


Abstract
Human hydroxysteroid sulfotransferase or (HUMAN)SULT2A1 catalyzes the sulfonation of procarcinogen xenobiotics, hydroxysteroids, and bile acids and plays a dynamic role in hepatic cholesterol homeostasis. The treatment of primary cultured human hepatocytes with a peroxisome proliferator-activated receptor α (PPARα)-activating concentration of ciprofibrate (10- 4 M) increased (HUMAN)SULT2A1 mRNA, immunoreactive protein, and enzymatic activity levels by ∼2-fold. By contrast, expression of (RAT)SULT2A3, the rat counterpart to (HUMAN)SULT2A1, was induced by treatment of primary hepatocyte cultures with an activator of the pregnane X receptor, but not PPARα. In HepG2 cells, transient transfection analyses of luciferase reporter constructs containing upstream regions of the (HUMAN)SULT2A1 gene implicated a candidate peroxisome proliferator response element (PPRE) at nucleotides (nt) -5949 to -5929 relative to the transcription start site. Site-directed mutagenesis and electrophoretic mobility shift assay studies confirmed that this distal PPRE (dPPRE), a direct repeat nuclear receptor motif containing one intervening nt, represented a functional PPRE. Chromatin immunoprecipitation analysis indicated that the (HUMAN)SULT2A1 dPPRE was also a functional element in the context of the human genome. These data support a major role for the PPARα transcription factor in the regulation of hepatic (HUMAN)SULT2A1. Results also indicate that important species differences govern the transactivation of SULT2A gene transcription by nuclear receptors.
The human hydroxysteroid sulfotransferase enzyme, recently designated under a proposed standardized nomenclature as (HUMAN)SULT2A1 (Blanchard et al., 2004), catalyzes the transfer of a sulfonyl moiety from the physiological sulfate donor 3′-phosphoadenosine-5′-phosphosulfate to a variety of endogenous and exogenous substrates, including α-hydroxysteroid hormones, β-hydroxysteroids such as the androgen precursor dehydroepiandrosterone, bile acids, benzylic alcohol procarcinogens, and other hormonal and xenobiotic substrates (Falany et al., 1995; Glatt et al., 1995). Sulfonation generally produces stable and relatively polar conjugates that are recognized and eliminated from the cell (detoxication) through the facilitated action of ATP-binding cassette membrane transporters expressed on the canalicular membranes of hepatocytes (Chu et al., 2004). However, the generation of unstable sulfonate esters represents a clear risk factor for mutagenesis (intoxication). For example, recombinant (HUMAN)SULT2A1 is capable of bioactivating hydroxymethyl polycyclic aromatic hydrocarbons (Falany et al., 1995) and benzylic alcohol procarcinogens (Glatt et al., 1995) to reactive DNA-damaging species.
(HUMAN)SULT2A1 is most abundantly expressed in the liver, but it is also expressed in other metabolically active tissues, such as the intestine and adrenal gland (Falany et al., 1995; Chen et al., 2003). The elevated levels of (HUMAN)SULT2A1 in the developing fetal adrenal gland underscore the involvement of (HUMAN)SULT2A1 in early steroidogenesis (Rehman et al., 2003). Unlike the rat, which expresses three known hepatic SULT2A family members (Yamazoe et al., 1994), humans express one major SULT2A enzyme (Falany et al., 1995).
Increasing evidence indicates that the SULT2A enzymes play a pivotal role in the maintenance of hepatic bile acid homeostasis. In concert with abnormalities of sex hormone metabolism, significant aberrations in bile acid hydroxylation and sulfonation occur as clinical hallmarks of pregnancy-associated intrahepatic cholestasis and are believed to contribute to disease progression (Reyes and Sjovall, 2000). In addition to serving as end-products of cholesterol catabolism, bile acids are now appreciated for their important roles in cellular signaling. In the rat, the expression of hepatic “bile acid sulfotransferase” or (RAT)SULT2A3 (Blanchard et al., 2004) is regulated by bile acid-activated members of the nuclear receptor superfamily. Primary bile acids, such as chenodeoxycholic acid and cholic acid, and their respective taurine and glycine conjugates, have been revealed as functional ligands for the hepatic FXR transcription factor (Wang et al., 1999). The hepatic accumulation of the secondary bile acid, LCA, which is formed in the small intestine through microflora-catalyzed 7α-dehydroxylation of chenodeoxycholic acid, can cause cholestasis (Staudinger et al., 2001). However, activation of the PXR by LCA (Staudinger et al., 2001) triggers a defensive adaptation that includes increased SULT2A-mediated sulfonation, which facilitates the elimination of LCA. The transcription of (RAT)SULT2A3 reporter genes can be activated by both the FXR (Song et al., 2001) and the PXR (Runge-Morris et al., 1999; Sonoda et al., 2002) transcription factors, supporting the concept that SULT2A enzyme activity plays an important physiological role in the maintenance of normal hepatic bile acid homeostasis.
The PPARs constitute another major class of nuclear receptors that function in the regulation of hepatic lipid metabolism. Thus, in the hepatocyte, PPARα is well known to regulate the expression of myriad genes involved in fatty acid metabolism. In addition, several recent studies have implicated PPARα as an important regulator of bile acid metabolism. For example, PPARα activation has been reported to cause up-regulation of murine bile acid-CoA thioesterase activity and protein content, with simultaneous suppression of peroxisomal bile acid-CoA:amino acid N-acyltransferase activity, thereby favoring the accumulation of hepatotoxic unconjugated bile acids (Solass et al., 2004). PPARα has also been implicated as a positive regulator of hepatic murine multidrug resistance transporter 2 expression and associated increases in bile flow (Gbaguidi and Agellon, 2004), as well as of human UGT2B4 and murine UGT2B, enzymes that catalyze the glucuronidation and detoxification of bile acids (Barbier et al., 2003).
Therefore, in view of the evidence that both SULT2A and PPARα play important roles in the control of hepatic bile acid homeostasis, the present study was designed to test the hypothesis that the hepatic (HUMAN)SULT2A1 gene is a target gene for regulation by the PPARα transcription factor. The results show that the PPARα activators ciprofibrate and Wy-14,643 induce (HUMAN)SULT2A1 expression in primary cultured human hepatocytes, but they do not exert such an effect in primary cultured rat hepatocytes. The evidence also indicates that PPARα transactivates (HUMAN)SULT2A1 gene transcription via a PPRE located in the distal portion of the 5′-flanking region of the (HUMAN)SULT2A1 gene.
Materials and Methods
Primary Cultured Rat and Human Hepatocytes. Rat hepatocytes (male Sprague-Dawley) were prepared, plated onto 60-mm dishes coated with 1.5 mg of Matrigel and maintained in supplemented Williams' medium E, as described previously (Runge-Morris et al., 1996). Primary cultured human hepatocyte preparations were obtained from the Liver Tissue Procurement and Distribution System of the University of Minnesota, in collaboration with Dr. Stephen Strom (University of Pittsburgh, Pittsburgh, PA) and maintained as described previously (Kocarek et al., 2002).
Northern Blot, Western Blot, and Enzyme Activity Analyses. The cDNA probes for rat SULT2A (Runge-Morris et al., 1996) and (HUMAN)SULT2A1 (Duanmu et al., 2002) were prepared as described previously. Because there is one known human SULT2A family member, the cDNA probe specifically detects (HUMAN)SULT2A1 mRNA content on Northern blots. The rat SULT2A cDNA probe is expected to cross-react with all rat SULT2A mRNAs. A CYP4A1 cDNA, which cross-reacts with multiple rat CYP4A subfamily mRNAs, has been described previously (Shenoy et al., 2004). Total RNA was prepared from primary cultured rat and human hepatocytes using TRIzol reagent (Invitrogen, Carlsbad, CA). Samples of total RNA were fractionated on denaturing agarose gels, transferred onto nylon filters, and hybridized with 32P-labeled cDNA probes (Runge-Morris et al., 1996). To normalize Northern blots for subtle differences in RNA loading and transfer, filters were stripped of radiolabeled probe after autoradiography and rehybridized with a 32P-labeled 7S cDNA probe. The autoradiographs for Northern blots were quantified using a scanning laser densitometer and the ImageQuant software package (Amersham Biosciences Inc., Piscataway, NJ).
For Western blot analysis, human hepatocyte monolayers (2-3 T25s per treatment group) were washed with ice-cold phosphate-buffered saline, scraped (replicates pooled) into Matrisperse (Collaborative Research, Bedford, MA), mixed end-over-end at 4°C for ∼30 min to remove traces of the Matrigel overlay, and centrifuged. The cell pellets were homogenized by sonication in buffer (20 μl per flask of hepatocytes) consisting of 50 mM Tris-HCl, 25 mM sucrose, 1 mM EDTA, and 1× Halt protease inhibitor (Pierce Chemical, Rockford, IL), pH 7.4. Homogenates were centrifuged at 20,000g at 4°C for 20 min, and supernatants were used for Western blot analysis. Enhanced chemiluminescence Western blot analysis of (HUMAN)SULT2A1 immunoreactive protein content was accomplished, essentially as described previously (Duanmu et al., 2002), using 30 μg of sample protein and a commercially available polyclonal antibody directed against (HUMAN)SULT2A1 (PanVera Corp., Madison, WI). Expressed recombinant (HUMAN)SULT2A1 was obtained from Dr. Charles Falany (University of Alabama at Birmingham, Birmingham, AL) and used as a protein standard.
For measurement of cytosolic SULT2A enzymatic activity, human hepatocytes (eight T25s per treatment group) were harvested, as described above. The cell pellets were homogenized by sonication in buffer (50 μl per flask of hepatocytes) consisting of 50 mM Tris-HCl, 10% glycerol, and 1.5 mM dithiothreitol, pH 7.4. Homogenates were centrifuged at 20,000g at 4°C for 20 min, and supernatants were centrifuged at 105,000g at 4°C for 1 h. SULT2A enzyme activities were measured in the cytosolic fractions, as described previously (Falany et al., 1989).
Preparation of (HUMAN)SULT2A1 Reporter Plasmids. A luciferase reporter plasmid containing 1463 nt of the (HUMAN)SULT2A1 5′-flanking region has been described previously (Duanmu et al., 2002). The (HUMAN)SULT2A1 region corresponding to nt -492 to +48 was amplified by PCR, using the aforementioned plasmid as template, and ligated into the MluI and XhoI sites of pGL3-Basic (Promega, Madison, WI), creating the recipient (HUMAN)SULT2A1 promoter-luciferase reporter plasmid that was used for the ligation of additional upstream (HUMAN)SULT2A1 fragments. A series of fragments (∼1000 nt each) of the (HUMAN)SULT2A1 5′-flanking region, extending to ∼8.8 kilobases upstream from the transcription start site, were amplified by PCR, using available sequence information (GenBank NT_011109) and human genomic DNA as template. Primer positions and sequences are provided in Table 1. Amplified fragments were ligated into the KpnI and MluI sites of the (HUMAN)SULT2A1 promoter-luciferase reporter plasmid.
Primer sets for the preparation of (HUMAN)SULT2A1 5′-flanking region-luciferase reporter constructs
The underscored bases of the forward and reverse primers are KpnI and MluI sites, respectively.
Dinucleotide mutations were introduced into two candidate PPRE sequences in the (HUMAN)SULT2A1 gene using the QuikChange II XL site-directed mutagenesis kit, according to the manufacturer's instructions (Stratagene, La Jolla, CA). The mutagenic primer pairs corresponded to nucleotides -5962 to -5911(forward primer, 5′-AAGATAAGTATATGTAAAATAGGTGAACTGTAAGGATCAATCCAGAGGAAG-3; complementary reverse primer; the underscored nucleotides indicate the changes in the candidate PPRE located at nt -5949 to -5929) and -332 to -277 (forward primer, 5′-GATTTTGTGTTTATGCTTGATGAAAAGAGTTGTTCTTGTTTTTAAGTTTGCACTCA-3′; complementary reverse primer; the underscored nucleotides indicate the changes in the candidate PPRE located at nt -291 to -310).
(HUMAN)SULT2A1 promoter-luciferase reporter plasmids containing one, two, or three copies of a candidate PPRE (nt -5949 to -5929) were prepared by PCR, using the oligonucleotide 5′-GATAAGTATATGTAAAATAGGTGAAAGGTAAGGATCAATCCA-3′ as template, a series of forward primers of general sequence GCG-restriction site-GATAAGTATAT (where restriction site is KpnI, EcoRI, or PstI), and a series of reverse primers of the general sequence GCG-Restriction Site-TGGATTGATC (where Restriction Site is MluI, EcoRI, or PstI). For a single copy, PCR was performed using the KpnI-forward primer and the MluI-reverse primer. The product was digested with KpnI and MluI and ligated into the corresponding sites of the (HUMAN)SULT2A1 promoter-luciferase reporter plasmid. For two copies, two PCR reactions were performed, one set using the KpnI-forward primer and the EcoRI-reverse primer, and the second set using the EcoRI-forward primer and the MluI reverse primer. The individual products were digested with EcoRI, ligated together, digested with KpnI and MluI, and ligated into the reporter plasmid. For three copies, three PCR reactions were performed, one set using the KpnI-forward primer and the PstI-reverse primer, the second set using the PstI-forward primer and the EcoRI-reverse primer, and the third set using the EcoRI-forward primer and the MluI-reverse primer. The products were digested and ligated into the reporter plasmid. The sequences of all constructs were verified using the resources of the Applied Genomics Technology Center at Wayne State University.
Transient Transfection Analysis. Approximately 250,000 HepG2 cells were seeded into the wells of 12-well plates and cultured in 2 ml of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Invitrogen), at 37°C under a humidified atmosphere of 95% air, 5% CO2. The following day, culture medium was replaced with 0.6 ml of Opti-MEM (Invitrogen) containing a premixed complex of 6.25 μl of Lipofectin reagent (Invitrogen), 800 ng of a reporter plasmid, 10 ng of mouse PPARα expression plasmid (provided by Dr. Eric Johnson, Scripps Research Institute, La Jolla, CA), 1.25 ng of pRL-SV40 (Promega; to normalize for differences in transfection efficiency), and sufficient pBluescript II KS+ (Stratagene) to adjust the total amount of DNA to 1 μg. A luciferase reporter plasmid containing the peroxisome proliferator-responsive region of the CYP4A1 gene (Kocarek and Mercer-Haines, 2002) was used as a positive control. After 5-h incubation, transfection medium was replaced with fresh, unsupplemented Opti-MEM (i.e., serumfree). The following day, cultures were incubated with Opti-MEM containing either 0.1% DMSO or 10-4 M ciprofibrate and were harvested 24 h later for the measurement of firefly and Renilla reniformis luciferase activities using the dual luciferase reporter assay system (Promega) and a Dynex model MLX Luminometer.
EMSA. EMSA was applied to either HepG2 nuclear extracts, prepared using the nuclear and cytoplasmic extraction reagents (NEPER) kit (Pierce Chemical), or in vitro transcribed and translated PPARα and RXR. The latter were prepared using the TNT T7-coupled reticulocyte lysate system (Promega), mouse PPARα expression plasmid (described above), and human RXR expression plasmid (provided by Dr. Steven Kliewer, University of Texas Southwestern, Dallas, TX). The following complementary oligonucleotide pairs were used as probes: 1) consensus PPRE: 5′-TCCAAAACTAGGTCAAAGGTCATCGATCG-3′ and 5′-TCCGATCGATGACCTTTGACCTAGTTTTG-3′, 2) dPPRE, corresponding to nt -5949 to -5929: 5′-TCGTAAAATAGGTGAAAGGTAAGCGATCG-3′ and 5′-TCCGATCGCTTACCTTTCACCTATTTTAC-3′, 3) mutant dPPRE: 5′-TCGTAAAATAGGTGAACTGTAAGCGATCG-3′ and 5′-TCCGATCGCTTACAGTTCACCTATTTTAC-3′ and 4) pPPRE, corresponding to nt -309 to -291: 5′-TCAAAAACAAGAACAAAGCTTTTCGATCG-3′ and 5′-TCCGATCGAAAAGCTTTGTTCTTGTTTTT-3′. Each an-nealed oligonucleotide pair contained a TC overhang at each 5′ end, and the probes were 3′ end-labeled by filling in with Klenow DNA polymerase and [α-32P]dATP. The HepG2 nuclear extract proteins (2.5 μg) or in vitro transcribed/translated proteins (5 μl) were incubated with 50,000 cpm of a labeled probe for 20 min at room temperature in buffer containing 12 mM HEPES, pH 7.9, 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 12% glycerol, and 2 μg of poly(dI-dC), in a total volume of 15 μl. The DNA-protein complexes were separated on native 4% polyacrylamide gels in 0.5× TBE running buffer, at 30 mA, for 1.5 h. For competition analysis, incubations included various amounts of the unlabeled PPRE double-stranded oligonucleotides. For supershift analysis, 1 μl of an antibody directed against PPARα (either H-98 or N-19; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was added to the binding reaction.
ChIP. ChIP analysis was performed using the ChIP-IT kit, according to the manufacturer's instructions (Active Motif, Carlsbad, CA). HepG2 cells were grown in 150-mm dishes to ∼70-80% confluence in Dulbecco's modified Eagle's medium containing 10% charcoal-stripped fetal bovine serum and were then incubated with Opti-MEM containing either 0.1% DMSO or 10-4 M ciprofibrate for either 0, 2.5, or 24 h. After treatment, medium was changed to fixing medium containing 1% formaldehyde, and the cultures were incubated for 10 min. Chromatin samples (average length ∼500 to 1000 nt) were prepared and processed according to the manufacturer's instructions. Before immunoprecipitation, chromatin samples were precleared, first, using protein G beads only, and second, using normal rabbit IgG (supplied with the ChIP-IT kit) with protein G beads. The precleared chromatin samples were then incubated with one of the following rabbit polyclonal antibodies (all from Santa Cruz Biotechnology, Inc.): anti-PPARα (H-98), anti-RXRα (D-20), anti-CBP (A-22), or anti-PGC-1 (H-300). The normal rabbit IgG supplied with the ChIP-IT kit served as a negative control. PCR was performed using 5 μl of the DNA extracted from each immunoprecipitation reaction and using a primer pair flanking the (HUMAN)SULT2A1 distal PPRE: forward, 5′-TTAGGGGCTGGAGTGATGTTA-3′ (-6026 to -6006); reverse, 5′-TCCTCCCTTTGATGCATGT-3′ (-5894 to -5876). A primer pair corresponding to a more upstream region of the (HUMAN)SULT2A1 5′-flanking region was used as a negative control: forward, 5′-GGTGCTAGCGCTCAAGG-3′ (-11,748 to -11,732); reverse, 5′-AGCCAAGAAACCGCAGTT-3′ (-11,588 to -11,605). PCR amplifications were performed for 33 to 36 cycles, and products were visualized on ethidium bromide-stained 2.5% agarose gels.
Results
To determine the relative roles of liver-enriched nuclear receptor transcription factors in the regulation of (HUMAN)SULT2A1 gene transcription, primary cultured human hepatocytes were treated with a panel of known nuclear receptor-activating agents (Fig. 1A). Consistent with our previous findings (Duanmu et al., 2002), induction of (HUMAN)SULT2A1 mRNA content occurred after treatment with a PXR-activating concentration of rifampin (Fig. 1A). Treatment with a PPARα-activating concentration of ciprofibrate (10-4 M) also substantially increased (HUMAN)SULT2A1 mRNA content, by ∼2-fold, relative to DMSO-treated control (Fig. 1A). Treatment of human hepatocytes with ciprofibrate or Wy-14,643 (10-4 M), another peroxisome proliferating drug, also induced the amounts of (HUMAN)SULT2A1 immunoreactive protein (Fig. 1B) and enzymatic activity (Fig. 1C). By contrast, test activators of the FXR (chenodeoxycholic acid), liver X receptor [24(S), 25-epoxycholesterol] or constitutive androstane receptor (phenobarbital) nuclear receptors failed to produce appreciable changes in hepatocellular (HUMAN)SULT2A1 mRNA content (Fig. 1A). As demonstrated previously (Runge-Morris et al., 1999), expression of the rat counterparts to (HUMAN)SULT2A1 was induced in primary cultured rat hepatocytes after treatment with a PXR-activating concentration of dexamethasone (Fig. 1D). However, although ciprofibrate and Wy-14,643 treatments markedly increased the levels of CYP4A mRNA, a known PPARα target in the rat, these treatments failed to produce detectable increases in SULT2A mRNA content (Fig. 1D).
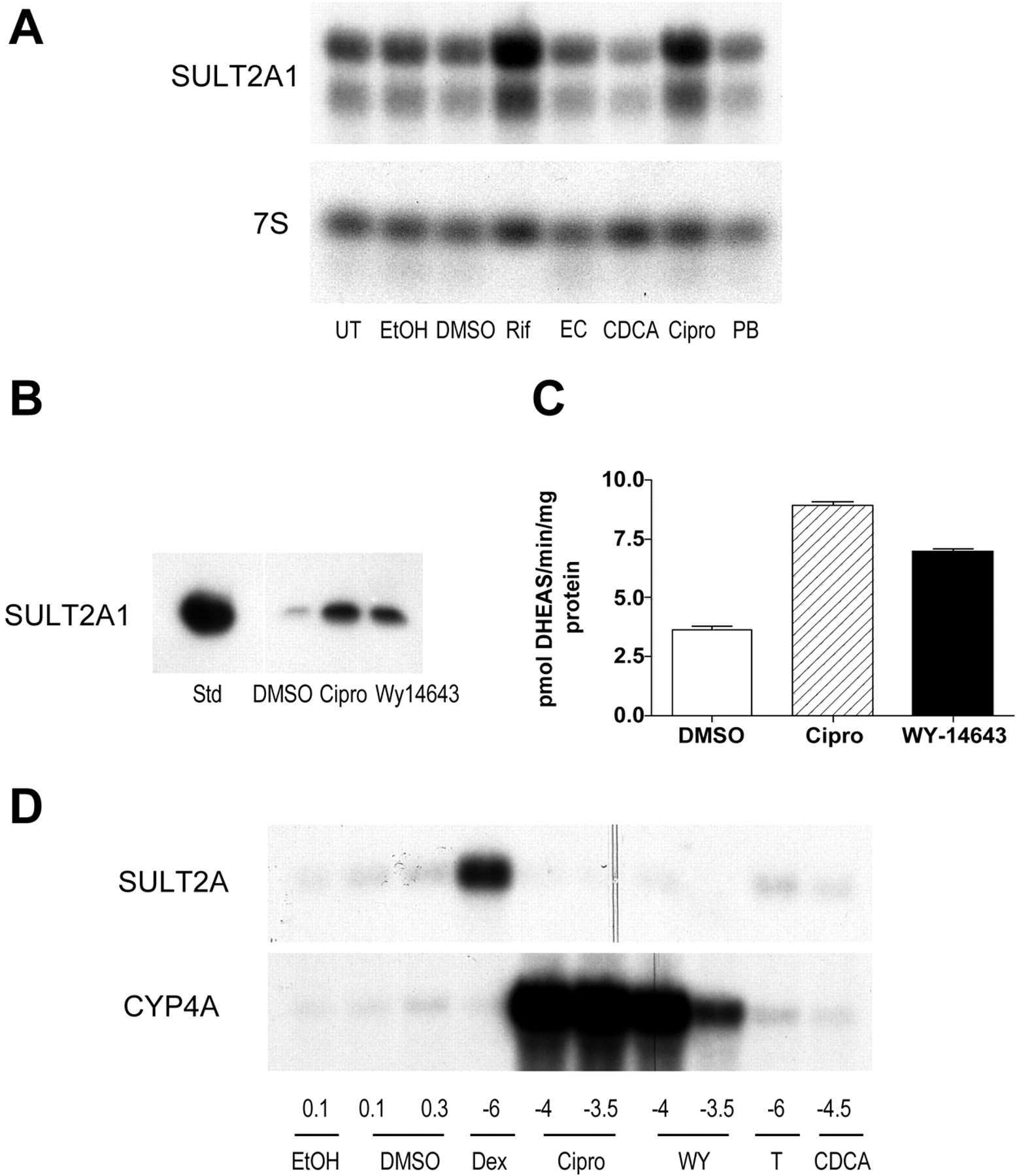
Effects of peroxisome proliferator treatments on SULT2A1 expression in primary cultured hepatocytes. A, Northern blot analysis of (HUMAN)SULT2A1 mRNA content in primary cultured human hepatocytes. Human hepatocyte cultures were incubated for 48 h with either medium alone (UT) or with medium containing 0.1% ethanol (EtOH), 0.1% DMSO, 5 × 10-5 M rifampin (Rif), 5 × 10-5 M 24(S),25-epoxycholesterol (EC), 5 × 10-5 M chenodeoxycholic acid (CDCA), 10-4 M ciprofibrate (Cipro), or 10-4 M phenobarbital (PB). After treatment, hepatocytes were harvested for the preparation of total RNA, and levels of (HUMAN)SULT2A1 mRNA and 7S RNA were measured by northern blot hybridization. Two of the three (HUMAN)SULT2A1 mRNA species that have been described previously were detected on the Northern blot. B, Western blot analysis of (HUMAN)SULT2A1 immunoreactive protein content in primary cultured human hepatocytes. Human hepatocyte cultures were incubated for 48 h with medium containing 0.1% DMSO, 10-4 M ciprofibrate (Cipro) or 10-4 M Wy-14,643. After treatment, hepatocytes were harvested for the preparation of postmitochondrial supernatant fractions, and levels of (HUMAN)SULT2A1 immunoreactive protein were measured by Western blot hybridization. For comparison, immunoreactivity to an expressed recombinant (HUMAN)SULT2A1 standard (Std) is shown. C, effect of 48-h treatment with 0.1% DMSO, 10-4 M ciprofibrate (Cipro) or 10-4 M Wy-14,643 on SULT2A enzymatic activity, as measured by the conversion of dehydroepiandrosterone to dehydroepiandrosterone sulfate (DHEAS), in cytosolic fractions prepared from primary cultured human hepatocytes. Each column represents the mean ± S.D. of triplicate determinations. D, Northern blot analysis of SULT2A mRNA content in primary cultured rat hepatocytes. Forty-eight-hour-old cultures of primary rat hepatocytes were incubated for 24 h with medium containing EtOH, DMSO, dexamethasone (Dex), Cipro, Wy-14,643 (WY), T0901317 (T), or CDCA, at the indicated concentrations (volume percentages are indicated for EtOH and DMSO; log molar concentrations are indicated for the other treatments). After treatment, hepatocytes were harvested for the preparation of total RNA, and levels of SULT2A and CYP4A mRNAs were measured by Northern blot hybridization. All data are representative of results observed in two independent human or rat hepatocyte culture experiments.
The mechanism underlying ciprofibrate-inducible regulation of the (HUMAN)SULT2A1 gene was evaluated by performing transient transfection studies in HepG2 cells with a series of luciferase reporter plasmids, which contained progressively more upstream ∼1-kilobase regions ligated in cis with the first 492 nt of the (HUMAN)SULT2A1 promoter (Fig. 2A). As a reference parameter for these studies, a PPARα-responsive rat CYP4A1 luciferase reporter plasmid was used as a positive control. For these studies, the HepG2 cells were cotransfected with a PPARα expression plasmid. After transfection, the cells were treated for 24 h with DMSO or 10-4 M ciprofibrate. As shown in Fig. 2A, ciprofibrate treatment caused an ∼1.7-fold increase in CYP4A1-PPRE-driven luciferase expression, relative to DMSO-treated control. A similar ciprofibrate-mediated increase in reporter activity (∼2.0-fold) was observed for cells transfected with the construct containing nt -6832 to -5812 (-6832:5812) of the (HUMAN)SULT2A1 5′-flanking region (Fig. 2A), suggesting that this portion of the (HUMAN)SULT2A1 5′-flanking region may harbor a functional PPRE. A ciprofibrate-mediated increase in luciferase activity (∼1.7-fold) was also demonstrated for the -7626:6626 fragment (Fig. 2A). We also noted that cells transfected with plasmids containing these ciprofibrate-responsive regions had higher basal luciferase activities (i.e., DMSO-treated activities) than did those transfected with reporters that did not display ciprofibrate responsiveness. Additional experiments confirmed that transfection of HepG2 cells with the PPARα expression plasmid alone (without ciprofibrate treatment) was sufficient to produce some increase in reporter activity in ciprofibrate-responsive transfectants (data not shown). Such an effect has been documented previously (Barrero et al., 2003).

Identification of a peroxisome proliferator-responsive region in the 5′-flanking region of (HUMAN)SULT2A1. A and B, HepG2 cells were transiently cotransfected with plasmid expressing mouse PPARα and each of the indicated reporter plasmids. Transfected cells were treated for 24 h with 0.1% DMSO or 10-4 M ciprofibrate (Cipro) and harvested for measurement of luciferase activities. For comparison, effects on expression from a peroxisome proliferator-responsive CYP4A1 luciferase reporter construct are shown. Each bar represents the mean ± S.D. of normalized (firefly/R. reniformis) luciferase measurements (three wells per treatment group).
Because our initial transfection studies primarily implicated information contained within the -6832:5812 fragment as responsible for mediating ciprofibrate-inducible expression, but secondarily suggested some involvement by sequences located within the -7626:6626 fragment, subsequent studies were focused on the entire -7624:5817 region (Fig. 2B). In addition, we noted that an additional construct designed to contain the overlap region between constructs -5825:4807 and -6832:5812 (construct -6042:5542) displayed elevated activity after transfection with PPARα, which was further enhanced by ciprofibrate treatment (Fig. 2B). These findings suggested that the region common to constructs -6832:5812 and -6042:5542 (indicated by dotted lines) probably contained a ciprofibrate-responsive element. Systematic deletion of the overlap region from the -7624: 5817 construct indicated that a marked loss of luciferase expression occurred when nt -5956 through -5905 were removed (Fig. 2B). A computer-based analysis (MatInspector; Quandt et al., 1995) of the -5956:5905 region identified a sequence, at nt -5949 to -5929, with significant similarity (100% core; 83.3% overall) to the peroxisome proliferator responsive element matrix (V&PERO/PPARA.01). The sequence of this candidate (HUMAN)SULT2A1 PPRE (termed dPPRE for distal PPRE) differed from consensus by 4 nt, two of which were contained within the direct repeat of AGG-TCA with one intervening nt (DR1) portion of the motif (Fig. 3A). Evaluation of the 492 nt proximal region of the (HUMAN)SULT2A1 promoter identified a second sequence, at -291 to -310, with significant similarity (100% core; 71.2% overall) to the PPRE matrix, although this sequence (termed pPPRE for proximal PPRE) contained four mismatches with the consensus PPRE within the DR1 region (Fig. 3A). To evaluate the importance of the dPPRE in mediating ciprofibrate-inducible (HUMAN)SULT2A1 transcription, the effect of introducing a dinucleotide mutation (AG→CT) into the core region of this element (in the downstream hexameric half-site of the DR1 motif) was evaluated in transient transfections. For comparison, a comparable mutation was introduced into the pPPRE. In addition, both mutations were evaluated within the contexts of both the -6832:5812 fragment and the longer -7624:5817 fragment. As shown in Fig. 3B, mutation of the dPPRE largely abolished PPARα transfection-mediated luciferase expression, as well as the ciprofibrate-induced enhancement, within the contexts of both reporter constructs. By contrast, mutation of the pPPRE had no effect on ciprofibrate/PPARα-inducible reporter expression, and when introduced in combination with the dPPRE mutation, did not modify the suppression that was attributable to the dPPRE (Fig. 3B), indicating that the pPPRE is not a functional PPRE. To establish whether the (HUMAN)SULT2A1 dPPRE functions as an enhancer element, the effects of PPARα activation were examined in (HUMAN)SULT2A1 reporter plasmids containing either one, two, or three concatemerized copies of the dPPRE (Fig. 3C). The results demonstrated that increasing the copy number of the dPPRE in the (HUMAN)SULT2A1 promoter-luciferase reporter plasmid produced a dose-dependent increase in ciprofibrate/PPARα-inducible (HUMAN)SULT2A1 luciferase activity (Fig. 3C).
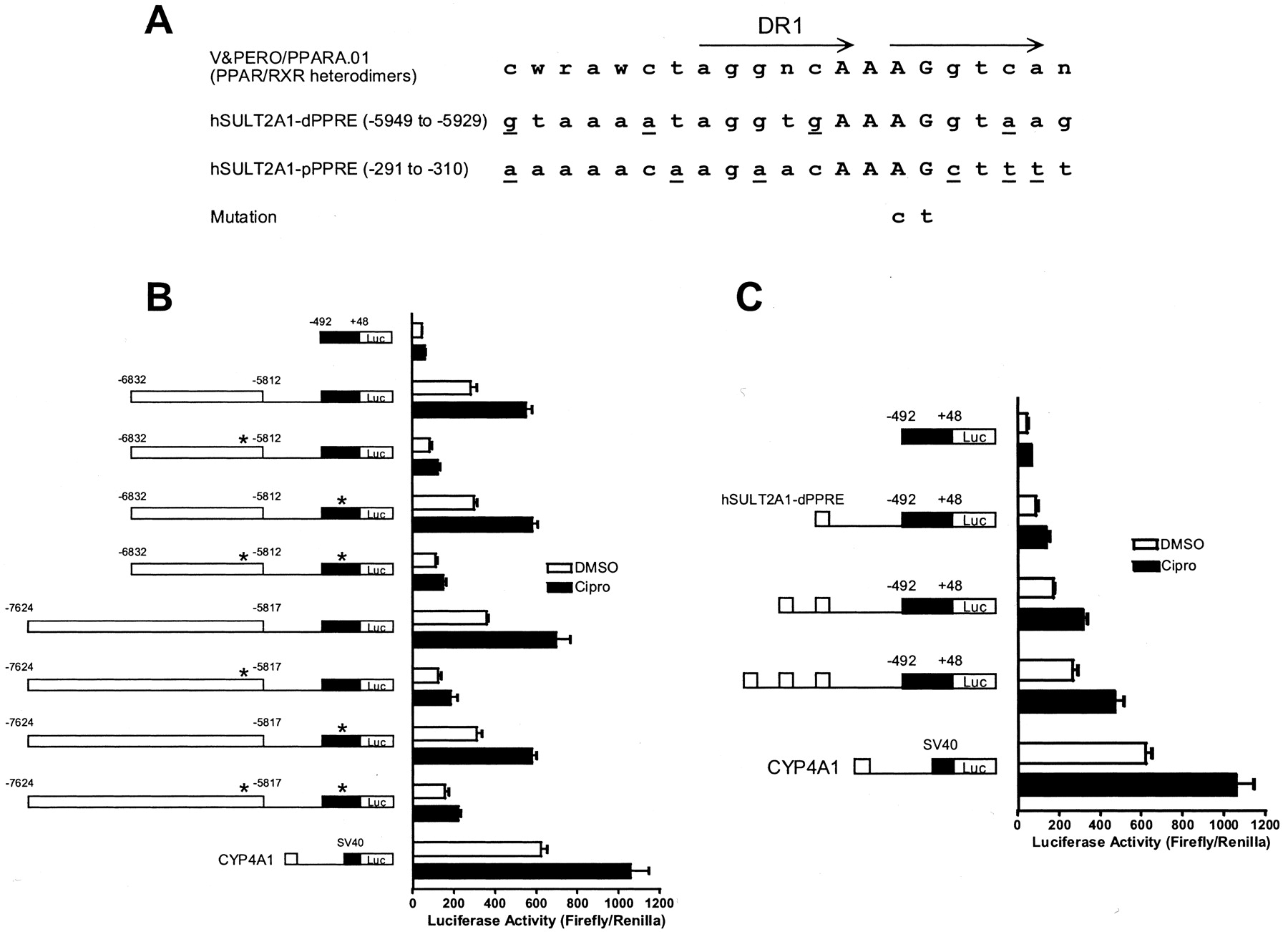
Identification of a functional distal PPRE in the 5′-flanking region of (HUMAN)SULT2A1. A, sequence of the consensus PPRE (i.e., TRANSFAC matrix V&PERO/PPARA.01) is indicated, as are the sequences and locations of two candidate PPREs contained within the (HUMAN)SULT2A1 5′-flanking region. The more upstream (i.e., distal) PPRE is designated hSULT2A-dPPRE and the more downstream (i.e., proximal) PPRE is designated hSULT2A-pPPRE. Core regions of the PPREs are capitalized, and SULT2A PPRE nucleotides differing from consensus are underlined. The location of the DR-1 motif is marked with arrows. Nucleotide changes introduced by site-directed mutagenesis are indicated. B, candidate distal and proximal PPREs were mutated within the contexts of two different (HUMAN)SULT2A1 5′-flanking region constructs. Mutations are indicated with asterisks. HepG2 cells were transiently cotransfected with plasmid expressing mouse PPARα and the indicated reporter plasmids. Transfected cells were treated for 24 h with 0.1% DMSO or 10-4 M ciprofibrate (Cipro) and harvested for measurement of luciferase activity. For comparison, effects on expression from a peroxisome proliferator-responsive CYP4A1 luciferase reporter construct are shown. Each column represents the mean ± S.D. of normalized (firefly/R. reniformis) luciferase measurements (three wells per treatment group). C, one, two, or three copies of the (HUMAN)SULT2A1 distal PPRE were ligated upstream of the 492 nt (HUMAN)SULT2A1 promoter fragment. HepG2 cells were transiently cotransfected and analyzed, as described above.
As further support that the (HUMAN)SULT2A1 dPPRE represents a functional PPRE, EMSA analysis of HepG2 nuclear extracts demonstrated that the dPPRE competed for protein binding to a consensus PPRE probe, albeit with slightly less affinity than did the consensus competitor (Fig. 4A). As expected, a dPPRE competitor containing the same dinucleotide mutation that abolished ciprofibrate/PPARα responsiveness in the transfection assays, failed to compete for binding to the PPRE probe (Fig. 4A). These results were reenforced by EMSA studies using in vitro transcribed/translated PPARα and RXR. Thus, the PPARα-RXR heterodimer bound directly to both the consensus PPRE probe and to a dPPRE probe, as indicated by the emergence of a more slowly migrating band, but not to the mutated dPPRE probe (Fig. 4B). The presence of PPARα in this more slowly migrating band was confirmed by coincubation of EMSA binding reactions with either of two commercially available antibodies directed against the N terminus of PPARα. Thus, antibody H-98 reduced the intensity of the shifted band and caused the clear appearance of a supershifted band (Fig. 4C, antibody 1), whereas antibody N-19 primarily caused the retardation and broadening of the shifted band, with the slight appearance of a supershifted band (Fig. 4C, antibody 2). As seen for the HepG2 nuclear extract EMSAs, the (HUMAN)SULT2A1 dPPRE competed for the binding of consensus PPRE probe to in vitro transcribed/translated PPARα-RXR with slightly less affinity than did the consensus PPRE, whereas the mutant dPPRE did not compete at all (Fig. 4C). Also consistent with our functional data, in vitro transcribed/translated PPARα-RXR did not bind to a probe corresponding to the pPPRE (data not shown).

EMSA analysis of protein binding to (HUMAN)SULT2A1 PPREs. A, in vitro binding reactions contained 32P-labeled consensus PPRE double-stranded oligonucleotide probe without (-) or with (+) HepG2 nuclear extract. For competition experiments, reactions also contained the indicated amounts of unlabeled oligonucleotide corresponding to consensus PPRE, the (HUMAN)SULT2A1 distal PPRE, or the (HUMAN)SULT2A1 distal PPRE containing the mutations indicated in Fig. 3A (Mut). B, binding reactions contained 32P-labeled double-stranded oligonucleotide probe corresponding to consensus PPRE, the (HUMAN)SULT2A1 distal PPRE, or the (HUMAN)SULT2A1 distal PPRE containing the mutations indicated in Fig. 3A. The indicated reactions also contained in vitro transcribed/translated mouse PPARα, human RXR, or PPARα and RXR in combination. For comparison, reactions using the consensus PPRE probe also were performed without lysate (first lane) and containing HepG2 nuclear extract (NE). Positions of PPARα-RXR-shifted bands are indicated. C, binding reactions contained 32P-labeled consensus PPRE double-stranded oligonucleotide probe without (-) or with (+) in vitro transcribed/translated mouse PPARα and human RXR. For competition experiments, reactions also contained the indicated amounts of unlabeled oligonucleotide corresponding to consensus PPRE, the (HUMAN)SULT2A1 distal PPRE, or the (HUMAN)SULT2A1 distal PPRE containing the mutations indicated in Fig. 3A (Mut). For supershift experiments, reactions were supplemented with either of two antibodies (designed 1 and 2) directed against PPARα. Positions of PPARα-RXR-shifted bands and supershifted bands are indicated.
To determine whether the dPPRE is a functional element within the context of the endogenous (HUMAN)SULT2A1 gene, a ChIP experiment was performed to investigate the association of that element with PPARα and RXR, as well as with PGC-1 and CBP, two coactivators that are expected to be associated with a functional PPRE after receptor activation (Mukherjee et al., 2002). In this experiment, HepG2 cells were treated with DMSO or 10-4 M ciprofibrate for either 24 h (according to our treatment regimen in the transfection experiments), or for a shorter time, 2.5 h, based on the knowledge that cofactor association can occur rapidly after ligand treatment, and is a highly dynamic process, in which complexes are continuously assembled and disassembled (Shang et al., 2000). Immunoprecipitation with antibody directed against each of the aforementioned proteins, but not nonspecific IgG, resulted in the amplification of a clear PCR product when primers flanking the dPPRE were used, but no product when primers targeting a further upstream region of the (HUMAN)SULT2A1 gene were used (Fig. 5). The ChIP analysis indicated that PPARα and RXR were associated with the dPPRE in the absence or presence of ciprofibrate treatment, as would be expected for this heterodimeric transcription factor, which is known to be nuclear and associated with PPRE motifs independent of ligand treatment (Dowell et al., 1999). Although these cultures were not transfected with supplemental PPARα, association was clearly evident, consistent with our ability to detect PPARα-RXR in untransfected HepG2 nuclear extracts by EMSA (Fig. 4A). Ciprofibrate treatment, for either 2.5 or 24 h, did not increase the amount of PPARα or RXR that was associated with the dPPRE. For the coactivators, ciprofobrate treatment of HepG2 cells for 24 h resulted in detectably greater association of CBP and PGC-1 with the dPPRE than was evident in the corresponding DMSO-treated controls, whereas such a difference was not evident in the 2.5-h treated cultures (Fig. 5). These results indicate that the (HUMAN)SULT2A1 dPPRE serves as a functional response element within the context of the human genome.

Chromatin immunoprecipitation analysis of transcription factor and coactivator association with the (HUMAN)SULT2A1 dPPRE in HepG2 cells. HepG2 cells were incubated with Opti-MEM containing 0.1% DMSO (-) or 10-4 ciprofibrate (+) for either 0, 2.5, or 24 h and were fixed and harvested for the preparation of sheared chromatin. Immunoprecipitations were performed using an antibody directed against PPARα, RXR, CBP, or PGC-1, or with nonspecific IgG, as a negative control. After immunoprecipitation, associated DNA was amplified with a primer pair flanking the (HUMAN)SULT2A1 dPPRE, or with a negative control primer pair. Ethidium bromide-stained agarose gels of the PCR products are shown. For comparison, the amplifications derived from unprecipitated chromatin are also shown (input).
Discussion
Relative to what is known about (RAT)SULT2A3 regulation, the molecular mechanisms that govern the expression of hepatic (HUMAN)SULT2A1 are less well defined. Transient transfection analyses suggest that the rat and human SULT2A genes share certain aspects of regulation in response to bile acid-sensitive nuclear receptor transcription factors. For example, in primary cultured rat hepatocytes, reporter constructs of both the rat and human SULT2A genes display inducible transcription after treatment with activators of PXR (Runge-Morris et al., 1999; Duanmu et al., 2002). In addition, the loss of PXR expression in mice abolishes the inducible expression of mouse hepatic SULT2A after treatment with the PXR ligand pregnenolone-16α-carbonitrile (Sonoda et al., 2002). Likewise, transient cotransfection assays in HepG2 cells and 3T3 fibroblasts implicate a role for FXR in the transcriptional regulation of rat SULT2A (Song et al., 2001). The present study demonstrates that treatment of primary cultured human hepatocytes with a prototypical activator of PPARα (i.e., ciprofibrate or Wy-14,643) induces the mRNA and protein levels, as well as the enzymatic activity, of (HUMAN)SULT2A1. Subsequent transfection analyses demonstrated that a PPRE in the upstream (HUMAN)SULT2A1 5′-flanking region was responsible for PPARα-inducible (HUMAN)SULT2A1 gene transcription. In support of these findings, binding studies indicated that the PPARα-RXR heterodimer binds to the distal PPRE, although somewhat less avidly than to a consensus PPRE. By contrast, functional and EMSA analyses indicated that a more proximal sequence with significant similarity to a consensus PPRE in the (HUMAN)SULT2A1 5′-flanking region binds weakly, if at all, to PPARα.
In rodents, activation of PPARα produces peroxisome proliferation together with a cascade of cellular events, including excessive hydrogen peroxide generation, that ultimately lead to hepatocarcinogenesis (Rao and Reddy, 1996). In contrast, activation of PPARα in humans is not a potent inducer of peroxisome proliferation, nor is it a significant risk factor for the development of hepatocellular carcinoma (Frick et al., 1987). Recent studies in “humanized” transgenic mice that have been specifically engineered to express the human PPARα in liver have clearly demonstrated that both wild-type and humanized PPARα mice respond similarly to treatment with PPARα activators and demonstrate analogous increases in peroxisome proliferation and fatty acid metabolizing enzyme gene expression (Cheung et al., 2004). However, unlike wild-type mice, PPARα humanized mice activate hepatocellular proliferation pathways with a markedly reduced efficiency, suggesting that the oxidant stress produced by peroxisome proliferators requires cell proliferation to tip the balance of events toward hepatocarcinogenesis (Cheung et al., 2004).
Insights into the physiological function of human PPARα suggest that it plays a role in the regulation of compensatory insulin secretion in the pancreas and represents a central coordinator of hepatic lipid signaling in the liver (Sugden and Holness, 2004). The induction of the bile acid-conjugating enzyme UGT2B4 by PPARα ligands in HepG2 cells was found to be transactivated through a PPRE located in the 5′-flanking region of the human UGT2B4 gene (Barbier et al., 2003). Hepatic SULT2A1 enzyme activity is essential for the detoxication of deleterious bile acids, such as the hydrophobic bile acid LCA, which is subject to conjugation by SULT2A1 (Staudinger et al., 2001). Likewise, UGT2B4 and UGT2B7 in the human intestine efficiently catalyze the glucuronidation of hyodeoxycholic acid (Barbier et al., 2003; Strassburg et al., 2000). Given that both the UGT2B4 and (HUMAN)SULT2A1 enzymes are actively engaged as bile acid detoxicating enzymes in human liver and gastrointestinal tissues, it is reasonable to hypothesize that the genes that encode these enzymes share some common aspects of regulation.
There seems to be a dynamic exchange in communication that occurs between the regulatory matrices that control various branches of lipid metabolism. Bile acids, the endproducts of cholesterol metabolism, represent natural ligands for the FXR transcription factor, which in turn regulates the expression of cholesterol and bile acid biosynthesis enzymes (Wang et al., 1999). The targeted disruption of FXR in mice produced elevations not only in bile acid concentrations, but also in cholesterol and triglyceride levels (Sinal et al., 2000). The PPARα transcription factor potentiates fatty acid catabolism (Lee et al., 2003), and in the liver where both the FXR and PPARα receptors are abundantly expressed, there is considerable opportunity for regulatory overlap. For example, deoxycholic acid induces the transcription of the human PPARα gene via an FXR-mediated mechanism (Torra et al., 2003). In turn, the PPARα transcription factor mediates the transcriptional repression of cholesterol 7α-hydroxylase (CYP7A1) and sterol 27-hydroxylase (CYP27), key enzymes in bile acid biosynthesis (Post et al., 2001). (RAT)SULT2A3 gene transcription has been shown to be inducible by the FXR in cell-based assays (Song et al., 2001). However, our present work in primary cultured human hepatocytes (Fig. 1A; lack of chenodeoxycholic acid-inducible expression) suggests that this mode of regulation by FXR may not be conserved in (HUMAN)SULT2A1.
A recent report on the effects of clofibric acid treatment on global gene expression in primary cultured rat, mouse, and human hepatocytes, determined that the expression of the HNF1α transcription factor was singularly increased in human but not rodent hepatocytes (Richert et al., 2003). Although this suggests that certain genes in human hepatocytes may be regulated by peroxisome proliferator treatment through indirect effects on intermediary transcription factors, transient transfection, EMSA and ChIP analyses in the current study indicate that (HUMAN)SUTL2A1 is a direct target for regulation by the PPARα transcription factor in cultured human hepatocytes.
Our assessment of coactivator recruitment to the distal PPRE in SULT2A1 suggests that ligand-activated PPARα mediates SULT2A1 induction through a mechanism that includes the recruitment of CBP and PGC-1. Some association of both CBP and PGC-1 with the dPPRE was observed in untreated HepG2 cells. For PGC-1, this finding is consistent with a previous report that the in vitro binding of PGC-1 to PPARα occurs in a ligand-influenced manner, with some interaction occurring in the absence of ligand, which is then increased in the presence of ligand (Vega et al., 2000). In addition, PGC-1, through its N-terminal region, has been reported to interact with the CBP/p300 coactivator (Wallberg et al., 2003), providing a possible mechanism for the ligand-independent association of CBP with the dPPRE.
In summary, the present investigation demonstrates that ligand-activated PPARα transcription factor transactivates (HUMAN)SULT2A1 gene transcription through a cis-acting PPRE located in the distal portion of the 5′-flanking region of the SULT2A1 gene. The results suggest that, like UGT2B4, the (HUMAN)SULT2A1 gene represents an important linchpin in the control of lipid homeostasis in the human liver.
Acknowledgments
We thank Amy Weckle for dedicated technical assistance.
Footnotes
-
This work was conducted with support from National Institutes of Health grants ES05823 (to M.R.-M.), HL50710 (to T.A.K.), and DK92310 (Liver Tissue Procurement and Distribution System, to S.C.S.), and services provided by the Cell Culture Facility Core and the Imaging and Cytometry Facility Core of Environmental Health Sciences Center grant ES06639.
-
ABBREVIATIONS: SULT, sulfotransferase; FXR, farnesoid X receptor; LCA, lithocholic acid; PXR, pregnane X receptor; PPAR, peroxisome proliferator-activated receptor; UGT, UDP-glucuronosyltransferase; Wy-14,643, pirinixic acid; 4-chloro-6-(2,3-xylidino)-2-pyrimidinyl)thioacetic acid; nt, nucleotide; PCR, polymerase chain reaction; PPRE, peroxisome proliferator response element; dPPRE, distal peroxisome proliferator response element; DMSO, dimethyl sulfoxide; EMSA, electrophoretic mobility shift assay; RXR, retinoic acid receptor; ChIP, chromatin immunoprecipitation; PGC-1, peroxisome proliferator-γ coactivator-1; CBP, cAMP response element-binding protein-binding protein.
- Received July 26, 2004.
- Accepted January 5, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}