


Abstract
The mitogen-activated protein kinase (MAPK) cascade is stimulated by both receptor tyrosine kinases and G protein-coupled receptors. We show that recombinant human dopamine D3 receptors expressed in Chinese hamster ovary cells transiently activate MAPK via pertussis toxin-sensitive Gi and/or Go proteins. The involvement of D3 receptors was confirmed by use of the D3agonists PD 128,907 and (+)-7-hydroxy-2-dipropylaminotetralin, which mimicked the response to dopamine (DA). Furthermore, haloperidol and the selective D3 receptor antagonists S 14297 and GR 218,231 attenuated DA-induced MAPK activation; however, when tested alone, S 14297 weakly stimulated MAPK activity, suggesting partial agonist activity. The transduction mechanisms by which hD3receptors activate MAPK were explored with specific kinase inhibitors. Genistein and lavendustin A, inhibitors of tyrosine kinase activity, did not reduce DA-induced MAPK activation. In contrast, PD 98059, an inhibitor of MAPK kinase, and Ro 31–8220 and Gö 6983, inhibitors of protein kinase C (PKC), blocked DA-induced MAPK activation. However, MAPK activation was insensitive to PKC down-regulation by phorbol esters, indicating the involvement of an “atypical” PKC. Furthermore, MAPK activation involved phosphatidylinositol 3-kinase inasmuch as its inhibition by LY 294002 and wortmannin reduced DA-induced MAPK activation. In conclusion, this study demonstrates that stimulation of hD3 receptors activates MAPK. This action is mediated via an atypical isoform of PKC, possibly involving cross-talk with products of phosphatidylinositol 3-kinase activation.
Extracellular signal-regulated kinases ERK1 and ERK2, also known as mitogen-activated protein kinases (MAPK), are involved in the control of cell growth and differentiation by growth factors (Schlessinger and Ullrich, 1992). Recently, various G protein-coupled receptors (GPCRs) have also been shown to stimulate MAPK, although activation cascades differ according to receptor subtype and G protein family (Lopez-Ilasaca, 1998).
Intracellular mechanisms leading to MAPK activation by growth factor receptors are now well defined, and mainly involve Ras GTP-binding protein activation via SH2 and SH3 domain adaptor proteins and subsequent Raf/MAPK kinase activation (Schlessinger and Ullrich, 1992;Pawson, 1995). In contrast, the nature of G protein coupling to MAPK activation is less well characterized, although both α and βγ subunits of G proteins are implicated. It has, thus, been shown that Gβγ subunits stimulate Ras protein via Src-like proteins, possibly involving other intermediates, such as phosphatidylinositol 3-kinase (PI 3-kinase), and subsequent tyrosine phosphorylation of Shc and recruitment of the Grb2-Sos complex (Faure et al., 1994, van Biesen et al. 1995; Hawes et al., 1995; Igishi and Gutkind, 1998). As concerns αo and αq subunits, their potential involvement in MAPK activation remains unclear, but may involve protein kinase C (PKC) and Pyk2 activation, respectively (van Biesen et al. 1995; Dikic and al., 1996; Igishi et al., 1998; Berts et al., 1999).
The dopamine D2-like receptor family includes D2, D3, and D4 receptors. Although they all inhibit adenylyl cyclase, their respective transduction mechanisms differ and those of dopamine D3 receptors are still poorly understood (Robinson and Caron, 1997; Watts and Neve, 1997). D3 receptors display marked sequence homology with D2 receptors and pharmacological similarity in their in vitro ligand-binding profiles (Levant, 1997; Missale et al., 1998). However, we have shown recently that D3 receptors activate pertussis sensitive Gi/Go proteins less effectively than D2 receptors and may couple to G protein subtypes different than those of D2 receptors, including Gq/11 proteins (Newman-Tancredi et al., 1999), suggesting that the intracellular activation cascades engaged by D3 versus D2 receptors may differ. Indeed, D2 receptors expressed in C6 glioma cells stimulate MAPK and thymidine incorporation via the Ras protein (Luo et al., 1998) and, when expressed in Chinese hamster ovary (CHO) cells, D2 receptors activate MAPK via PI-3 kinase (Welsh et al., 1998). D4 receptors expressed in SK-N-MC human neuroblastoma cells stimulate a pathway involving Ras activation via Shc/Grb2/Sos complex and the tyrosine kinase Src (Zhen et al., 1998). In contrast, little comparative information is available concerning D3 receptors, although they stimulate expression of the immediate early gene c-fos in cultured neurons (Pilon et al., 1994) and mediate stimulation of mitogenesis (Pilon et al., 1994; Sautel et al., 1995). These data suggest that D3 receptors may couple to serine/threonine kinase pathways and activate MAPK, but the demonstration of such coupling has not, as yet, been documented. To investigate whether D3 receptors couple to p42/p44MAPK, we examined the effect of D3 ligands on the phosphorylation state of p42/p44MAPK in CHO cells expressing human D3 (hD3) receptors. In addition, to elucidate the signal transduction pathways involved, we used specific inhibitors of key factors potentially involved in the MAPK stimulation pathway.
Materials and Methods
Cell Culture and Cellular Extract Preparations.
CHO cells expressing hD3 receptors were grown as previously described (Newman-Tancredi et al., 1999). For MAPK determinations, cells were grown in 6-well plates until 90% confluent. The cells were then washed once with serum-free medium and incubated overnight in this medium. Drugs were diluted in the serum-free medium and added to cells to obtain the appropriate final concentration. Cells were preincubated 5 min with antagonists at indicated concentrations and then stimulated with either dopamine (DA) (100 nM) or fibroblast growth factor (FGF) (20 ng/ml) for 5 min. Kinase inhibitors [wortmannin; [2-(4-morpholinyl)-8-phenyl-4H-1-benzopiran-4-one] (LY 294002); 3-[1-[3-(amidinothio)propyl-1H-indol-3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide (Ro 31–8220); 12-(2-cyanoethyl)-6,7,12,13,- tetrahydro-13-methyl-5-oxo-5H-indolo[2,3-a]pyrrolo[3,4-c]-carbazol (Gö6976); 3-[1-(3-dimethylamino-propyl)-5-methoxy-1H-indol-3-yl] 4-(1H-indol-3-yl)pyrrolidine-2,5-dione (Gö 6983); 4′,5,7-trihydroxyisoflavone (genistein); 5-amino-[(N-2,5-dihydroxybenzyl)-N′-2-hydroxybenzyl]salicylic acid (lavendustin A); and 2′-amino-3′-methoxyflavone (PD 98059) purchased from France Biochem, Meudon, France] were preincubated 30 min with cells at indicated concentrations before adding DA (100 nM) for 5 min or phorbol-12-myristate-13-acetate (PMA) (1 μM) for 30 min. Desensitization of PKC by PMA was achieved by overnight treatment of cells by PMA at 1 μM. At the end of the incubation period, 0.25 ml/well of Laemmi sample buffer containing 200 mM dithiotreitol was added. Whole-cell lysates were boiled for 3 min at 95°C. In experiments with pertussis toxin (PTX), cells were treated overnight in serum-free medium with a concentration of 100 ng/ml PTX.
Immunoblotting.
Cell extract (14 μl) was loaded on 15-well 10% polyacrylamide gels and “fully” activated MAPK was revealed with a monoclonal antibody specifically raised against the phosphorylated pp42mapk (ERK 2) and pp44mapk (ERK 1) forms on both threonine and tyrosine residues (NanoTools, Denzlingen, Germany), followed by enhanced chemiluminescence (ECL) detection with horseradish peroxidase as a secondary antibody (Amersham Corp., les Ulis, France). Total MAPK immunoreactivity was determined with antibody raised against unphosphorylated and phosphorylated forms of p42mapk and p44mapk (Santa Cruz Biotechnologies, Santa Cruz, CA) and ECL detection. Immunoblots shown are from representative experiments repeated at least three times with comparable results.
Results
Activation by Dopamine of MAPK in CHO-hD3 Cells.
DA (1 μM) elicited a transient phosphorylation of p42 (ERK 2) forms of MAPK, reaching a maximum at ∼5 min and returning to basal levels after 20 min of treatment (Fig. 1A). The p44 form of MAPK (ERK 1) was weakly phosphorylated and was visible only on overexposed film (Fig. 2A). Control experiments showed that the levels of both p42mapk and p44mapk were unchanged in cellular extracts at each of the different incubation times (Fig. 1B). In all subsequent experiments, cells were stimulated with DA for 5 min. Activation of MAPK by DA was concentration-dependent, maximal activation being observed at ∼100 nM DA (Fig. 2A). Stimulation of MAPK induced by DA (100 nM) was completely blocked in CHO cells pretreated with PTX (Fig. 2B). PTX also abolished the stimulation induced by the preferential D3agonists (+)-7-hydroxy-2-(di-n-propylamino)tetralin [(+)7-OH-DPAT] (100 nM), and (+)-(4aR,10bR)-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-[1-]benzopyrano-[4,3-b]-1,4-oxazin-9-ol (PD 128,907) (100 nM), suggesting the involvement of Gi and/or Go proteins in hD3 receptor coupling to MAPK.

Time course of dopamine-induced MAPK activation in CHO-hD3 cells. A, CHO-hD3 cells were incubated in the presence of DA (1 μM) for the indicated times. Cell extracts were prepared as described in Materials and Methods. Proteins were loaded on 10% polyacrylamide gels and fully activated pp42 MAPK was revealed with a monoclonal antibody specifically raised against phosphorylated forms on both threonine and tyrosine residues. B, p42 and p44 MAPK from the same cell extracts were detected with antibodies raised against both their native and phosphorylated forms.

Concentration-dependence of DA-induced MAPK activation and effect of cell treatment by PTX. A, CHO-hD3cells were incubated in the presence of DA (0 to 1 μM) for 5 min and phosphorylated MAPK was detected by immunoblotting. B, CHO hD3 cells were treated overnight with or without PTX (100 ng/ml) and then incubated for 5 min with DA (100 nM) or with the D3 preferential agonists (+)7-OH-DPAT (100 nM) and PD 128,907 (100 nM).
Antagonism of DA-Induced MAPK Activation in CHO-hD3Cells.
Pretreatment of cells for 5 min with dopaminergic antagonists attenuated subsequent DA-induced MAPK activation. These antagonists included the antipsychotic agent haloperidol and the selective D3 antagonists 2(R,S)-(dipropylamino)-6-(4-methoxyphenylsulfonylmethyl)-1,2,3,4-tetrahydronaphtalene (GR 218,231) and (+)-[7-(N,N-dipropylamino)-5,6,7,8-tetrahydro-naphtho-(2,3b)dihydro-2,3-furane] (S 14297) (Millan et al., 1995) (Fig.3B). Each of these ligands inhibited DA (100 nM)-induced MAPK activation. However, although haloperidol and GR 218,231 did not induce MAPK activation when tested alone, S 14297 showed weak stimulation of MAPK relative to DA, suggesting partial agonist properties (Fig. 3A). In control experiments, haloperidol and GR 218231 did not inhibit FGF-induced MAPK activation, indicating the absence of effect on this tyrosine kinase receptor (Fig. 3C).

Effect of dopaminergic antagonists on DA and FGF-induced MAPK activation. A, CHO-hD3 cells were incubated for 5 min with or without haloperidol (1 μM), GR 218,231 (1 μM), or S 14297 (1 μM). B, cells were treated as in (A) and dopamine (100 nM) was then added for 5 min. C, cells were treated as in (A) and FGF (20 ng/ml) was then added for 5 min. Phosphorylated MAPK was detected by immunoblotting.
Effect of Kinase Inhibitors on hD3 Receptor-Mediated MAPK Activation.
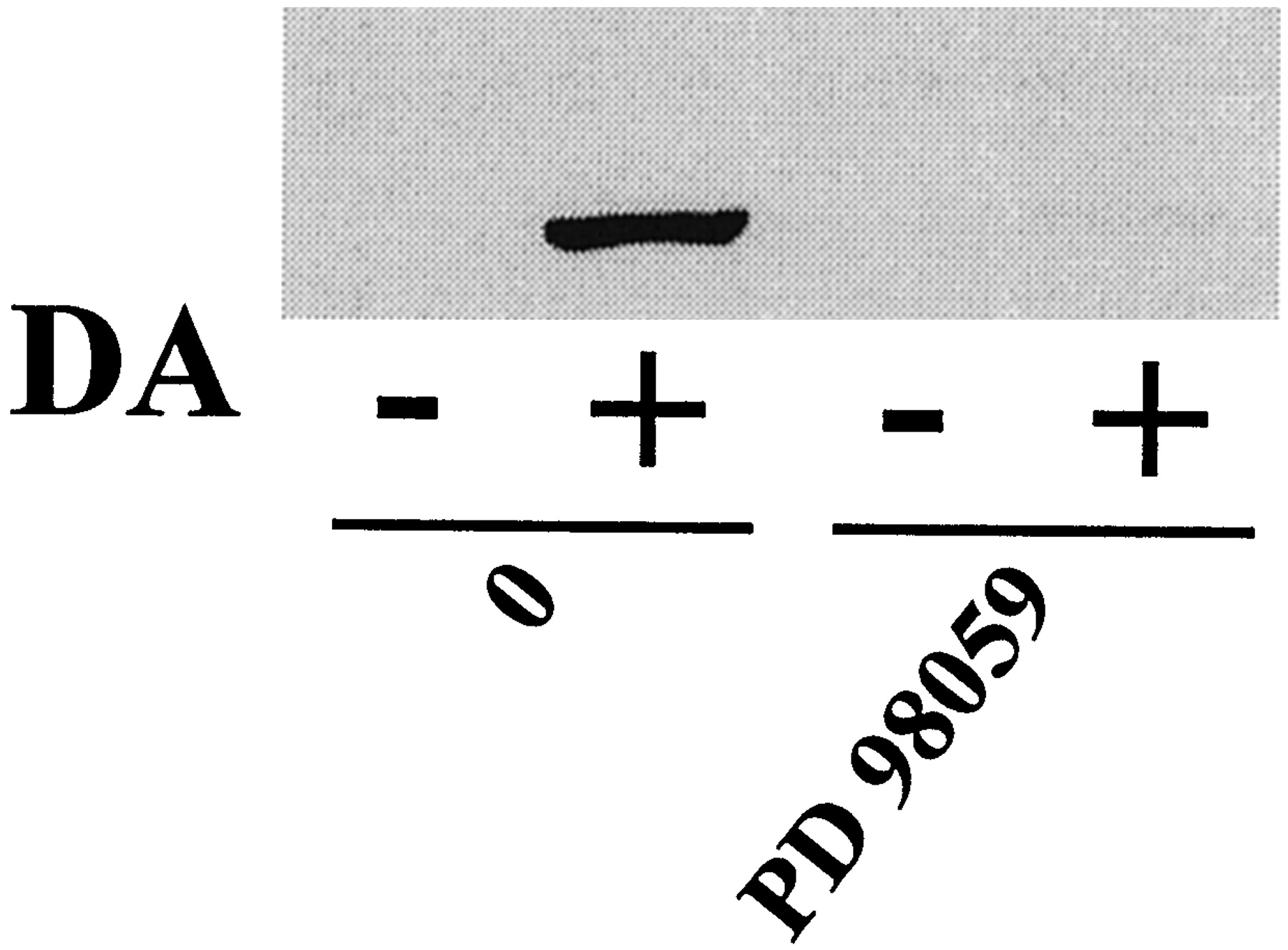
To characterize the pathways leading to MAPK activation by DA, we investigated the roles of different kinases potentially involved. Cell treatment for 30 min with wortmannin (1 μM) and LY 294002 (30 μM), two inhibitors of PI 3-kinase activity, reduced, but did not abolish, MAPK activation induced by DA (Fig.4A). Indeed, residual MAPK phosphorylation was observed even with a higher dose of wortmannin (10 μM) (Fig. 4B). In contrast, two inhibitors of protein tyrosine kinase (PTK) activity, genistein and lavendustin A, did not block stimulation of MAPK by DA (Fig. 4A). PD 98059 (50 μM), an inhibitor of MAPK kinase (MEK) activation that directly controls the MAPK activity, abolished the DA-induced MAPK activation (Fig.5).

Effect of kinase inhibitors on DA-induced MAPK activation. A, CHO-hD3 cells were preincubated for 30 min with or without the PI 3-kinase inhibitors wortmannin (1 μM) and LY 294002 (30 μM), or the protein tyrosine kinase inhibitors genistein (100 μM) and lavendustin A (50 μM). MAPK activation was then induced by dopamine (100 nM) for 5 min. B, CHO-hD3 cells were preincubated for 30 min with different concentrations of wortmannin (0 to 10 μM) and MAPK activation was then induced by DA (100 nM) for 5 min. Phosphorylated MAPK was detected by immunoblotting.

Effect of the MEK inhibitor PD 98059 on DA-induced MAPK activation. CHO-hD3 cells were preincubated for 30 min with PD 98059 (50 μM) and MAPK stimulation was then induced by DA (100 nM) for 5 min. Phosphorylated MAPK was detected by immunoblotting.
Role of PKC on hD3 Receptor-Mediated MAPK Activation.
The involvement of PKC in DA-mediated MAPK activation was first evaluated by depletion of endogenous PKC by overnight pretreatment of CHO-hD3 cells with phorbol ester (PMA, 1 μM). Therefore, subsequent activation of MAPK by PMA (30-min incubation) was suppressed in treated cells but retained in untreated cells (Fig. 6A). However, DA-mediated MAPK activation was unaffected in either treated or untreated cells (Fig. 6A). Nevertheless, the PKC inhibitor Ro 31–8220 abolished DA-mediated MAPK activation (Fig. 6B), indicating that a PKC-dependent mechanism mediated DA-induced MAPK activation, and that the PKC isoform was not sensitive to PMA, suggesting the involvement of an atypical PKC (aPKC). Moreover, we checked that PMA-mediated MAPK activation also was abolished by the PKC inhibitor Ro 31–8220 (Fig. 6B). Fig.7 shows the concentration-dependent effect of two other inhibitors of PKC, Gö 6976 and Gö 6983, that can differentially inhibit several PKC isoforms (Martiny-Baron et al., 1993; Gschwendt et al., 1996). Gö 6983, which blocks the activity of the aPKC isoform PKC-ζ, (IC50 = 60 nM; Gschwendt et al., 1996), blocked DA-induced MAPK activation (Fig.7). In contrast, Gö 6976, which is ineffective against PKC-ζ (IC50 > 20 μM; Martiny-Baron et al., 1993), did not, when tested at a concentration of 5 μM.

Role of PKC in DA-induced MAPK activation. A, CHO-hD3 cells were preincubated overnight with or without phorbol ester (PMA, 1 μM). MAPK stimulation was then induced by DA (100 nM) for 5 min or by PMA (1 μM) for 30 min. B, CHO-hD3 cells were preincubated for 30 min with or without the PKC inhibitor Ro 31–8220 (10 μM). MAPK activation was then induced by DA (100 nM) for 5 min or by PMA (1 μM) for 30 min. Phosphorylated MAPK was detected by immunoblotting.

Differential effects of the PKC inhibitors Gö6976 and Gö 6983. CHO-hD3 cells were preincubated 30 min with increasing concentration of the PKC inhibitors Gö 6976 and Gö 6983 (0 to 5 μM). MAPK activation was then induced by DA (100 nM) for 5 min and phosphorylated MAPK was detected by immunoblotting.
Discussion
As noted in the introduction, both dopamine D2 and D4 receptor subtypes are known to stimulate MAPK in cultured cells (Luo et al., 1998; Welsh et al., 1998; Zhen et al., 1998) and the present study demonstrates that dopamine D3 receptors activate the MAPK pathway in CHO cells stably transfected with hD3receptors (CHO-hD3). Indeed, haloperidol and the selective D3 receptor antagonist GR 218,231 blocked DA-mediated MAPK activation, whereas they did not modify FGF stimulation of MAPK phosphorylation, indicating the pharmacological specificity of their actions. Furthermore, the preferential D3 agonists (+)7-OH-DPAT and PD 128,907, like DA, triggered MAPK activation. It is interesting that stimulation was similar for the three agonists, whereas in our recent study of G protein activation in CHO-hD3 cell membranes, both (+)7-OH-DPAT and PD 128,907 exhibited partial agonist properties (Newman-Tancredi et al., 1999). This difference suggests possible signal amplification at the level of kinase cascade following G protein activation. In this respect, the selective D3receptor “antagonist” S 14297, which does not stimulate [35S]GTPγS binding (Newman-Tancredi et al., 1999), partially stimulated MAPK activity on entire cells. The present measure of MAPK phosphorylation induced by hD3receptor activation may, thus, more readily detect weak agonist actions.
The activation of MAPK phosphorylation by hD3receptors exhibits certain features common to other GPCRs. For example, DA stimulated MAPK phosphorylation via hD3receptors in a time-dependent manner with a peak of activation occurring at 5 min and a rapid return to the basal level, and similar time courses have been observed with hD2receptors as well as other GPCRs (Welsh et al., 1998). Another similarity between hD3 receptors and other GPCRs concerns the importance of Gi and/or Go proteins for activation of MAPK. Indeed, PTX treatment of CHO-hD3 cells abolished MAPK stimulation by DA, (+)7-OH-DPAT, and PD 128,907, suggesting the exclusive involvement of Gi and/or Go proteins, analogous to that reported for D2 and 5-hydroxytryptamine1A receptors (Faure et al., 1994; Garnovskaya et al., 1996; Welsh et al., 1998), although a non-PTX sensitive pathway of MAPK stimulation by GPCRs also has been observed, mainly involving Gq/11 proteins (Hawes et al., 1995; Dikic et al., 1996; Igishi and Gutkind, 1998; Berts et al., 1999).
As in Pang et al. (1995), inhibition of MEK (located just upstream of the MAPK) by the specific inhibitor PD 98059 abolished MAPK activity in CHO-hD3 cells. This result excludes cross-talk between other kinase cascades potentially activated by D3 receptors that might lead to MEK-independent activation of MAPK. Furthermore, D3 receptor activation in CHO-hD3 cells does not elicit p38 and Junk kinase activation (D.C., unpublished data) as observed in the case of α1A adrenergic receptor activation in PC12 cells (Williams et al., 1998; Berts et al., 1999).
Despite these similarities, further investigation revealed distinctive features in the activation of the MAPK cascade by hD3 receptors versus D2 and D4 receptors and other GPCRs. First, in the case of other Gi/Go-coupled receptors, βγ dimers derived from G protein dissociation are involved in the stimulation of the proto-oncogene Ras protein via PTK activation such as Src-like proteins, and subsequent tyrosine phosphorylation of Shc and recruitment of the Grb2-Sos complex (Faure et al., 1994; van Biesen et al., 1995; Igishi and Gutkind, 1998). However, in the present study, the tyrosine kinase inhibitors genistein and lavendustin A did not prevent DA-induced MAPK activation, suggesting that hD3 receptors do not engage this signal transduction pathway.
Second, DA-induced MAPK activation in CHO-hD3cells was not due to cross-talk between D3 and tyrosine kinase receptors known to be present on these cells (FGF and insulin receptors), in contrast to certain other GPCRs (lysophosphatidic acid or bradykinin) (Daub et al., 1996; Zwick et al., 1997). Thus, as mentioned above, the tyrosine kinase inhibitors genistein and lavendustin A had no effect on DA-induced MAPK activation in CHO-hD3 cells.
Third, D3 receptors differentially modulated PI 3-kinase activity compared with other GPCRs, although similarity with D2 dopamine receptors was noted (Welsh et al., 1998). Indeed, in both cases, the PI 3-kinase inhibitors wortmannin and LY 294002 attenuated DA-induced MAPK activation. The γ isoform of PI 3-kinase is directly activated by βγ subunits released from G protein activation (Stephens et al., 1997; Lopez-Ilasaca et al., 1997). Several points arise from these observations. βγ Subunits have been suggested to mediate PI 3-kinase-dependent Shc phosphorylation via a Src-like tyrosine kinase (Gutkind et al., 1990; Touhara et al., 1995;Lopez-Ilasaca et al., 1997). However, in the present study, hD3 receptor coupling to MAPK was not sensitive to PTK inhibitors, so the involvement of a putative Src-like protein in PI 3-kinase-dependent MAPK activation may be excluded. Another, somewhat speculative, possibility suggested by Welsh et al. (1998) for dopamine D2 receptors is that wortmannin and LY 294002 could indirectly affect MAPK activation by perturbing the interaction of PI 3-kinase with an active form of Ras (Rodriguez-Viacana et al., 1994; Rubio et al., 1997). However, a more probable hypothesis is that of a reciprocal interaction between the PI 3-kinase pathway and other proteins involved in MAPK activation. For example, products of PI 3-kinase activity, such as phosphatidylinositol-3,4-diphosphate (PIP2) and phosphatidylinositol-3,4,5-triphosphate (PIP3), are able to activate different PKC isoforms (Nakanishi et al., 1993;Liscovitch and Cantley, 1994).
Fourth, in this context, we show that the PKC inhibitor Ro 31-8220 abolished DA-induced MAPK activation, indicating that PKC is a key element in hD3 receptor coupling to MAPK pathway. Classically described mechanisms of PKC activation are mediated by Gq via phospholipase C activation and diacylglycerol generation. This pathway is not pertinent to the present system because hD3 receptor activation of MAPK is abolished by PTX, implicating Gi/Go proteins and not Gq. Nevertheless, PTX-sensitive Go proteins also can stimulate the MAPK pathway in a Ras-independent and PKC-dependent mechanism, probably involving a direct phosphorylation of Raf-1 by PKC (Kolch et al., 1993; van Biesen et al., 1996). In fact, this previously described PKC activation by Go proteins was sensitive to PMA (van Biesen et al., 1996), suggesting involvement of “classical” and/or “novel” isoforms of PKC, which are sensitive to diacylglycerol and phorbol ester (Casabona, 1997). In contrast, the PKC involved in hD3 coupling to MAPK was not sensitive to PMA, suggesting that it belongs to the aPKC family. These aPKCs includes the PKC-λ, ι, and ζ isoforms that are insensitive to classical cofactors of PKC such as calcium, diacylglycerol, and phorbol ester (Ono et al., 1989; Casabona, 1997). The coupling of D3 receptors to aPKC was not due to a lack of other isoforms of PKC because MAPK phosphorylation in CHO-hD3 cells can be induced by PMA cell treatment (Fig. 6A). The PKC-ζ isoform interacts directly with the effector binding domain of Ras (Diaz-Meco et al., 1994) and has been shown to be critical for mitogenic signal transduction in fibroblasts and the maturation of Xenopus oocytes (Berra et al., 1993). Gö 6983, which is a PKC-ζ inhibitor (Gschwendt et al., 1996), blocked DA-induced MAPK activation in CHO-hD3cells, whereas Gö 6976, which is not an inhibitor of PKC-ζ (Martiny-Baron et al., 1993), did not. These findings suggest that PKC-ζ may be involved in CHO-hD3-mediated MAPK activation, although the effects of these inhibitors on other aPKC isoforms and kinases have not, as yet, been characterized in detail. Interestingly, PKC-ζ is strongly activated by PIP3 and, to a lesser extent, by PIP2 (Nakanishi et al., 1993), both of which are products of PI 3-kinase activity, which, as described above, is involved in D3-induced MAPK activation. Thus, D3 receptors may mediate aPKC activation via PI 3-kinase. However, the partial effect of the PI 3-kinase inhibitors wortmannin and LY 294,002 suggests additional modes of aPKC regulation, possibly involving a Gαi/o subunit-dependent membrane relocalisation of aPKC to the proximity of PI 3-kinase, as has been suggested in the case of PKC-ζ-Ras interaction (Diaz-Meco et al., 1994) (Fig.8). However, the exact nature of the aPKC isoform regulated by hD3 receptors via Go and/or Gi proteins and whether this aPKC can phosphorylate a specific Raf protein (Raf-1, A/B-Raf) remains to be established.

Proposed model of D3 receptor-mediated MAPK activation. Convergent pathways of PKC activation are shown. One involves PI 3-kinase γ isoform activation via βγ subunits from Gi and/or Go dissociation, and subsequent activation of an aPKC via PIP3 and/or PIP2 (see text). The other involves aPKC activation via αi and/or αo subunits. The activation of both pathways may be necessary to fully stimulate aPKC and MAPK. Dotted arrows indicate multiple or uncharacterized steps in the pathway.
In conclusion, the present study demonstrates that hD3 receptors activate MAPK activity. Although the MAPK activation pathway bears some similarities to previously characterized systems for other GPCRs (such as dopamine D2 receptors) a distinctive pattern of intracellular kinase cascade activation is implicated. Thus, hD3 mediated MAPK phosphorylation involves the activation of PI 3-kinase and an atypical isoform of PKC. The present data raise the possibility that MAPK stimulation may be relevant to regulation of diverse cellular events mediated by D3 receptors, such as neuronal plasticity, morphological differentiation, and synaptic transmission.
Footnotes
- Received June 11, 1999.
- Accepted August 5, 1999.
-
Send reprint requests to: Adrian Newman-Tancredi Ph.D., Department of Psychopharmacology, Institut de Recherches Servier, 125, Chemin de Ronde, 78290 Croissy-sur-Seine (Paris), France. E-mail: newman_tancredi{at}hotmail.com
Abbreviations
- MAPK
- mitogen-activated protein kinase
- GPCR
- G-protein coupled receptor
- PI 3-kinase
- phosphatidylinositol 3-kinase
- PKC
- protein kinase C
- CHO
- chinese hamster ovary
- hD3
- human dopamine D3 receptors
- DA
- dopamine
- FGF
- fibroblast growth factor
- PMA
- phorbol-12-myristate-13-acetate
- PTX
- pertussis toxin
- ECL
- enhanced chemiluminescence
- (+)-7-OH-DPAT
- (+)-7-hydroxy-2-dipropylaminotetralin
- PTK
- protein tyrosine kinase
- MEK
- mitogen-activated protein kinase kinase
- aPKC
- atypical protein kinase C
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}