


Abstract
The dopamine transporter (DAT) terminates dopamine (DA) neurotransmission by reuptake of DA into presynaptic neurons. Regulation of DA uptake by D2 dopamine receptors (D2R) has been reported. The high affinity of DA and other DAT substrates for the D2R, however, has complicated investigation of the intracellular mechanisms mediating this effect. The present studies used the fluorescent DAT substrate, 4-[4-(diethylamino)-styryl]-N-methylpyridinium iodide (ASP+) with live cell imaging techniques to identify the role of two D2R-linked signaling pathways, extracellular signal-regulated kinases 1 and 2 (ERK1/2), and phosphoinositide 3 kinase (PI3K) in mediating D2R regulation of DAT. Addition of the D2/D3 receptor agonist quinpirole (0.1–10 μM) to human embryonic kidney cells coexpressing human DAT and D2 receptor (short splice variant, D2SR) induced a rapid, concentration-dependent and pertussis toxin-sensitive increase in ASP+ accumulation. The D2/D3 agonist (S)-(+)-(4aR, 10bR)-3,4,4a, 10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano-[4,3-b]-1,4-oxazin-9-ol hydrochloride (PD128907) also increased ASP+ accumulation. D2SR activation increased phosphorylation of ERK1/2 and Akt, a major target of PI3K. The mitogen-activated protein kinase kinase inhibitor 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059) prevented the quinpirole-evoked increase in ASP+ accumulation, whereas inhibition of PI3K was without effect. Fluorescence flow cytometry and biotinylation studies revealed a rapid increase in DAT cell-surface expression in response to D2R stimulation. These experiments demonstrate that D2SR stimulation increases DAT cell surface expression and therefore enhances substrate clearance. Furthermore, they show that the increase in DAT function is ERK1/2-dependent but PI3K-independent. Our data also suggest the possibility of a direct physical interaction between DAT and D2R. Together, these results suggest a novel mechanism by which D2SRautoreceptors may regulate DAT in the central nervous system.
Dopamine (DA) is the predominant catecholamine neurotransmitter in the central nervous system. Dysregulation of DA neurons has been implicated in the pathogenesis of Parkinson's disease, schizophrenia, and drug addiction (Sotnikova et al., 2006). Extracellular DA levels are primarily regulated by the DA transporter (DAT), an integral membrane protein that is a member of the Na+/Cl–-dependent cotransporter gene family (Amara and Kuhar, 1993). By removing extracellular DA and recycling it back to the neuron, DAT plays an essential role in terminating DA signaling.
Receptor and second-messenger-linked signal transduction pathways modulate DAT activity. Activation of protein kinase C (PKC) decreases transport capacity and promotes the redistribution of DAT from the plasma membrane to intracellular compartments (Loder and Melikian, 2003). More recently, modulation of DAT function and cell surface expression by extracellular signal-regulated kinases 1 and 2 (ERK1/2) and phosphoinositide 3 kinase (PI3K) has been reported. Activation of ERK1/2 increases transport capacity, and similar effects are observed in response to PI3K activation (Carvelli et al., 2002; Moron et al., 2003).
In addition to their location postsynaptically or as heteroreceptors on the terminals of other neurons, dopamine D2 receptors (D2R) are located on DA nerve terminals in which their activation inhibits DA synthesis and release. They are also located on DA cell bodies in which they function to inhibit cell firing. Studies in which voltammetric methods have been used to monitor the rate of DA clearance in the striatum have shown that D2/D3 receptor agonists increase the Vmax value of DA transport (Meiergerd et al., 1993; Batchelor and Schenk, 1998), presumably through their action at presynaptic D2Rs. Conversely, D2R antagonists decrease DA clearance (Meiergerd et al., 1993). This effect is absent in mice lacking the gene encoding D2R (Dickinson et al., 1999), suggesting a specific role of D2R in regulating DAT function.
In Xenopus laevis oocytes heterologously expressing DAT and the long form of the D2R (D2LR), D2LR-induced up-regulation of DA uptake occurs via a voltage-independent mechanism that is Gi/Go-dependent (Mayfield and Zahniser, 2001). However, D2LRs are primarily postsynaptic (Khan et al., 1998), whereas the short form of D2R(D2SR) is located on DA terminals and serves an autoreceptor function Therefore, fundamental questions remain as to the intracellular mechanisms by which D2Rs regulate DAT. Indeed, increasing evidence suggests that the short and long D2R isoforms may, at least in part, differ in their downstream signaling pathways and may have different functions in vivo (Choi et al., 1999; Usiello et al., 2000).
D2Rs signal via pertussis toxin-sensitive G proteins to a number of downstream effectors, including adenylyl cyclase (Choi et al., 1999; Neve et al., 2004). Studies in striatal primary cultures and heterologous expression systems have shown that their stimulation also induces phosphorylation of ERK1/2 (Brami-Cherrier et al., 2002; Beom et al., 2004). D2SR-mediated phosphorylation of protein kinase B (Akt), a member of the serine/threonine kinase family and a major target of PI3K, has also been reported (Burgering and Coffer, 1995; Brami-Cherrier et al., 2002), although a recent report has also demonstrated a novel arrestin-mediated mechanism for D2R-induced inhibition of Akt (Beaulieu et al., 2005). Recent work has shown that the PI3K pathway (Carvelli et al., 2002) and the mitogen-activated protein kinase pathway (Moron et al., 2003) modulate DAT function by altering cell surface expression, but their potential role in mediating the functional interaction of D2SR with DAT is unknown.
Studies of DAT function typically assess [3H]DA uptake or clearance of exogenous DA. It is noteworthy, however, that DA binds with very high affinity to DA receptors (Sokoloff et al., 1990) Therefore, its use in studies investigating D2R regulation of DAT is not without confound because the concentration range of DA typically used will result in D2R activation. The present studies used the fluorescent DAT substrate, 4-[4-(diethylamino)-styryl]-N-methylpyridinium iodide (ASP+), which we show does not activate D2sR, in combination with live cell imaging techniques (Schwartz et al., 2003, 2005; Mason et al., 2005) to determine whether D2SR activation up-regulates DAT in heterologous expression systems and to identify the role of ERK1/2 and PI3K signaling pathways in mediating this effect. Flow cytometry and biotinylation studies were conducted to determine whether up-regulation is associated with an increase in DAT cell surface expression. BRET and coimmunoprecipitation techniques were used to explore whether D2SR and DAT may be located in close proximity in cells coexpressing these two proteins.
Materials and Methods
Materials. PD98059, LY294002, PD128907, and quinpirole hydrochloride were obtained from Tocris Cookson Inc. (Ellisville, MO). ASP+, S(–)-eticlopride hydrochloride, sulpiride, and pertussis toxin were obtained from Sigma-Aldrich (St. Louis, MO).
Cell Culture. Flp-in T-rex 293 cells (Invitrogen, Carlsbad, CA) stably expressing FLAG-tagged D2sR were used for studies assessing the ability of DAT substrates to activate D2sR. All other experiments were conducted in EM4 cells, human embryonic kidney 293 cells stably expressing a macrophage scavenger receptor to increase their adherence to tissue culture plastic (R.A. Horlick, Pharmacopeia, Cranberry, NJ), or Neuro-2a cells (N2a; American Type Culture Collection, Manassas, VA). EM4 cells were maintained in DMEM/Ham's F-12 medium (50:50; Mediatech, Inc., Herndon, VA) supplemented with 10% FBS. N2a cells were maintained in minimum essential medium (Mediatech) supplemented with 10% FBS. They were grown in a humidified atmosphere at 37°C and 5% CO2. Twenty four hours after plating, cells were transiently transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Experiments were performed 48 h after transfection when cells were 70 to 90% confluent.
Cell-Based ELISA for Phosphorylated ERK1/2 Activation. A phosphospecific cell-based ELISA (modified from Versteeg et al., 2000) for phosphorylated ERK1/2 (pERK) was used to determine the activation of D2SR by the DAT substrates: DA (1 pM to 10 μM), tyramine (1 pM to 100 μM), and ASP+ (1 pM to 100 μM). Cells (50,000 cells/cm2) were seeded in 96-well plates in DMEM media supplemented with 10% FBS and grown at 37°C and 5% CO2 for 24 h. Tetracycline (0.1 μM) was added for an additional 16 to 20 h to induce D2SR expression. Cells were serum-starved in serum-free media for 2 h before substrate addition (100 μl). After 3 min, media were aspirated, and cells were immediately fixed by adding 150 μlof 4% formaldehyde PBS solution for 30 min. Cells were permeabilized by the addition of 0.1% Triton X-100 PBS solution (3 × 10 min). After blocking with 10% BSA in Triton/PBS solution for 1 h, cells were washed with Triton/PBS solution. Primary pERK monoclonal antibody (Cell Signaling Technology, Danvers, MA) was diluted 1:400 in Triton/PBS solution containing 5% BSA, and cells were incubated with primary antibody for 1 h at room temperature. After washing cells three times with PBS/Triton solution and 1 h incubation with horseradish peroxidase-conjugated goat anti-mouse secondary antibody (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) in PBS/Triton solution containing 5% BSA, cells were again washed with Triton/PBS solution. Pierce supersignal ELISA Pico substrate (100 μl) was added to each well for 2 min, and the luminescence signal was measured with a BMG POLARstar reader (BMG Labtech GmbH, Offenburg, Germany). The values from each experiment were normalized to the basal level for that experiment, and the means and S.E.M. are shown for these determinations from three independent experiments, each performed in triplicate. The data were analyzed with GraphPad Prism software using a one-site sigmoidal dose-response curve (GraphPad Software Inc., San Diego, CA).
ASP+Uptake. For ASP+ experiments, cells were seeded on day 1 at 1 × 105 cells/35-mm Delta T Petri dish (Bioptechs, Butler, PA). Between 40 and 100 cells were used for each experiment unless otherwise stated. These cells were pooled from at least three separate transfections, with a minimum of two dishes analyzed per transfection. EM4 or N2a cells were transiently transfected with human D2SR tagged with either an N-terminal FLAG epitope (FLAG-D2SR) or an N-terminal myc epitope (myc-D2SR) and/or yellow fluorescent protein-tagged human DAT (YFP-hDAT). The addition of these tags does not alter transporter-mediated [3H]DA uptake or D2SR function (Guo et al., 2003; Khoshbouei et al., 2003). For the N-terminal truncation studies, FLAG-tagged wild-type human DAT (WT FLAG-hDAT) (Saunders et al., 2000) and a functional N-terminally truncated FLAG-tagged hDAT lacking the first 55 amino acids (ΔN1–55 FLAG-hDAT; N. Sen, J.A. Javitch, unpublished results) were used. A human D2SR with a yellow fluorescent protein at its C terminus (YFP-D2SR) was used in these studies to enable cell visualization.
Immediately before experiments, growth media were removed, and cells were washed two times in Krebs-Ringer-HEPES (KRH; 130 mM NaCl, 1.3 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 10 mM HEPES, and 1.8 g/l glucose, pH 7.4) buffer. After washing, fresh KRH was added to the culture dish, which was then mounted onto an UltraVIEW LCI spinning-disc confocal microscope (PerkinElmer Life and Analytical Sciences, Boston, MA).
Time-resolved quantification of DAT function in single cells was achieved using the fluorescent, high-affinity DAT substrate ASP+. Use of this fluorescent analog in conjunction with a fluorescently tagged transporter allowed monitoring of DAT and norepinephrine transporter function in single cells (Schwartz et al., 2003, 2005; Mason et al., 2005). A within-cell design was used to assess the effects of D2SR stimulation on ASP+ uptake. The microscope was focused on the center of a monolayer of cells, and background autofluorescence was determined by collecting an image immediately before replacement of the KRH buffer with that containing ASP+ (10 μM). The rate (slope of the linear accumulation function) of ASP+ uptake was then measured for 1 min immediately before drug addition. Vehicle or agonist (quinpirole, 0.1–10 μM; PD128907, 10 μM) was added, and the slope of ASP+ accumulation was again determined over a 1-min period. Control studies showed that ASP+ uptake by DAT was linear over the first 10 min after ASP+ addition, was temperature-dependent, and inhibited by the DAT substrates amphetamine and cocaine (Kivell et al., 2004; A. Zapata, B. Kivell, Y. Han, J. Javitch, E. Bolan, D. Kuraguntla, V. Jaligam, L. Jayanthi, D. Samuvel, S. Ramamoorthy, et al., submitted). Images were collected every 20 s for 10 min to enable capture of ASP+ fluorescence (excitation, 488 nm; emission, 607–652 nm). The influence of the D2/D3 receptor antagonist eticlopride (0.1 μM), the MEK inhibitor PD98059 (10 μM; Alessi et al., 1995), and the PI3K inhibitor LY294002 (10 μM; Vlahos et al., 1994) on quinpirole (10 μM)-evoked ASP+ uptake was assessed by preincubating cells with vehicle or inhibitor for 15 min at 37°C. ASP+ was then added, and intracellular accumulation was measured before and after the addition of vehicle or agonist as described above. The concentrations of inhibitors used were those shown previously to be selective for the specific kinase examined (Vlahos et al., 1994; Alessi et al., 1995).
Image Analysis. ASP+ fluorescence accumulation was determined from the average pixel intensity at each time point accumulated within the cell using the UltraView software package (PerkinElmer Life and Analytical Sciences), the boundaries of which were determined from a reference picture at the initial time point of YFP fluorescence (indicating the presence of YFP-hDAT or YFP-D2SR at the plasma membrane). Values are expressed as the percentage of change in ASP+ accumulation rate after the addition of drug. The resultant data were analyzed using a one-way ANOVA followed by the Student Newman-Keuls test (SNK) or using Student's t test. Tests were performed using the Prism version 4.0 statistical software package (Graph Pad Software). A value of p ≤ 0.05 was considered statistically significant.
Immunoblotting. EM4 cells were transfected as described above with FLAG-D2SR and YFP-hDAT and serum-starved in serum-free media for 16 to 18 h before the assay. On the day of the assay, cells were treated for 15 min at 37°C with the kinase inhibitor or vehicle before the addition of quinpirole (10 μM) for 1 min at 37°C. A 1-min time point was selected based on the results of ASP+ experiments. After incubation, media were aspirated, and boiling Laemmli buffer (62.5 mM Tris, pH 6.8, 20% glycerol, 2% SDS, 5% β-mercaptoethanol, and 0.01% bromphenol blue) was added directly to the wells. Lysates were then collected and boiled for 10 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis (4–20% Duramide gradient gel; Cambrex Bio Science Walkersville, Walkersville, MD) and blotted onto polyvinylidene difluoride membranes. The membranes were then blocked for 1 h at room temperature in Tris-buffered saline/Tween 20 containing 5% nonfat milk. pERK was detected using a rabbit polyclonal antibody specific for ERK1/2 phosphorylated at residues threonine 202 and tyrosine 204. Phosphorylated Akt was detected using a rabbit polyclonal antibody, which detects levels of Akt only when phosphorylated at serine 473 (p-Akt). To control for differences in protein loading, blots were stripped with 2% SDS, 100 mM β-mercaptoethanol in 62.5 mM Tris, pH 6.8, for 1 h at 50°C, and reprobed with a polyclonal antibody that recognizes total ERK1/2 or total Akt. All kinase antibodies were purchased from Cell Signaling Technology. Blots were visualized using a horseradish peroxidase-conjugated secondary antibody (Upstate Biotechnology, Lake Placid, NY) with enhanced chemiluminescence reagents.
For quantification, films were scanned at high resolution using an HP Scanjet 7400c (Hewlett Packard, Palo Alto, CA), and band densities were quantified using MetaMorph software (Molecular Devices, Sunnyvale, CA). The relative amounts of phosphorylated kinases were normalized against those of the corresponding total kinase. Data were analyzed by one-way ANOVA followed by the Student Newman-Keuls test or using Student's t test with GraphPad Prism version 4.0. The significance level for all analyses was p ≤ 0.05.
[3H]Tyramine Uptake. Uptake studies with [3H]tyramine were performed as described previously (Hastrup et al., 2001). Tyramine was used for these studies because it exhibits much lower potency at D2SR than does DA (Fig. 1). Hemagglutinin (HA) epitope-tagged hDAT (HA 2EL hDAT) was constructed from a synthetic gene encoding the human dopamine transporter mutated to include an HA epitope tag in the second extracellular loop (Sorkina et al., 2006). This construct allowed FACS analysis to measure surface DAT (see below). EM4 cells were transiently transfected with FLAG-D2SR and HA 2EL hDAT. One day after transfection, the cells were split and plated in poly(d-lysine)-coated 96-well plates. Forty-eight hours after transfection, when the cells were confluent, they were washed twice with 200 μl of buffer (130 mM NaCl, 1.3 mM KCl, 10 mM HEPES, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.2 mM CaCl2, and 10 mM glucose, pH 7.4). Cells were incubated with quinpirole (10 μM) or sulpiride (10 μM) + quinpirole (10 μM) for 1 min before the addition of 66 nM [3H]tyramine. Nonspecific uptake was determined in the presence of 2 mM tyramine and was similar to that detected in the presence of a DAT blocker. After 1-min incubation at room temperature, uptake was terminated by aspiration and washing twice with ice-cold buffer. Cells were lysed with 50 μl of 1% Triton X-100 for 15 min. Two hundred microliters of Optiphase supermix scintillation fluid was added, and radioactivity was determined in a Wallac 1450 microbeta counter (PerkinElmer Life and Analytical Sciences).

Activation of D2SR by DAT substrates. HEK 293 cells stably expressing FLAG-D2SR were incubated with the indicated concentrations of the DAT substrates, DA, tyramine, or ASP+ for 3 min at 37°C. Phosphorylation of ERK1/2 (pERK1/2) as determined in an intact cell ELISA was used to quantify D2SR activation as described under Materials and Methods. Data are expressed as the mean fold increase ± S.E.M in pERK1/2 from three experiments, each conducted in triplicate.
DAT Trafficking. Biotinylation and flow cytometry were used to determine whether D2SR stimulation alters DAT cell surface expression. Cell surface biotinylation and immunoblot analyses were performed as described previously (Li et al., 2004) to quantify the amount of plasma membrane DAT protein. EM4 cells (100,000 cells/well) were seeded into 12-well plates containing DMEM/Ham's F-12 medium supplemented with 10% FBS and penicillin (100 U/ml) and streptomycin (100 μg/ml) in an atmosphere of 5% CO2 and 95% humidified atmosphere at 37°C. After 24 h, cells were transfected with different expression plasmids together with DAT and myc-D2SR receptor cDNA plasmids (0.9 μg of D2SR receptor and 0.3 μg of DAT) using Lipofectamine 2000 transfection reagent according to manufacturer's protocols. In all wells, the total amount of plasmid DNA was adjusted with corresponding empty vector. Cells were treated with vehicle or quinpirole (10 μM) for 1 min after 48 h of transfection. At the end of the treatment, cells were washed two times with ice-cold PBS/calcium-magnesium buffer (138 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 9.6 mM Na2HPO4, 1 mM MgCl2, and 0.1 mM CaCl2, pH 7.3) and incubated with EZ link sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (1 mg/ml) in 1× PBS/calcium-magnesium buffer for 30 min at 4°C. The reaction was quenched by two washes with ice-cold 100 mM glycine in PBS/calcium-magnesium buffer and further incubation with 100 mM glycine in PBS/calcium-magnesium buffer at 4°C for 20 min. Then the cells were lysed in 500 μl of radioimmunoprecipitation assay buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, and 0.1% sodium deoxycholic acid) containing protease inhibitors (1 μM pepstatin A, 250 μM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, and 1 μg/ml aprotinin) for 1 h at 4°C with constant shaking. Lysates were centrifuged at 25,000g for 30 min at 4°C, and clear supernatants were incubated with streptavidin beads (100 μlof beads/400 μl of cell lysates from one well) for 1 h at room temperature. Beads were washed three times with radioimmunoprecipitation assay buffer, and bound proteins were eluted with 50 μl of Laemmli buffer (62.5 mM Tris, pH 6.8, 20% glycerol, 2% SDS, 5% β-mercaptoethanol, and 0.01% bromphenol blue) for 30 min at 22°C. Aliquots from total cell lysates (50 μl) and unbound fractions (100 μl) and all (50 μl) of the avidin-bound samples were analyzed by immunoblotting with DAT-specific antibody. To validate the surface localization of biotinylated DAT protein, blots were striped and reprobed with calnexin antibody (Stressgen Biotechnologies, Victoria, BC, Canada). Band intensities were quantified using NIH ImageJ (version 1.32j). Exposures were precalibrated to ensure quantitation within the linear range of the film, and multiple exposures were taken to validate linearity of quantitation. DAT densities from total, nonbiotinylated (representing the intracellular pool), and biotinylated (representing the surface pool) fractions were normalized using levels of calnexin in the total extract, and values were averaged across four experiments. The data were analyzed by ANOVA followed by SNK.
Flow cytometry was performed in cells transiently transfected with cDNA for HA 2EL hDAT and empty vector or HA 2EL hDAT and FLAG-D2SR. Cells were washed with PBS and incubated in the presence or absence of quinpirole (10 μM) for 2 min at 37°C. After fixing the cells with 2% platelet-activating factor, surface expression of HA 2EL hDAT was quantified by FACS analysis using an anti-HA as described previously (Costagliola et al., 1998). The fluorescence of 10,000 cells/tube was determined using a Guava Easy Cite cytoflurometer (Guava Technologies, Hayward, CA). Cells transfected with empty vector served as negative controls. The effect of quinpirole was compared with that of vehicle treatment for each experiment, and the mean ± S.E.M. values are shown for these determinations from three independent experiments, each performed in triplicate. The Student's t test was used for statistical analysis.
Coimmunoprecipitation. Coimmunoprecipitation experiments were conducted as described previously (Gomes et al., 2003). EM4 cells were transiently transfected with cDNA for myc-D2SR, WT FLAG-hDAT, or ΔN1–55 FLAG-hDAT either alone or in combination (Guo et al., 2003). Receptor-transporter complexes were solubilized in radioimmunoprecipitation buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, and 0.1% sodium deoxycholate) containing 10 mM iodoacetamide and protease inhibitor cocktail (Sigma-Aldrich) for 1 h at 4°C and centrifuged for 15 min in a microcentrifuge at 14,000 rpm. Lysates were immunoprecipitated with polyclonal anti-myc antibodies (rabbit, clone A-14; Santa Cruz Biotechnology). The anti-myc antibodies were pulled down with protein A-linked agarose beads, washed several times, and eluted in Laemmli sample buffer. The immunoprecipitate was subjected to SDS-polyacrylamide gel electrophoresis as described above and subsequently immunoblotted with monoclonal anti-FLAG antibodies (mouse, M2; Sigma-Aldrich) to detect DAT. In other studies, FLAG-D2SR was transfected as above with and without YFP-hDAT. Complexes were immunoprecipitated with the M2 monoclonal anti-FLAG antibodies as described above and pulled down with protein G-linked agarose beads. Proteins were visualized with a monoclonal DAT antibody (rat, MAB369; Chemicon, Temecula, CA). In addition, myc-D2SR was transfected as above with and without YFP-hDAT. Complexes were immunoprecipitated with polyclonal anti-myc antibodies, pulled down with protein A-linked agarose beads, and proteins were visualized with the monoclonal DAT antibody as described above.
BRET Assay. Human embryonic kidney cells (HEK-293) were transfected with either human D2SR with a luciferase on the C terminus (2 μg; D2SR-Luc) alone or in combination with YFP-hDAT (2 μg) using Lipofectamine (Invitrogen) according to the manufacturer's protocol. Cells were detached from plates 48 h after transfection by incubation with PBS containing 1 mM EDTA for 2 min, collected by centrifugation, washed with PBS, and resuspended to ∼1 to 2 × 106 cells/ml with PBS containing 1 mM EDTA. Cells (2 ml) were placed in a cuvette, coelenterazine h was added to 5 μM final concentration, and light emission was monitored from 420 to 590 nm at 5-nm intervals for 0.5 s using a FluoroMax-2 spectrometer (Jobin Yvon Spex Instruments, Edison, NJ). For BRET assays using the Fusion Microplate Reader, transfected cells (1 × 105 cells/well) were plated in complete media onto a 96-well microplate (Optiplate-96; PerkinElmer Life and Analytical Sciences). The next day, wells were washed three times with PBS, treated with 5 μM coelenterazine h, and readings were collected using a Fusion Universal Microplate Reader (PerkinElmer Life and Analytical Sciences). BRET ratios were calculated as described by Angers et al. (2000). As a positive control, a concurrent set of cells was transfected with luciferase-tagged κ-opioid receptors (2 μg; κ-Luc) alone and in combination with YFP-tagged δ-opioid receptors (2 μg). Functional interactions between κ- and δ-opioid receptors have been reported previously, and the receptors have been shown to be in close proximity (<100 Å) using coimmunoprecipitation and BRET (Gomes et al., 2002). In contrast to κ- and δ-opioid receptors, previous studies have shown no association of D2R with the chemokine receptor CCR5. Therefore, negative control studies were also conducted in cells cotransfected with human CCR5 tagged on the C terminus with YFP (2 μg; YFP-CCR5) and D2SR-Luc.
Results
DA and Tyramine, but Not ASP+, Activate D2SR. Addition of DA to cells expressing D2SR resulted in a concentration-dependent increase in pERK1/2 (EC50 = 5 nM; Fig. 1). The DAT substrate tyramine showed a similar maximal effect but a much lower potency (EC50 = 1 μM; Fig. 1). In contrast, no increase in pERK1/2 was seen with ASP+ at concentrations as high as 100 μM (Fig. 1).
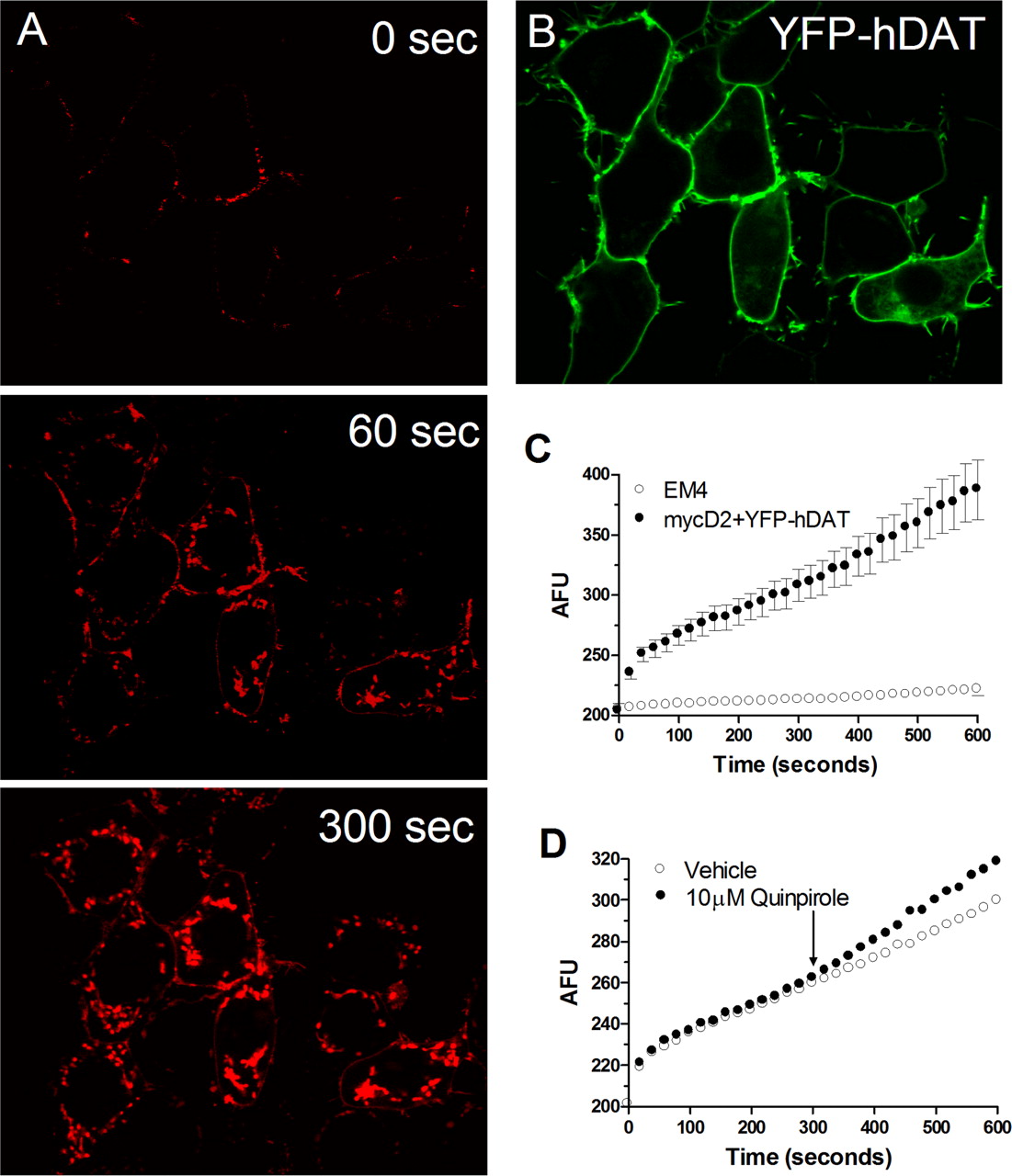
Activation of D2SR Up-Regulates DAT Function. ASP+ rapidly accumulated in the cytoplasm of EM4 cells coexpressing D2SR and DAT (Fig. 2, A and B). In contrast, little accumulation was observed in cells lacking DAT. Consistent with previous studies with norepinephrine transporters (Schwartz et al., 2003), two distinct phases of incorporation of the ASP+ signal were observed. Binding of ASP+ to transporters located on the cell surface was rapid (milliseconds) and was followed by a second, slower (seconds per minute) phase of accumulation of ASP+ signal in the cytoplasm (Fig. 2C). The addition of quinpirole to EM4 cells coexpressing FLAG-D2SR and YFP-hDAT increased the rate of ASP+ accumulation (Figs. 2C and 3A). This effect was significant in response to 10 μM quinpirole. Increased ASP+ accumulation was also observed in response to PD128907, a structurally different D2/D3 receptor agonist (Fig. 3A). Cells were pretreated with vehicle or eticlopride, a D2R antagonist, before the addition of quinpirole (10 μM) to determine whether the increase in accumulation was D2SR-mediated. Eticlopride (100 nM) prevented the quinpirole-evoked increase in ASP+ accumulation (ANOVA: F3,113 = 12.4; p ≤ 0.0001; SNK: p ≤ 0.001). ASP+ accumulation in quinpirole-treated cells was increased by 31.4 ± 6.7% (n = 35) from predrug levels compared with 3.4 ± 3% in eticlopride + quinpirole-treated (n = 41) cells (data not shown). Incubation of cells with either vehicle (2.7 ± 4%; n = 25) or eticlopride (–0.8 ± 2.6%; n = 30), alone did not alter ASP+ accumulation. In EM4 cells transiently transfected with WT FLAG-hDAT and D1 DA receptors (D1R), the addition of the selective D1R agonist B135 (10 μM) did not alter ASP+ uptake, indicating that in contrast to D2SR, D1R activation is ineffective in modulating DAT activity (A. Zapata, B. Kivell, Y. Han, J. Javitch, E. Bolan, D. Kuraguntla, V. Jaligam, L. Jayanthi, D. Samuvel, S. Ramamoorthy, et al., submitted). To determine whether D2SR activation up-regulates DAT function in a neuroblastoma cell line, ASP+ accumulation was quantified in N2a cells coexpressing FLAG-D2SR and YFP-hDAT. Quinpirole (1 μM) induced a significant increase in ASP+ accumulation relative to vehicle-treated cells (Fig. 3B).

ASP+ Uptake in EM4 cells transiently cotransfected with myc-D2SR and YFP-hDAT. A, ASP+ rapidly accumulates in the intracellular compartment of EM4 cells coexpressing D2SR and hDAT. B, accumulation is negligible in cells not expressing hDAT. C, the time course of ASP+ uptake in a representative within cell experiment is shown. Note the initial rapid binding phase after ASP+ addition followed by the linear uptake phase. D, the addition of quinpirole (10 μM) increases the rate of ASP+ uptake relative to predrug values. The increase in DAT function is measured as a change in the slope of uptake measured within 1 min before and after addition of drug.

Activation of D2SR up-regulates DAT function. A, EM4 cells were transiently cotransfected with FLAG-D2SR and YFP-hDAT. DAT function was measured as accumulation of ASP+ fluorescence over time in cells treated with vehicle, quinpirole (0.1–10 μM), or PD128907 (10 μM). Data are expressed as the mean percentage increase ± S.E.M. ***, significant difference of 10 μM quinpirole from vehicle-treated cells (ANOVA: F3,182 = 8.45, p ≤ 0.0001; SNK: p ≤ 0.001) and of 10 μM PD128907 versus vehicle group (Student's t test: t = 4.08, df = 41; p ≤ 0.001). B, N2a cells were transfected as above and quinpirole (0.1 μM, 1 μM)-evoked ASP+ accumulation was quantified. Data represent the mean percentage increase ± S.E.M. **, significant difference from vehicle-treated cells (ANOVA: F2,92 = 6.13, p ≤ 0.01; SNK: p ≤ 0.01).
D2SRs are G protein-coupled receptors that signal primarily through the pertussis toxin-sensitive Gi/Go class of heterotrimeric G proteins (Choi et al., 1999). To test whether the interaction of D2SR with DAT is Gi/Go-dependent, EM4 cells expressing FLAG-D2SR and YFP-hDAT were incubated with pertussis toxin (100 ng/ml) for 16 to 24 h and the ability of quinpirole (10 μM) to increase ASP+ uptake was assessed. Preincubation with pertussis toxin prevented the quinpirole-evoked increase in ASP+ uptake (Fig. 4) suggesting that the D2SR-mediated increase in DAT function requires coupling to Gi/Go proteins. No difference between vehicle and pertussis toxin-treated cells in the rate of ASP+ accumulation before the addition of quinpirole was seen. The slopes normalized for DAT expression levels were 0.00072 ± 0.00005 and 0.00064 ± 0.00004, respectively, for vehicle- and pertussis toxin-treated cells (data not shown).
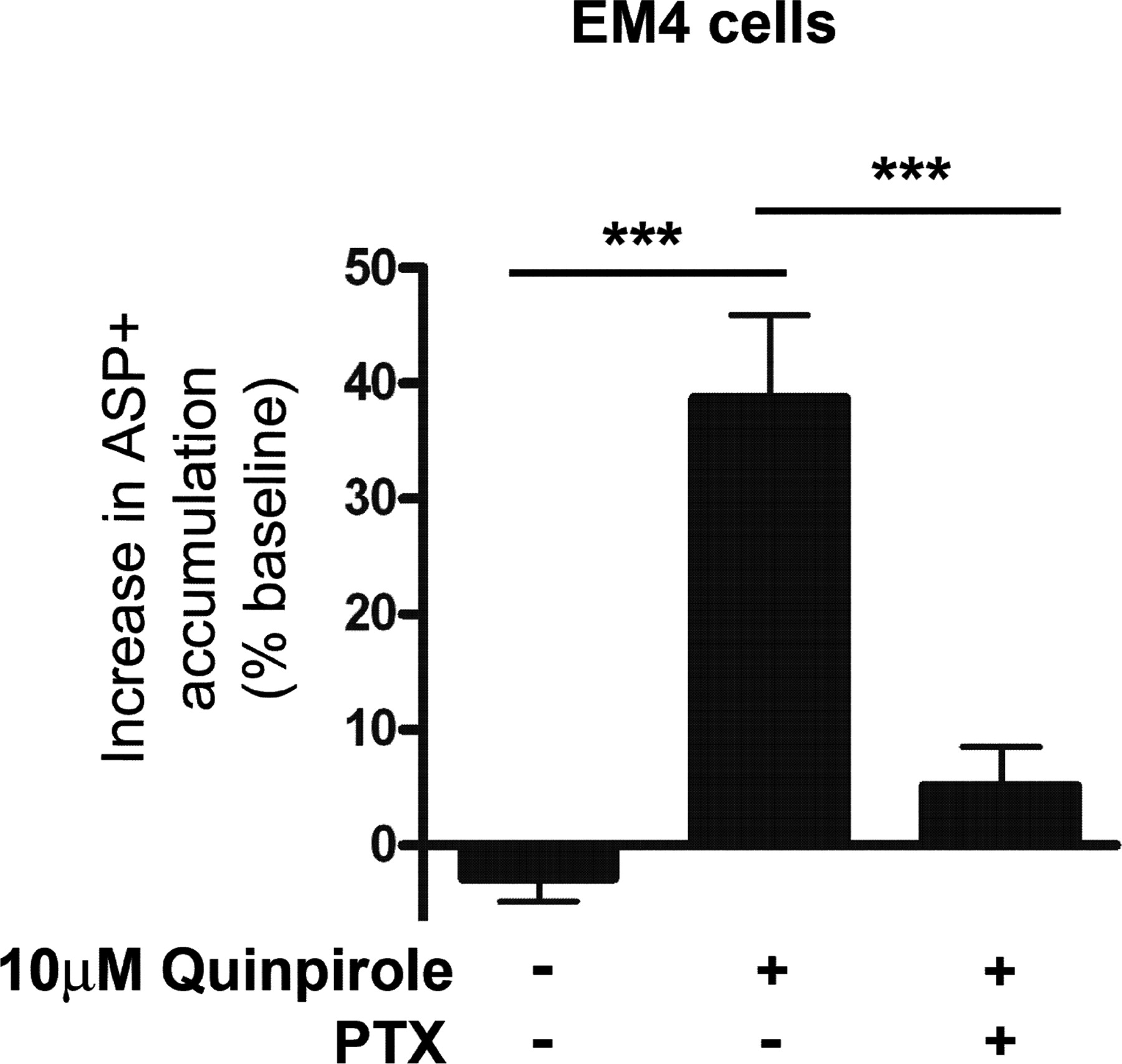
The D2SR-mediated up-regulation of DAT function is pertussis toxin-sensitive. EM4 cells transiently cotransfected with FLAG-D2SR and YFP-hDAT were pretreated 16 to 24 h with pertussis toxin (PTX, 100 ng/ml) and quinpirole (10 μM)-evoked ASP+ accumulation was quantified. Data are expressed as mean percentage increase ± S.E.M. (ANOVA: F2,107 = 10.60, p ≤ 0.0001; SNK: ***, p ≤ 0.001).

The D2SR-mediated up-regulation of DAT function is ERK1/2-sensitive and PI3K-insensitive. A, D2SR signals via both ERK1/2 and PI3K. Left, immunoblot analysis of pERK1/2 in EM4 cells transiently expressing FLAG-D2SR and YFP-hDAT. Cells were preincubated for 15 min at 37°C with vehicle or PD98059 (10 μM) before stimulation with quinpirole (10 μM) for 1 min. Data are expressed as the mean ± S.E.M. fold increase in phosphorylated to total ERK1/2 ratio from vehicle (ANOVA: F3,27 = 23.84, p ≤ 0.0001; SNK: ***, p ≤ 0.001). Right, immunoblot analysis of pAkt in EM4 cells preincubated for 15 min at 37°C with LY294002 (10 μM) before the addition of quinpirole. Data represent the mean ± S.E.M. fold increase in phosphorylated to total Akt ratio from vehicle (ANOVA: F3,31 = 25.33, p ≤ 0.001; SNK: ***, p ≤ 0.001). Representative immunoblots of at least three experiments are shown. B, inhibition of ERK1/2 but not PI3K attenuates the quinpirole-evoked increase in ASP+ accumulation. EM4 cells were preincubated for 15 min with vehicle, PD98059 (10 μM), or LY294002 (10 μM) before the addition of quinpirole (10 μM). ASP+ uptake was determined as described under Materials and Methods. ANOVA: F3,108 = 12.23, p ≤ 0.0001; SNK: ***, p ≤ 0.001).
Dopamine D2SR Modulates DAT Function Via an ERK1/2-Dependent, PI3 Kinase-Independent Mechanism. D2SRs have been shown to signal via ERK1/2 and PI3K in brain and various cell lines (Brami-Cherrier et al., 2002; Beom et al., 2004). Similar to our results in the intact cell ELISA with DA (Fig. 1), quinpirole (10 μM, 1 min, 37°C incubation) evoked a 2.8-fold increase in phosphorylated ERK1/2 in EM4 cells coexpressing FLAG-D2SR and YFP-hDAT as detected by immunoblotting (Fig. 5A). Preincubation with the MEK inhibitor PD98059 (10 μM, 15 min, 37°C; Fig. 5A) prevented the quinpirole-evoked ERK1/2 phosphorylation in these cells. In contrast, the PI3K inhibitor LY294002 (10 μM, 15 min, 37°C preincubation) was without effect (data not shown, Student's t test, t = 0.532, p = 0.605 df = 11).
Akt, a serine/threonine protein kinase, is a major target of PI3K via its pleckstrin homology domain (Burgering and Coffer, 1995). To determine whether D2SRs couple to PI3K in EM4 cells, phosphorylation of the Ser473 residue of Akt was assessed by immunoblotting (Fig. 5B). Quinpirole evoked a 2.5-fold increase in phosphorylated Akt in EM4 cells coexpressing FLAG-D2SR and YFP-hDAT relative to that observed in vehicle-treated cells coexpressing the two proteins. This increase was blocked by preincubation with LY294002 (10 μM, 15 min, 37°C; Fig. 5B), confirming its mediation by PI3K. In contrast, preincubation of cells with PD98059 (10 μM, 15 min, 37°C) was without effect on Akt phosphorylation (data not shown, Student's t test, t = 0.291, p = 0.776, df = 12). These findings confirm that D2SRs activate ERK1/2 and PI3K and show that D2SR activation of ERK1/2 occurs independently of PI3K in the EM4 cell line.
To determine the involvement of ERK1/2 and PI3K in mediating DAT regulation by D2SR, EM4 cells expressing FLAG-D2SR and YFP-hDAT were incubated with either PD98059, LY294002, or vehicle for 15 min at 37°C before the addition of quinpirole (10 μM). ASP+ accumulation was measured before and after addition of quinpirole, as detailed under Materials and Methods. Preincubation with PD98059 (10 μM, 15 min, 37°C) prevented the quinpirole-induced increase in DAT activity (Fig. 5C). In contrast, the PI3K inhibitor LY294002 (10 μM, 15 min, 37°C preincubation) was without effect (Fig. 5C). Concentrations of LY294002 as high as 30 μM were also without effect (data not shown).
Influence of D2SR Activation on ASP+Accumulation in Cells Expressing N-Terminally Truncated DAT. The N terminus of DAT contains a number of serine and threonine residues that are potential phosphorylation sites for ERK1/2. To determine whether the first 55 amino acids of DAT are necessary for the functional interaction of D2SR with DAT, EM4 cells were cotransfected with YFP-D2SR and either WT FLAG-hDAT or ΔN1–55 FLAG-hDAT. ASP+ accumulation was measured before and after addition of 10 μM quinpirole. Quinpirole significantly increased the rate of ASP+ accumulation in cells expressing YFP-D2SR and WT FLAG-hDAT (vehicle, 6.2 ± 2.5, n = 67; quinpirole, 33.3 ± 2.8, n = 81; t = 7.1, df = 146, p ≤ 0.0001; data not shown). The magnitude of this effect did not differ from that observed in cells transfected with FLAG-D2SR and YFP-hDAT (Fig. 2D). A similar D2SR-mediated increase in ASP+ accumulation was observed in cells expressing ΔN1–55 FLAG-hDAT (vehicle, 9.6 ± 1.6, n = 72; quinpirole, 30.2 ± 3.8, n = 63; t = 5.2, df = 133, p ≤ 0.0001 versus vehicle group ΔN1–55 FLAG-hDAT), indicating that D2SR regulation of DAT does not require the first 55 amino acids of DAT.

D2SR stimulation increases tyramine uptake and DAT cell surface expression. EM4 cells were cotransfected with FLAG-D2SR and HA 2EL hDAT (▪) or with HA 2EL and empty vector (□). A, after 48 h cells were treated with or without quinpirole (10 μM) in the presence of absence of sulpiride (10 μM) for 1 min. [3H]Tyramine uptake was determined as described under Materials and Methods. Data are expressed as the percentage of vehicle-treated uptake for the respective transfection conditions (mean ± S.E.M., n = 5). Drug treatments were not significantly different from vehicle treatment for hDAT alone (ANOVA: F3,16 = 0.05, p > 0.98). In cells coexpressing D2SR and hDAT, quinpirole induced a sulpiride-reversible increase in uptake (ANOVA: F3,16 = 7.01, p = 0.003; SNK: **, p ≤ 0.01 versus vehicle; †, p < 0.05 versus quinpirole). B, cells were treated with or without quinpirole (10 μM) for 2 min and then were fixed, and surface labeling was quantified with an anti-HA antibody and FACS analysis as described under Materials and Methods. Data are expressed as the percentage of vehicle-treated fluorescence for the respective transfection conditions (mean ± S.E.M., n = 3). Quinpirole significantly increased fluorescence in cells coexpressing D2SR and hDAT (t = 2.49, df = 4, *, p < 0.05).
D2SR Activation Increases Transport Activity of DAT. To determine whether D2SR regulation of DAT can be observed with radiolabeled substrate, the influence of quinpirole (10 μM) on [3H]tyramine uptake was determined in EM4 cells transiently transfected with FLAG-D2SR and HA 2EL hDAT, Incubation of cells with 10 μM quinpirole significantly increased [3H]tyramine uptake relative to that of vehicle-treated cells (Fig. 6A). (ANOVA: F3,16 = 7.01, p = 0.003; SNK: p ≤ 0.01 versus vehicle). Pretreatment with sulpiride prevented the qunipirole-evoked increase in [3H]tyramine uptake (p < 0.05 versus quinpirole) (Fig. 6A). Quinpirole failed to alter [3H]tyramine uptake in cells expressing hDAT but not D2SR (Fig. 6A), and sulpiride was also without significant effect on uptake in these cells (ANOVA: F3,16 = 0.05, p > 0.98).
D2SR Activation Regulates DAT Cell Surface Expression. In view of the results obtained with ASP+ and [3H]tyramine, experiments were conducted to determine whether the D2SR-mediated increase in DAT function was associated with altered surface expression of DAT. FACS analysis was used to detect DAT cell surface immunofluorescence in EM4 cells coexpressing FLAG-D2SR and HA 2EL hDAT (Fig. 6B). Addition of 10 μM quinpirole for 2 min significantly increased DAT cell surface immunofluorescence relative to vehicle-treated cells (fold increase, 1.60 ± 0.24; t = 2.49, p < 0.05, df = 4). In cells that were preincubated with sulpiride before the addition of quinpirole, no alteration of cell surface fluorescence was seen (data not shown). In contrast, in cells expressing hDAT but not expressing D2SR, quinpirole was without significant effect (Fig. 6B) (fold increase, 1.06 ± 0.05; t = 1.11, p = 0.2, df = 4).

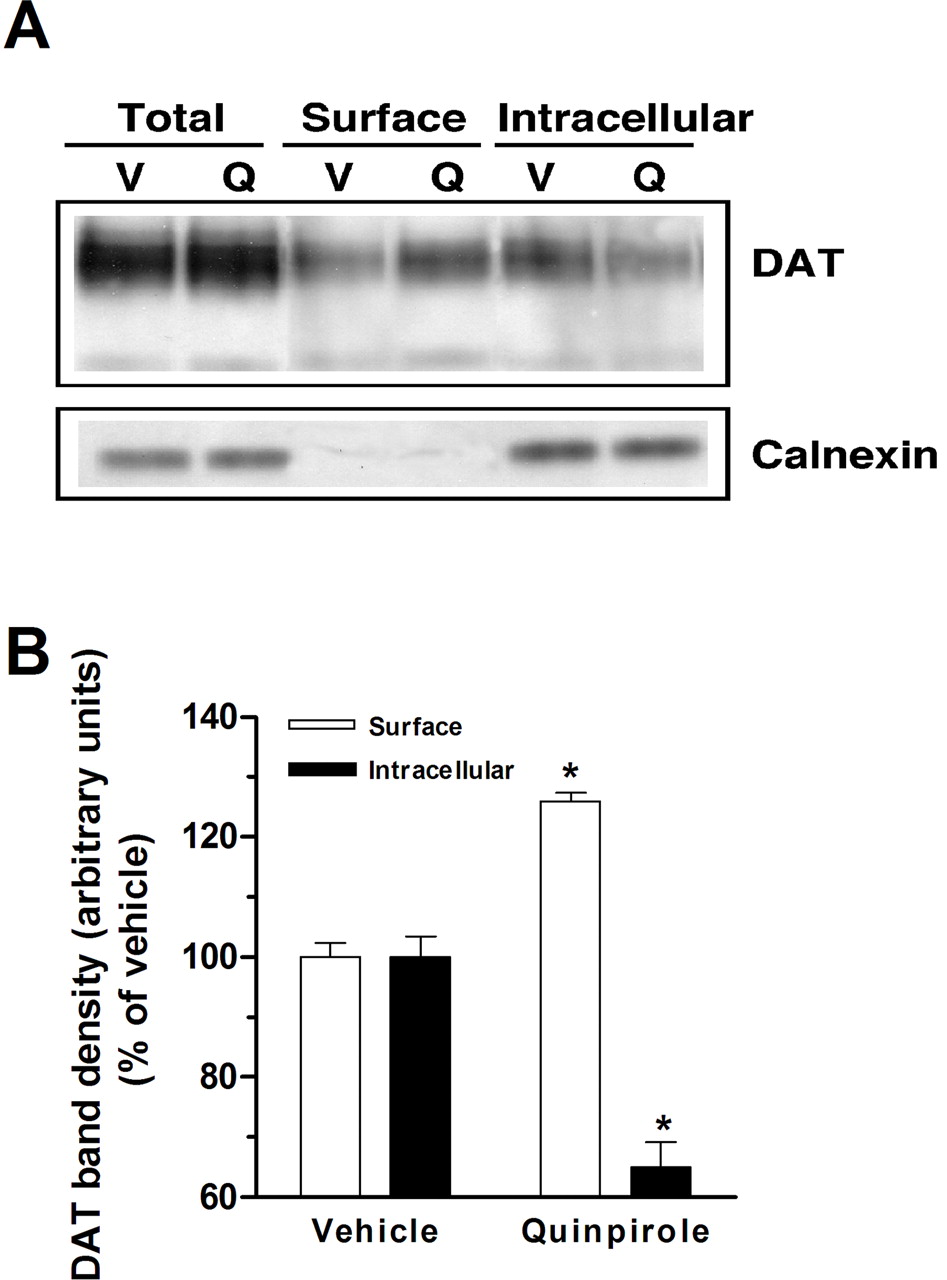
D2SR stimulation increases DAT cell surface expression. A, EM4 cells were cotransfected with FLAG-hDAT and myc-D2SR. After 48 h, cells were washed once with KRH buffer and incubated with biotinylating reagent with or without quinpirole (10 μM) for 1 min. Isolation of biotinylated DAT and detection of DAT were performed as described under Materials and Methods. Western blots of DAT and calnexin from total lysates, avidin beads eluates, and avidin beads unbound fractions are shown at the top. Each blot is a representative of four separate experiments. Quantitative analysis of DAT band densities for four experiments (means ± S.E.M.) is shown at the bottom. *, significant changes in biotinylated DAT compared with vehicle treatment (p < 0.05, one-way ANOVA with Bonferroni post hoc analysis).
Biotinylation studies, which permit quantification of cell surface and intracellular DAT, were conducted in EM4 cells coexpressing myc-D2SR and FLAG-hDAT. Consistent with ASP+ uptake, tyramine uptake, and FACS analysis, incubation of cells for 1 min with quinpirole (10 μM) significantly increased biotinylated DAT (surface) (126 ± 1.3%) and reduced nonbiotinylated DAT (intracellular) (65 ± 4.2%: F1,7 = 499; p ≤ 0.0001; SNK: p ≤ 0.0001), indicating an increase in cell surface DAT (Fig. 7). Treatment with quinpirole did not alter the total amount of DAT protein as measured by immunoblotting. Less than 0.5% of total calnexin was present in streptavidin-bound fractions, indicating that cells were intact and intracellular proteins were not significantly biotinylated.
Coimmunoprecipitation and BRET Studies to Explore Potential Proximity of D2SR and DAT. To determine whether DAT and D2SR complexes could be isolated and to assess the involvement of the N terminus of DAT in the potential interaction with D2SR, solubilized lysates were prepared from EM4 cells coexpressing either myc-D2SR and WT FLAG-hDAT or myc-D2SR and NΔ1–55 FLAG-hDAT or from a mixture of cells individually expressing myc-D2SR and WT FLAG-hDAT or myc-D2SR and NΔ1–55 FLAG-hDAT and subjected to immunoprecipitation with an anti-myc antibody. When myc-D2SR was coexpressed with either WT FLAG-hDAT or NΔ1–55 FLAG-hDAT, immunoblotting with the anti-FLAG antibody revealed a ∼100-kDa band corresponding to the molecular mass of hDAT (Fig. 8, lanes 2 and 4). This indicates that the N terminus is not necessary for its participation in the immunocomplex with myc-D2SR. No specific bands were observed with the FLAG antibody in the mixture of cells individually expressing the proteins (Fig. 8, lanes 1 and 3) or with untransfected EM4 cells (Fig. 8, lane 5), indicating that the presence of the myc-D2SR/WT FLAG-hDAT or myc-D2SR/NΔ1–55 FLAG-hDAT complexes required expression in the same cell and was not simply an artifact of the solubilization and immunoprecipitation process. A faint 150-kDa band was observed in all lanes on the blots and corresponded to the rabbit anti-myc immunoprecipitation antibody (Fig. 8). The band was not present when the anti-myc immunoprecipitation antibody was omitted (data not shown). Diffuse larger molecular mass species were observed in blots from coexpressing cells, suggesting that a substantial fraction of the myc-D2SR/WT FLAG-hDAT or myc-D2SR/NΔ1–55 FLAG-hDAT complexes were resistant to SDS/β-mercaptoethanol (Fig. 8, lanes 2 and 4). Under the same conditions described above, YFP-hDAT also coimmunoprecipitated with myc-D2SR or FLAG-D2SR selectively when coexpressed in cells, suggesting that the presence of different epitope tags does not interfere with the interaction (data not shown; see Materials and Methods for details).

D2SR coimmunoprecipitates both WT and N-terminally truncated DAT. Coimmunoprecipitation and immunoblot analysis of myc-D2SR with WT FLAG-hDAT or ΔN1–55 FLAG-hDAT. Lane 1, lysates from EM4 cells individually expressing myc-D2SR or WT FLAG-hDAT were mixed (mix) and immunoprecipitated (IP) with an anti-myc antibody and immunoblotted (IB) using an anti-FLAG antibody. Lane 2, lysate from EM4 cells coexpressing (co) both proteins (myc-D2SR and WT FLAG-hDAT) were subjected to the same procedure. Lane 3, lysates from EM4 cells individually expressing myc-D2SRor ΔN1–55 FLAG-hDAT were mixed (mix) and treated as above. Lane 4, lysate from EM4 cells coexpressing (co) both proteins (myc-D2SR and ΔN1–55 FLAG-hDAT) treated as above. Lane 5, lysate from EM4 cells subjected to the same procedure as above. A representative blot of n = 3 experiments is shown.
The BRET assay was used to determine whether in living cells D2SR and DAT might be in close enough proximity to interact (<100 Å; Fig. 9). Cells coexpressing either D2SR-Luc and YFP-hDAT or D2SR-Luc alone were treated with the luciferase substrate, coelenterazine h. Coexpression of D2SR-Luc and YFP-hDAT resulted in a positive energy transfer signal, as evidenced by the increased BRET ratio (Fig. 9, inset). The increased BRET ratio is comparable with that seen in experiments performed concurrently with cells coexpressing YFP-δ and κ-Luc opioid receptors (Fig. 9, inset), consistent with previous reports showing dimerization of these opioid receptor types (Jordan and Devi, 1999; Gomes et al., 2003). No energy transfer occurred when D2SR-Luc was coexpressed with YFP-CCR5 (Fig. 9). These data suggest that upon transient cotransfection, some fraction of D2SR and DAT appear to reside within 100 Å of each other (see below).

Energy transfer between D2SR and DAT in live cells. HEK-293 cells were transiently cotransfected with the following pairs: D2SR-Luc and YFP-hDAT; D2SR-Luc and YFP-CCR5, κ-Luc, and YFP-tagged δ-opioid receptors or transfected with D2SR-Luc or κ-Luc alone and subjected to BRET analysis (see Materials and Methods). Light emission spectra of the different conditions are shown. Inset, BRET ratios as determined from data obtained from the spectrophotometer or Fusion Microplate reader as described. Data are the mean ± S.E.M., n = 4.
Discussion
A live-cell fluorescent imaging technique was used to show that D2SRs modulate DAT function and to identify the intracellular mechanisms mediating this effect. Our studies demonstrate that activation of D2SR induces a rapid and pertussis toxin-sensitive increase in the rate of accumulation of the DAT substrate ASP+ in mammalian cell lines coexpressing D2SR and DAT. D2SR activation induced phosphorylation of ERK1/2 and Akt. Inhibition of ERK1/2 prevented the D2SR-mediated increase in ASP+ accumulation, whereas inhibition of PI3K was without effect. FACS and biotinylation studies revealed increased DAT cell surface expression in response to D2SR activation. Data obtained from coimmunoprecipitation and BRET studies show that some fraction of D2SR and DAT seems to be in close proximity.
D2Rs are enriched in striatum. Studies in which electrochemical techniques have been used to probe DAT function have provided evidence that D2-like receptors regulate DAT in this brain region (Hersch et al., 1997). Colocalization of D2 receptors and DAT in axonal terminals has been reported, providing an anatomical basis for the potential interaction of these proteins (Hersch et al., 1997). However, two splice variants of the D2 receptor, D2SR and D2LR, each with a distinct subcellular distribution, have been identified. D2SRs are enriched in presynaptic DA terminals, the site of DAT localization, whereas D2LRs are postsynaptic (Khan et al., 1998). Although D2LR modulation of DAT was reported in X. laevis oocytes (Mayfield and Zahniser, 2001), whether D2SRs produce similar effects is unknown. In vivo and ex vivo studies seeking to address this issue are precluded by the absence of ligands that discriminate between them.
ASP+ is a fluorescent high-affinity substrate of monoamine transporters (Schwartz et al., 2003). De Felice and coworkers have shown that this substrate can be used with live cell immunofluorescence confocal microscopy to monitor norepinephrine transporter and DAT function in individual cells (Mason et al., 2005; Schwartz et al., 2005). ASP+ accumulates rapidly in HEK cells expressing DAT but not in nontransfected cells. Accumulation is saturable and dependent on pH and temperature. It is blocked by DAT inhibitors and substrates. These findings, which have been replicated in our laboratory, are consistent with those of radiometric and electrochemical studies and indicate that ASP+ accumulation reflects a transporter-mediated process.
The D2/D3 receptor agonist quinpirole induced an eticlopride-reversible increase in ASP+ uptake in EM4 cells coexpressing DAT and D2SR. Increased uptake was observed in a response to a structurally distinct agonist, PD128907, and in N2a cells coexpressing D2SR and DAT. In contrast, D1 agonist treatment did not alter ASP+ accumulation in cells coexpressing DAT and D1R. These data provide the first demonstration that D2SR stimulation is sufficient to up-regulate DAT function.
Pertussis toxin treatment prevented the quinpirole-evoked increase in ASP+ accumulation. Using X. laevis oocytes, Mayfield and Zahniser (2001) reported a pertussis toxin-dependent increase of DA uptake in response to D2LR activation. Together, these data demonstrate that D2Rs regulate DAT via a Gi/Go-mediated process.
D2 activation induces pertussin toxin-sensitive phosphorylation of ERK1/2 and PI3K in striatum and heterologous cell systems (Choi et al., 1999; Brami-Cherrier et al., 2002; Kihara et al., 2002). Because these kinase cascades regulate DAT function and cell surface expression (Carvelli et al., 2002; Moron et al., 2003), we examined whether D2SR up-regulates DAT via activation of these cascades. Immunoblotting revealed a rapid, quinpirole-evoked induction of pERK1/2 in EM4 cells coexpressing D2SR and DAT, indicating that D2SR activates ERK1/2 in this cell line. Consistent with previous reports (Beom et al., 2004), the MEK inhibitor PD98059 prevented this effect. PD98059 pretreatment also prevented the increase in ASP+ accumulation produced by D2SR activation. These data provide the first demonstration that D2SR regulates DAT via an ERK1/2-dependent mechanism. The PI3K inhibitor LY294002 failed to modify the D2SR increase in ASP+ accumulation indicating a specific role of the ERK1/2 cascade in mediating the functional modulation of DAT by D2SR. The lack of effect of LY294002 cannot be attributed to the concentration used because the same treatment regimen prevented D2SR-mediated increases in Akt phosphorylation. Rather the differential effects of PI3K inhibition on Akt phosphorylation and ASP+ accumulation suggest a role of ERK1/2, but not PI3K, in mediating D2SR regulation of DAT. It is interesting that we have recently shown that D3 DA receptors also up-regulate DAT (A. Zapata, B. Kivell, Y. Han, J. Javitch, E. Bolan, D. Kuraguntla, V. Jaligam, L. Jayanthi, D. Samuvel, S. Ramamoorthy, et al., submitted). In contrast to D2SR, both PI3K and ERK1/2 inhibitors abolished the effect of D3 DA receptor activation on DAT function. Thus, although both D2 and D3 receptors regulate DAT function, they do so, at least in part, through different mechanisms.
The DAT N terminus contains serine and threonine residues which are potential phosphoacceptor sites for PKC and ERK1/2 (Lin et al., 2003). N-terminal serines of DAT are phosphorylated in response to PKC activation (Foster et al., 2002). Although this effect is not necessary for PKC-mediated DAT down-regulation (Granas et al., 2003), phosphorylation of these residues after treatment with DAT substrates (e.g., methamphetamine, amphetamine) is believed to be a mechanism by which psychostimulants promote transporter-mediated DA efflux (Cervinski et al., 2005). Data regarding the role of the DAT N terminus or phosphoacceptor sites therein in the regulation of DAT function by other kinases or G-protein coupled receptors are limited (Granas et al., 2003; Lin et al., 2003). However, truncation of the 55 N-terminal residues of DAT did not alter D2SR regulation of DAT, because quinpirole increased ASP+ accumulation similarly in cells expressing D2SR and either WT FLAG-hDAT or ΔN1–55 FLAG-hDAT. These data demonstrate that the DAT N terminus is not necessary for modulation of DAT by D2SR. Furthermore, they indicate that phosphorylation of serine or threonine residues contained therein does not mediate this effect.
Increased ASP+ uptake occurred within 1 min after agonist addition suggesting mediation by a post-translational mechanism. Constitutive DAT trafficking between the plasma membrane and intracellular compartments has been reported (Loder and Melikian, 2003). Under basal conditions, DAT traffics between the membrane and cytosol. After its internalization, DAT is degraded or recycled back to the membrane. Cell surface biotinylation experiments that used a protocol identical with that in ASP+ experiments showed that brief D2SR stimulation significantly increased cell surface DAT. This increase was accompanied by a significant decrease in intracellular DAT. Likewise, flow cytometry revealed increased DAT cell surface expression in cells expressing D2SR and an epitope-tagged DAT. These data suggest that D2SR activation increases DAT redistribution from the intracellular compartment to the cell surface, resulting in increased transport capacity. ERK1/2 activation increases cell surface DAT in heterologous expression systems (Moron et al., 2003). Our results suggest that D2sR increases DAT function and cell surface expression by activating this kinase cascade.
Coimmunoprecipitation studies suggest that, under some conditions, D2SR and DAT may form a complex. When coexpressed, D2SR can be immunoprecipitated with WT FLAG-hDAT or ΔN1–55 FLAG-hDAT. In contrast, no interaction occurred in a mixture of individually expressing cells. It is noteworthy that the DAT N terminus was not necessary for coimmunoprecipitation, just as it was not essential for D2SR-mediated modulation of DAT function. Although caution is necessary in the interpretation of coimmunoprecipitation of membrane proteins from detergent lysates (Salim et al., 2002), BRET revealed energy transfer between D2SR fused to luciferase and DAT fused to yellow fluorescent protein, suggesting that they are in close proximity. However, it is noteworthy that at high expression levels and single acceptor/donor ratios, substantial energy transfer can arise from random interactions within the membrane (James et al., 2006). Furthermore, it is unclear whether the interaction takes place on the cell surface and/or in internal membranes.
Although BRET and coimmunoprecipitation studies indicate that a fraction of D2SR and DAT may be associated under particular circumstances, D2SR regulation of DAT requires coupling to Gi/Go proteins and ERK1/2 activation. Therefore, coupling to intracellular signaling pathways rather than a direct physical interaction between D2SR and DAT most likely underlies D2SR-evoked DAT up-regulation. Furthermore, because receptor activation occurs at the plasma membrane, DAT that is recruited to the plasma membrane from intracellular vesicles/endosomes is unlikely to have been physically associated with activated receptors already at the plasma membrane. Additional studies are needed to determine whether an association of D2SR and DAT occurs in native tissue and what, if any, role the potential proximity of these two proteins might play in regulating DAT.
We were able to confirm the D2sR-mediated increase in transport using radiolabeled tyramine. However, the action of tyramine as a full agonist at D2sR complicates its use in experiments in which the D2sR is coexpressed, because some receptor activation will occur during the determination of uptake, particularly at high tyramine concentrations required for kinetic analysis. We show that the use of DA as a substrate is not feasible given its potency at D2sR. In contrast, ASP+ is inactive at the D2sR and permits time-resolved quantification of transporter function in living cells. These data highlight the utility of the ASP+ imaging technique for exploration of the coupling between receptor activation and transport activity.
In summary, the present studies describe activation of pERK1/2 as a mechanism by which D2SR autoreceptors modulates DAT cell surface expression and, consequently, the effective concentration of extracellular DA in the synapse. Additional studies addressing the mechanisms relevant for this receptor-transporter interaction will be important for understanding the diverse mechanisms that regulate transporter activity.
Acknowledgments
We thank Dr. U. Gether for the gift of HA 2EL hDAT, Dr. F. Ciruela for the gift of the YFP-D2SR cDNA, Dr. M. Bouvier for the gift of the YFP-CCR5 cDNA, Dr. O.W. Lindwasser for advice on FACS, and Drs. L. J. DeFelice and J. W. Schwartz for their helpful advice on the ASP+ uptake technique.
Footnotes
-
J.J., A.Z., and T.S.S. contributed equally to this work.
-
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Drug Abuse, and National Institutes of Health grants MH57324, MH54137, and DA11495 (to J.A.J.) and DA08863 and DA0019521 (to L.A.D.), and National Institute on Drug Abuse and National Institutes of Health grants P50DA015369 and MH062612 (to S.R.).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.106.027763.
-
ABBREVIATIONS: DA, dopamine; D2R, D2 dopamine receptor; DAT, dopamine transporter; D2SR, D2 receptor short splice variant; WT, wild type; BRET, bioluminescence resonance energy transfer; ERK1/2, extracellular signal-regulated kinases 1 and 2; PI3K, phosphoinositide 3 kinase; MEK, mitogen-activated protein kinase kinase; PKC, protein kinase C; D2LR, D2 receptor long splice variant; N2a, Neuro-2a cell; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; ELISA, enzyme-linked immunosorbent assay; PBS, phosphate-buffered saline; BSA, bovine serum albumin; YFP, yellow fluorescent protein; KRH, Krebs-Ringer-HEPES; ANOVA, analysis of variance; SNK, Student Newman-Keuls test; HA, hemagglutinin; FACS, fluorescence-activated cell sorting; HEK, human embryonic kidney; κ-Luc, luciferase-tagged κ-opioid receptor; HA 2EL hDAT, hemagglutinin epitope-tagged human dopamine transporter; PD98059, 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one; PD128907, (S)-(+)-(4aR, 10bR)-3,4,4a, 10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano-[4,3-b]-1,4-oxazin-9-ol hydrochloride; LY294002, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride; ASP+, 4-[4-(diethylamino)-styryl]-N-methylpyridinium iodide; B135, 3-(2-pyridyl)-5-(3-allyloxy-5-carboxyphenyl)-1,2,4-oxadiazole.
- Received June 8, 2006.
- Accepted January 31, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}