receptors decreases collagenase gene expression in interleukin-1–stimulated cultured fibroblast-like synoviocytes, an effect apparently mediated by cAMP elevation (Boyle et al., 1996). In contrast, A2Breceptors promoted the synthesis of IL-8 in HMC-1 mast cells by a cAMP-independent mechanism (Feoktistov and Biaggioni, 1995). It has been recently demonstrated that A2B receptors can also induce an increase in interleukin-6 mRNA levels and protein synthesis in the human astrocytoma cell line U373 MG (Fiebich et al., 1996a). The synergistic relationship between A2Breceptors and T-receptors, and generally between cAMP and protein kinase C pathways in gene expression, has been discussed in detail elsewhere (Fredholm, 1995; Fredholm et al., 1996a).
E. Regulation of Intestinal Tone and Secretion
The high levels of A2B receptor expression found in different parts of the intestinal tract motivated great interest in defining their function. In some studies, A2B receptor mRNA expression is greatest in intestinal tissue among all organs (Stehle et al., 1992). It appears that A2B receptors may be involved in modulation of intestinal tone as well as intestinal secretion. Adenosine elicits relaxation of dispersed guinea pig longitudinal muscle cells from small intestine via A2B receptors coupled to adenyl cyclase but produced contraction through A1receptors by increasing intracellular calcium (Murthy et al., 1995). The A2B-mediated relaxation was evident only after A1 receptor blockade, raising doubts as to their importance. However, blockade of A2receptors potentiated A1-mediated contraction, indicating that A2B receptors do provide a restraining function against intestinal contraction. In rat duodenum, A2B receptors cause relaxation of longitudinal muscle but contraction of muscularis mucosae (Nicholls et al., 1996). This is an unexpected result and the first example of an excitatory response by A2B receptors in a smooth muscle preparation. This tissue is also unusual in that A1 receptors were found to produce relaxation of duodenal longitudinal muscle (Nicholls et al., 1996). A2B receptors have also been shown to relax guinea pig taenia ceci, based on the pharmacological profile of agonists (Prentice and Hourani, 1997). The functional relevance of the intestinal relaxant actions of A2B receptors has not been defined. Of interest, adenosine A2Breceptors also mediate relaxation of other visceral smooth muscle such as rat urinary bladder (Nicholls et al., 1996) and rat vas deferens (Hourani et al., 1993).
The effect of A2B receptors on epithelial secretion has received particular attention because of its potential relevance to diarrheal processes. As part of the pathophysiology of these disorders, neutrophils are recruited into intestinal crypts, where they release a soluble “neutrophil-derived secretagogue” that then activates intestinal epithelium to stimulate chloride secretion. This chloride secretion has the net effect of producing isotonic fluid secretion, an important component of diarrheal diseases. This neutrophil-derived secretagogue has recently been identified as AMP (Madara et al., 1993), which is then converted to adenosine at the epithelial cell surface by ecto-5′-nucleotidase. It is adenosine that then acts as a paracrine mediator of chloride secretion (Madara et al., 1993). It was later demonstrated that neutrophil-derived adenosine elicits chloride secretion in the intestinal epithelial cell line T84 via activation of A2B receptors (Strohmeier et al., 1995), implying the possible involvement of this receptor subtype in the pathophysiology of diarrheal diseases. In contrast to these findings, A2B receptors reportedly inhibit intestinal fluid secretion induced by vasoactive intestinal peptide in rat jejunum (Hancock and Coupar, 1995). These results should be interpreted with caution, because receptor characterization was done by relative potency of agonists and antagonists injected intravenously in anesthetized rats. Definite receptor characterization cannot be completed until the tissue localization of these putative A2B receptors is determined and in vitro studies are performed (Hancock and Coupar, 1995). The presence of A2B receptors in epithelial cells of human intestine has been demonstrated by immunohistochemistry with an anti-A2B receptor antibody (Puffinbarger et al., 1995), but more studies are needed to define their role in intestinal secretion and diarrheal processes.
F. Adenosine and Asthma
Adenosine has been implicated in the pathophysiology of asthma (for review, see Church and Holgate, 1986; Feoktistov and Biaggioni, 1996), and several lines of evidence support this hypothesis. Inhaled adenosine, or its precursor AMP, provokes bronchoconstriction in asthmatic patients (Cushley et al., 1984). This effect is fairly specific for patients with asthma, and even high concentrations of inhaled adenosine fail to produce bronchoconstriction in the majority of normal subjects. Atopic subjects appear to be more responsive to inhaled AMP than they are to methacholine (Phillips et al., 1990), suggesting that adenosine may be a better discriminator of the disease. This preferential bronchoconstrictor effect in asthmatics is also observed with intravenous administration of adenosine (Drake et al., 1994) and in isolated human bronchi (Björck et al., 1992). Dipyridamole, a drug that blocks adenosine uptake and increases its extracellular concentrations, can also produce severe bronchospasm in asthmatic patients (Eagle and Boucher, 1989). Moreover, theophylline provides a better protection against adenosine-induced bronchoconstriction than against histamine-induced bronchoconstriction (Mann and Holgate, 1985).
The mechanism by which adenosine produces bronchoconstriction has been the focus of recent interest. In particular, it would be important to define the receptor type involved. Adenosine produces a direct constrictor action on isolated guinea pig trachea with an agonist profile consistent with A1 receptors (Ghai et al., 1987), and this process is not blocked by enprofylline (Farmer et al., 1988). In this same preparation, however, A2receptors were found to produce relaxation, but the receptor subtype was not identified. Based on rank order of potencies for agonists, it was found that A1 receptors also mediate bronchoconstriction in an allergic rabbit model in vivo (Ali et al., 1994a,b), and treatment with antisense oligodeoxynucleotide targeting the adenosine A1 receptor desensitized the allergic rabbits to subsequent challenge with either adenosine or allergen (Nyce and Metzger, 1997). Adenosine also constricts human bronchi isolated from asthmatics in vitro but not bronchi isolated from nonasthmatics (Björck et al., 1992). The contractile effect of adenosine was inhibited with 2-thio-[(1,3-dipropyl)-8-cyclopentyl]-xanthine, and this was taken as evidence of an A1-mediated effect.
The bronchoconstriction produced by inhaled adenosine in humans appears to be mediated through mast cell activation, because it can be blocked by specific antihistamines (Phillips et al., 1987; Rafferty et al., 1987) and prevented by cromoglycate and nedocromil sodium, drugs that inhibit mast cell degranulation (Phillips et al., 1989). Furthermore, a significant rise in plasma levels of histamine is detected after AMP challenge (Phillips et al., 1990). More recently, inhaled adenosine has been shown to increase levels of histamine, PGD2, and tryptase in bronchoalveolar lavage fluid from asthmatics but not from nonasthmatics (Polosa et al., 1995). Tryptase is a highly specific marker for mast cells (Schwartz, 1990) and provides strong evidence that these cells are activated by adenosine in vivo.
In summary, exogenous adenosine provokes asthma, potentiation of endogenous adenosine with dipyridamole also produces bronchoconstriction, and blockade of endogenous adenosine with theophylline is helpful in preventing asthma. The bronchoconstriction induced by inhaled adenosine is unique to asthmatics and not observed in nonasthmatics. Current evidence suggests that this phenomenon involves mast cell activation. It is important, therefore, to elucidate the adenosine receptor subtype that mediates this phenomenon.
G. Adenosine Receptors and Mast Cells
Marquardt et al. (1978) were the first to report that adenosine, although ineffective alone, potentiated histamine release induced by anti-immunoglobulin E (IgE), concanavalin A, compound 48/80, or the calcium ionophore A23187 in isolated rat mast cells. The mechanisms that mediate potentiation of these cells remain unclear. Stimulation of adenyl cyclase by adenosine was blocked by methylxanthines, but potentiation of histamine release was not, suggesting that these effects were mediated by different adenosine receptors (Church et al., 1986).
Because potentiation of rat peritoneal mast cells is insensitive to methylxanthines, the possibility was raised that this effect is mediated by A3 receptors, because the rat A3 receptor has remarkably low affinity for methylxanthines (Zhou et al., 1992). This possibility was examined in the rat basophilic leukemia cell line RBL 2H3, which has been used as a model for rat mast cells. Adenosine analogs stimulated phospholipase C, increased cytoplasmic calcium, and potentiated mediator release in these cells with a pharmacological profile consistent with A3 receptors (Ramkumar et al., 1993). Expression of A3 receptors in RBL 2H3 cells was confirmed by radioligand binding and detection of mRNA (Ramkumar et al., 1993). The effects mediated by A3 receptors in RBL 2H3 were blocked by pertussis toxin, suggesting a role of Gi-derived βγ subunits in the activation of β-phospholipase C. Coupling of A3 receptors to Gi2 and Gi3 proteins was recently reported (Palmer and Stiles, 1995). Of interest, A2A and A2B, but not A1 receptors, have also been found in RBL 2H3 cells (Marquardt et al., 1994; Ramkumar et al., 1995); however, their function has not been elucidated. It should also be noted that A1 receptors, to our knowledge, have not been found in other mast cell types (Marquardt et al., 1994).
It is important to consider that mast cells from different species, and even from different anatomical sites within the same species, can vary substantially in their morphological and biochemical characteristics and their response to pharmacological agents. There is increasing evidence that A2B receptors modulate mast cell function. Adenosine activates adenyl cyclase and protein kinase C and potentiates mediator release in mouse bone marrow-derived mast cells (Marquardt and Walker, 1990). It appears that the ability of adenosine to activate protein kinase C and thereby to augment mast cell degranulation are independent of changes in cAMP (Marquardt and Walker, 1994). Both A2A and A2Btranscripts were detected in mouse bone marrow-derived mast cells (Marquardt et al., 1994). The failure of the A2A-specific agonist CGS 21680 to enhance mediator release suggests that A2B receptors modulate degranulation of these mast cells (Marquardt et al., 1994).
A2B receptors have been shown to activate the human mast cell line HMC-1 (Feoktistov and Biaggioni, 1995). HMC-1 cells were derived from a patient with mast cell leukemia and their neutral proteases content is similar to that of human lung mast cells. These cells coexpress A2A and A2B receptors, and both are coupled to adenyl cyclase through Gs proteins. However, only A2B receptors activate HMC-1 cells, as indicated by stimulation of IL-8 secretion. Furthermore, this effect was not mediated by cAMP, but by coupling to phospholipase C through a cholera toxin- and pertussis toxin-insensitive G protein, presumably of the Gq family (Feoktistov and Biaggioni, 1995). A2B receptors not only produced direct stimulation of HMC-1 cells, but also potentiated phorbol 12-myristate 13-acetate-stimulated secretion of IL-8 (Feoktistov and Biaggioni, 1995). The expression of A2B receptors in HMC-1 cells was recently confirmed by immunoblotting and fluorescent immunostaining with a specific anti-A2B antibody (Feoktistov et al., 1996). Virtually identical findings have been reported in a canine BR mastocytoma cell line. Stimulation of A2B, but not A3 receptors, directly increased β-hexosaminidase release and also potentiated A23187-induced degranulation of mast cells. Also, these effects were not blocked by pertussis toxin (Auchampach et al., 1996).
In parenchymal human lung mast cells, obtained from normal sections of surgical specimens, adenosine does not directly evoke release of histamine and LTC4, but in micromolar concentrations it potentiates mediator release from immunologically activated cells (Peachell et al., 1991). The order of potency of adenosine analogs and the low affinity of this process suggests that the response of human lung mast cells to adenosine is mediated by A2B receptors. In support for this notion, the presence of A2B has been recently demonstrated in bronchoalveolar lavage mast cells by double immunostaining with specific anti-A2B and antitryptase antibodies (Feoktistov et al., 1996).
Given that inhaled adenosine affects only asthmatics but has no effect in nonasthmatics, there appears to be an intrinsic difference in the way adenosine interacts with mast cells from asthmatics. The in vitro response produced by A2B receptors in HMC-1 cells and in canine BR mastocytoma cells appears to mimic the in vivo responses to inhaled adenosine in asthmatics, inasmuch as adenosine provokes mast cells activation in these cell lines as it does in asthmatics. On the other hand, the in vitro response of mast cells from normal human lung to adenosine resembles the effect of A2B receptors in mouse bone marrow-derived mast cells, because in both cases adenosine potentiates mast cells activation but does not evoke direct activation. The molecular mechanisms behind these differential A2B-mediated responses in asthmatic versus normal mast cells, and in HMC-1 cells versus mouse bone marrow-derived mast cells, remain unknown.
Despite the direct bronchoconstricting action of A1 receptors observed in allergic rabbits (Nyce and Metzger, 1997), the bronchoconstriction induced by inhaled adenosine in humans is better explained by an A2Breceptor. Adenosine-induced bronchoconstriction appears to be mediated by mast cell activation, and A1 receptors have not been described in mast cells, whereas A2Breceptors are expressed in human mast cells. Moreover, enprofylline is an effective antiasthmatic and is a selective A2Bblocker at concentrations achieved clinically. Finally, A2B receptors have been shown to potentiate neurally mediated cholinergic bronchoconstriction through an enprofylline-sensitive process (Walday and Aas, 1991). The evidence presented so far does not preclude the contribution of more than one adenosine receptor in asthma, or the possibility that nonadenosinergic mechanisms contribute to the antiasthmatic effects of enprofylline and other methylxanthines.
VIII. A2B Receptors as Therapeutic Targets
The ability of adenosine to delay atrioventricular node conduction was the basis for its development as a therapeutic agent in the treatment of supraventricular arrhythmias (Belardinelli et al., 1989) and has become the drug of choice for the termination of that arrhythmia. Taking advantage of its profound vasodilatory effects, intravenous adenosine is used as a stress test in the diagnosis of myocardial ischemia (Verani et al., 1990). Adenosine was investigated as a hypotensive agent during anesthesia (Sollevi et al., 1984), where the reflex sympathetic activation induced by this agent (Biaggioni, 1992) is not observed. However, adenosine has been implicated in many other physiological and pathological processes. The biggest problem in translating this knowledge into therapeutic tools is perhaps the ubiquity of adenosine receptors, which often mediate contrasting effects. The challenge is how to develop drugs that will selectively target a receptor mediating a specific action. The ongoing development of selective agonists or antagonist represents a substantial advancement toward this goal. Nonetheless, even if specific agents can be developed for a given receptor subtype, the problem remains of selectively targeting the site of action. For example, A1-selective agonists could be developed for their antilipolytic potential. If given systemically, however, it is possible that atrioventricular conduction delay or bradycardia may be an undesirable, and perhaps limiting effect, given that these actions are also mediated by A1 receptors. In the development of useful therapeutic agents, therefore, care should be taken not only in the targeting of the receptor subtype, but also the site of action. This problem, of course, is not limited to adenosinergic systems and is common to others characterized by the widespread nature of their receptors.
Given that the functional role of A2B receptors is only now being addressed, a discussion of potential therapeutic opportunities arising from modulation of such receptors is necessarily speculative. There are, however, some promising areas that deserve further attention. The potential role of A2Breceptors in asthma can be used as an example. If confirmed, this mechanism would provide a novel approach for the treatment of this condition. Asthma continues to be a substantial medical problem that affects approximately 5 to 7% of the population. Despite advances in its treatment, the prevalence of asthma, emergency department visits, hospitalizations, and mortality related to the disease all appear to be on the rise (Gergen et al., 1988; Vollmer et al., 1992; Weiss et al., 1993). Theophylline continues to be an effective treatment in the prevention of asthma attacks, more than as an acute bronchodilator (Weinberger and Hendeles, 1996), but considerable plasma levels, in the range of 20–80 μm, are needed for it to be effective. Moreover, it has many side effects, which can be attributed to its nonspecificity. Its central actions contribute to theophylline’s side-effect profile and are of doubtful benefit for the treatment of asthma.
If indeed blockade of A2B receptors contributes to the antiasthmatic effects of theophylline, it would be possible to develop selective antagonists for this receptor subtype. Lipophobic compounds would have the advantage of not crossing the blood-brain barrier. Specific targeting to the site of action can also be accomplished if compounds that can be administered by inhalation are developed. This proposition is not unrealistic. For example, the xanthine antagonist DPSPX is approximately 100-fold more potent than theophylline as an A2B receptor antagonist. Because of a charge moiety in its molecule, this water-soluble xanthine does not penetrate cell membranes or cross the blood-brain barrier (Tofovic et al., 1991). It appears that the ionic p-sulfophenyl substituent in DPSPX may confer high A2B potency. The lack of l-alkyl substituents in the enprofylline molecule renders it an ineffective antagonist of other adenosine receptor subtypes. The systematic study of the structure-activity relationship for blockade of A2B receptors, considering the above-mentioned properties, could result in more potent and specific agents. Similarly, A2B receptor antagonists can be developed for the treatment of diarrheal processes, if adenosine is confirmed to play a role in this process. Targeting to the site of action could be achieved with compounds that are poorly absorbed, as long as they are able to reach the intestinal crypts involved in intestinal inflammatory processes.
Development of agonists to target A2B receptors, for example, to inhibit vascular smooth muscle growth, would be a greater challenge. Substantial progress would need to be made to develop an agonist potent enough to selectively activate the low-affinity A2B receptor while having negligible actions at other receptors. The actions of endogenous adenosine could be enhanced with uptake inhibitors, or adenosine-regulating agents (Mullane, 1993), but this approach would activate all adenosine receptors, perhaps others even more than A2Breceptors. Targeting the site of action is an additional problem that would have to be resolved for any drug that had to be administered systemically. The observation that A2B receptors mediate vasodilation of the rabbit corpus cavernosum (Chiang et al., 1994) raises the possibility that an agonist to this receptor type can be useful in impotence. Direct injections of adenosine into the corpus cavernosum of impotence patients produce a brief erection (Kilic et al., 1994), particularly if combined with prostaglandin E1 (Chiang et al., 1994). The short duration of effect is clearly related to the short half-life of adenosine in humans (Moser et al., 1989). If this effect is also mediated by A2B receptors in humans, it will be possible to develop stable and selective agonists that can be given locally.
IX. Concluding Remarks
Until recently, relatively little attention has been paid to A2B receptors. It is instructive that contemporary reviews about adenosine only briefly mention their existence. Conclusions about the selective nature of agonists or antagonists at specific adenosine receptors have often been made without testing these compounds on A2B receptors. The A2B receptor was often assumed to be simply a low-affinity variant of the A2A receptor. A2A and A2B receptors are frequently found in the same tissue, and, because both were thought to act by adenyl cyclase activation, it is easy to assume that A2B receptors will be of lesser physiological significance. The lack of selective pharmacological probes with which to study this receptor subtype has been the main obstacle in defining the role of A2B receptors.
Several factors can explain the increasing interest in A2B receptors. The cloning of adenosine receptors validated the belief that A2B receptors were distinct from A2A receptors. It also revealed its widespread and distinct distribution in tissues. The development of selective agonists and antagonists for other adenosine receptor types has indirectly improved our knowledge of A2Breceptors, but their pharmacological characterization is still mostly done by a method of exclusion, i.e., by the lack of efficacy of agonists and antagonists that are selective at other receptors. Also, other tools now are available for the study of this receptor. The amino acid sequence of the receptor, the nucleotide sequence encoding the receptor, and genomic information are now available. This opens the door for mutational or chimeric analysis of A2Breceptors to understand the molecular determinants for its unique coupling to G proteins. It is also possible to determine with precision the cellular localization of A2B receptors, particularly now that antibodies against this receptor have been generated.
Significant progress has been made using currently available tools. It is now clear that, in addition to adenyl cyclase, A2B receptors can also couple to other intracellular pathways, including calcium channels and phospholipase C. In that sense, A2B receptors have as much in common with A1 and A3receptors, as with A2A receptors. The functional relevance of A2B receptors is being defined, but much work is needed in this area. Given the ubiquitous nature of adenosine receptors, it is not surprising that more than one adenosine subtype is found in the same tissue, but it is now evident that they can be coexpressed in the same cell. In these situations, it would seem that the functional role of higher-affinity receptors would predominate over A2B receptors. Nonetheless, A2B receptors can play a role under these conditions by modulating events triggered by other receptor systems. Examples have been presented in this review of A2B receptors restraining the actions of A1 receptors, or potentiating the effects of thrombin and T-cell receptors. The relative importance of A2B receptors may be greater in situations characterized by substantial increases in interstitial levels of adenosine, as occurs during ischemia in metabolically active tissues. On the other hand, there are cellular systems in which the actions of A2B receptors appear to predominate. Such is the case, for example, of human mast cells, epithelial intestinal cells, and the regulation of vascular smooth muscle growth, among others.
Greater advances can now be made by the simple appreciation of the unique features of this receptor type. In the past, investigators often have failed to realize the potential involvement of A2B receptors in their experimental systems. However, the study of A2B receptors will be considerably improved by the introduction of specific pharmacological probes for this receptor type. Even though adenosine analogs have, in general, a poor affinity for A2B receptors, this is not the case for antagonists. Therefore, development of potent and selective A2B antagonists appears particularly promising and likely will further our knowledge of A2B receptors. Specific antagonists will be particularly useful in defining the role of A2Breceptors in physiological and pathological situations. The appreciation of the potential role of A2Breceptors in the pathogenesis of disease processes, including asthma, vascular remodeling, and diarrhea, raises the possibility that A2B receptors may become the target for future drug development.
Acknowledgments
The authors would like to thank Drs. Jack N. Wells and Lee Limbird, Department of Pharmacology, Vanderbilt University, for helpful discussions in the preparation of this manuscript. The authors would also like to thank the reviewers for their constructive suggestions.
Footnotes
-
↵FNa Address for correspondence: Italo Biaggioni, Division of Clinical Pharmacology, Vanderbilt University, Nashville, TN 37232-2195.
-
Dr. Feoktistov is the recipient of an American Lung Association Research Grant Award and an Asthma and Allergy Foundation of America Investigator Grant Award and is supported also by NIH grants RR00095 and R29HL55596.
- Abbreviations:
- alloxazine
- 2,4-dioxobenzo[g]pteridine
- ATP
- adenosine 5c-triphosphate
- cAMP
- adenosine 3c,5c-cyclic monophosphate
- cDNA
- complementary deoxyribonucleic acid
- CGS 21680
- 4-[(N-ethyl-5′-carbamoyladenos-2-yl)-aminoethyl]-phenylpropionic acid
- CHO
- Chinese hamster ovary
- CPA
- N6-cyclopentyladenosine
- DPCPX
- 1,3-dipropyl-8-cyclopentylxanthine
- DPSPX
- 1,3-dipropyl-8-(p-sulfophenyl)xanthine
- EC50
- concentration that produces half-maximal effect
- enprofylline
- 3-n-propylxanthine
- HEL
- human erythroleukemia
- IB-MECA
- N6-(3-iodobenzyl)-N-methyl-5′-carbamoyladenosine
- IgE
- immunoglobulin E
- IL-8
- interleukin-8
- KB
- dissociation constant of antagonist-receptor complex
- L-249313
- 6-carboxymethyl-5,9-dihydro9-methyl-2-phenyl-[1,2,4]-triazolo-{5,1-a}-[2,7]-naphthyridine
- L268605
- 3-(4-methoxyphenyl)-5-amino-7-oxo-thiazolo-[3,2]-pyrimidine
- l-NAME
- NG-nitro-l-arginine methyl ester
- mRNA
- messenger ribonucleic acid
- MRS 1067
- 3,6-dichloro-2′-isopropyloxy-4′-methylflavone
- MRS 1097
- 3,5-diethyl 2-methyl, 6-phenyl-4-[2-[phenyl-(trans)-vinyl]-1,4(±)dihydropyiridine-3,5-dicarboxylate
- MRS 1191
- 3-ethyl 5-benzyl 2-methyl-phenylethynyl-6-phenyl-1,4(±)dihydropyridine-3,5-dicarboxylate
- MRS 1222
- 3,5-diethyl 2-methyl-4-[2-(4-nitrophenyl)-(E)-vinyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate
- NECA
- 5′-N-ethylcarboxamidoadenosine
- NO
- nitric oxide
- R-PIA
- (R)-N6-phenylisopropyladenosine
- S-PIA
- (S)-N6-phenylisopropyladenosine
- SCH 58261
- 5-amino-7-(phenylethyl)-2-(1-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine
- WRC-0571
- C8-(N-methylisopropyl)-amino-N6-(5′-endohydroxy)-endonorbornan-2-yl-9-methyladenin
- XAC
- xanthine amine congener
- ZM 241385
- 4-(2-[7-amino-2-)2-furyl(triazolo {2,3-a}-[1,3,5]triazin-5-ylamino]ethyl)phenol
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
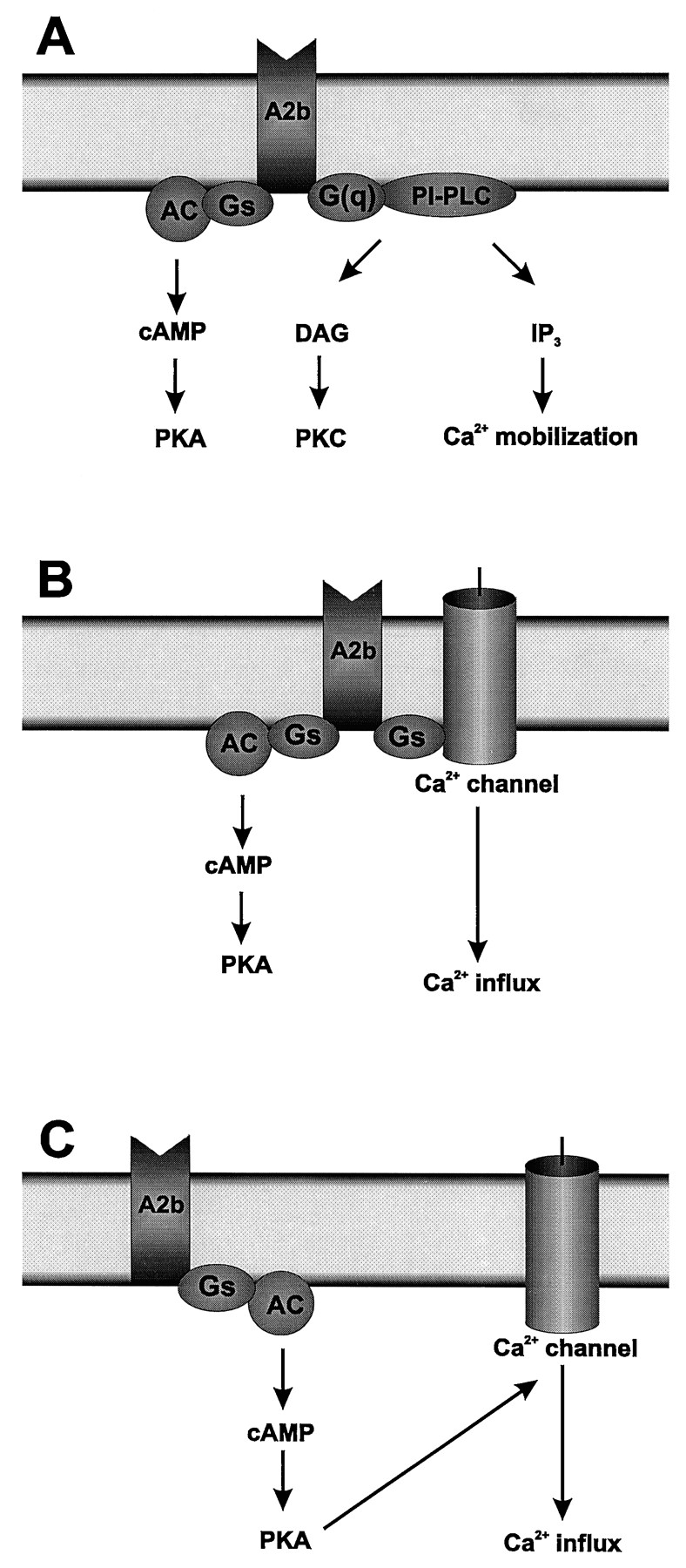{kind=link}
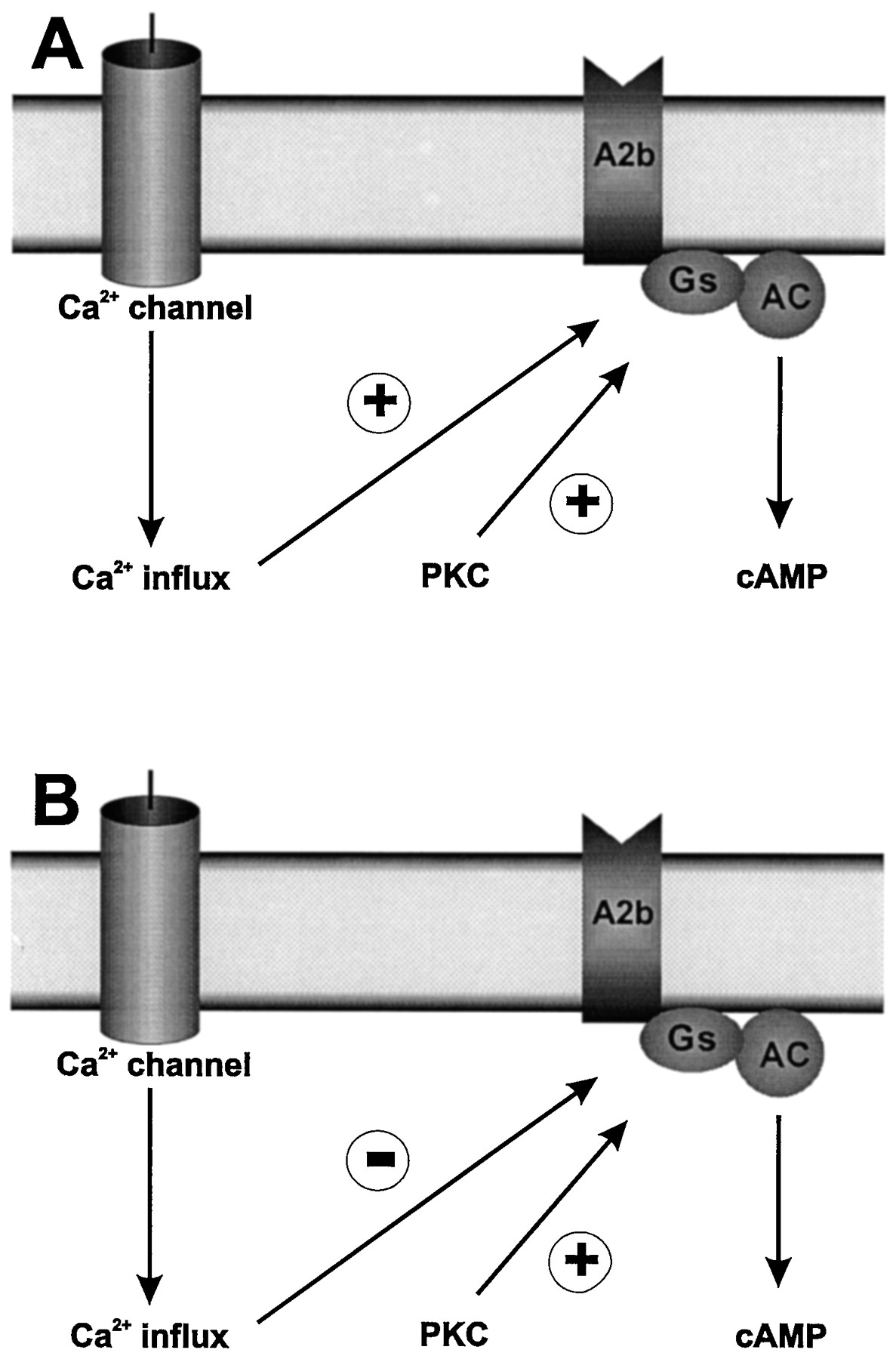{kind=link}
Jump to section
- Article
- I. Introduction
- II. Classification of Adenosine Receptors
- III. Molecular Characterization of A2B Receptors
- IV. Pharmacology of A2B Receptors
- V. Distribution of A2B Receptors
- VI. Intracellular Pathways Regulated by A2B Receptors
- VII. Physiological Functions of A2B Receptors
- VIII. A2B Receptors as Therapeutic Targets
- IX. Concluding Remarks
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters