


Abstract
Human immunodeficiency virus (HIV) pharmacotherapy, by combining different drug classes such as nucleoside analogs and HIV protease inhibitors (PIs), has increased HIV-patient life expectancy. Consequently, among these patients, an increase in non-HIV–associated cancers has produced a patient cohort requiring both HIV and cancer chemotherapy. We hypothesized that multidrug resistance protein 4/ATP binding cassette transporter 4 (MRP4/ABCC4), a widely expressed transporter of nucleoside-based antiviral medications as well as cancer therapeutics might interact with PIs. Among the PIs evaluated (nelfinavir, ritonavir, amprenavir, saquinavir, and indinavir), only nelfinavir both effectively stimulated MRP4 ATPase activity and inhibited substrate-stimulated ATPase activity. Saos2 and human embryonic kidney 293 cells engineered to overexpress MRP4 were then used to assess transport and cytotoxicity. MRP4 expression reduced intracellular accumulation of nelfinavir and consequently conferred survival advantage to nelfinavir cytotoxicity. Nelfinavir blocked Mrp4-mediated export, which is consistent with its ability to increase the sensitivity of MRP4-expressing cells to methotrexate. In contrast, targeted inactivation of Abcc4/Mrp4 in mouse cells specifically enhanced nelfinavir and 9-(2-phosphonylmethoxyethyl) adenine cytotoxicity. These results suggest that nelfinavir is both an inhibitor and substrate of MRP4. Because nelfinavir is a new MRP4/ABCC4 substrate, we developed a MRP4/ABCC4 pharmacophore model, which showed that the nelfinavir binding site is shared with chemotherapeutic substrates such as adefovir and methotrexate. Our studies reveal, for the first time, that nelfinavir, a potent and cytotoxic PI, is both a substrate and inhibitor of MRP4. These findings suggest that HIV-infected cancer patients receiving nelfinavir might experience both enhanced antitumor efficacy and unexpected adverse toxicity given the role of MRP4/ABCC4 in exporting nucleoside-based antiretroviral medications and cancer chemotherapeutics.
Introduction
The incidence of non-AIDS–defining cancers (e.g., Hodgkin’s lymphoma, lung, testicular germ-cell, breast) has increased significantly as patients with human immunodeficiency virus (HIV)/AIDS achieve longer life expectancy (Rudek et al., 2011; Deeken et al., 2012). These individuals are a therapeutic challenge because concurrent treatment with antineoplastic drugs and highly active antiretroviral therapy (HAART) might increase the potential for drug interactions (Rudek et al., 2011). The interactions between cancer chemotherapeutics and HAART drugs have the potential to increase the therapeutic benefit by increasing tumoricidal activity (De Clercq et al., 1999). Despite this, mechanistic evidence is lacking for direct interactions between cancer chemotherapeutics and drugs in the HAART regimen.
Acyclic nucleoside phosphonates like tenofovir and adefovir [PMEA; 9-(2-phosphonylmethoxyethyl) adenine] are acyclic nucleotide analogs of adenosine monophosphate that, due to their capacity to inhibit viral polymerases, are very effective against a variety of viruses (e.g., hepatitis B and HIV) and have become integral to the success of HAART regimens. Nonetheless, they also possess potent tumoricidal properties (De Clercq et al., 1999). Tenofovir is structurally similar to adefovir only differing by a methyl-group addition in the sugar-like aliphatic linker. In vitro studies and studies in knockout mice indicate that adefovir and tenofovir are exported by the ATP binding cassette (ABC) transporter, ATP binding cassette transporter 4/multidrug resistance protein 4 (Abcc4/Mrp4) (Ray et al., 2006; Imaoka et al., 2007; Takenaka et al., 2007). Notably, absence of Abcc4/Mrp4 enhances tenofovir toxicity, thereby indicating ABCC4/MRP4 export is crucial to preventing acyclic nucleoside phosphonate toxicity (Imaoka et al., 2007).
The HAART regimen typically includes HIV protease inhibitors (PIs). Although some PIs (ritonavir, nelfinavir) increase the toxicity of acyclic nucleoside phosphonates used in antiretroviral therapy (PMEA, adefovir, tenofovir) (Kiser et al., 2008), the basis for this is unknown. Because adefovir and tenofovir are substrates of MRP4, we hypothesized that PIs might inhibit MRP4 and increase not only their cytotoxicity but also cancer chemotherapeutics. We tested the possibility that PIs interact with ABCC4/MRP4 by assessing their impact on substrate-stimulated ATPase, inhibition of basal ATPase, and transport activity using genetic models of ABCC4/MRP4 overexpression and newly developed knockout cell lines. We show that the therapeutically important HIV PIs, nelfinavir (NFV) and ritonavir, modulate substrate-stimulated ATPase activity, which correlates with their potential as MRP4 substrates. These studies were extended to show that ABCC4/MRP4 overexpression reduces NFV uptake and protects against NFV cytotoxic effects. Moreover, absence of ABCC4/MRP4 renders cells more sensitive to NFV. Finally, because NFV is an ABCC4 substrate, we developed a pharmacophore to further identify potential substrates and/or inhibitors of ABCC4/MRP4. These findings suggest that inhibition of ABCC4/MRP4 by nelfinavir may alter antitumor efficacy among HIV-infected cancer patients.
Materials and Methods
Reagents
The following reagents were obtained through the AIDS Research and Reference Reagent Program (Division of AIDS, National Institutes of Health National Institute of Allergy and Infectious Diseases): nelfinavir, ritonavir, amprenavir, saquinavir, and indinavir. Generation of wild-type (WT) and Mrp4 knockout (KO) mouse embryo fibroblasts (MEFs) from C57BL/6J mouse embryos were described previously (Sinha et al., 2013).
ATPase Assays
ATPase activity of MRP4 in crude membranes (10 μg protein per assay) of insect cells was measured by the end-point Pi assay as previously described (Ambudkar, 1998; Sauna et al., 2004; Wu et al., 2005), with minor modifications. MRP4-specific activity was recorded as the beryllium fluoride–sensitive ATPase activity, where the amount of Pi released was quantified using a colorimetric method (Ambudkar, 1998) (Supplemental Methods).
Cell Proliferation Assay
Saos2, human embryonic kidney (HEK) 293, WT, or Abcc4/Mrp4 KO MEFs were incubated in 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) medium (Dulbecco’s modified Eagle’s medium containing 10% dialyzed serum) containing various concentrations of Bis(pivaloyloxymethyl [POM])-PMEA, NFV, or methotrexate (MTX) for 4–6 hours. After 3 days of culture in fresh MTT medium, cell proliferation was measured using the MTT assay according to the manufacturer’s instructions (Promega, Madison, WI).
Intracellular Accumulation of ABCC4 Substrates
Saos-2 and HEK293 stably expressing either vector or MRP4 and WT and ABCC4/Mrp4 KO MEFs were incubated with 10 μM Bis(POM)-PMEA (with a trace amount of [3H]Bis(POM)-PMEA) for 1–6 hours with 30 minutes preincubation with NFV or 3-([{3-(2-[7-chloro-2-quinolinyl]ethenyl)phenyl}-{(3-dimethylamino-3-oxopropyl)-thio-methyl]thio)propanoic acid (MK571) as indicated. The intracellular accumulation of PMEA was measured as previously described (Takenaka et al., 2007) (Supplemental Methods).
Intracellular amounts of nelfinavir in HEK293 cells expressing either vector or MRP4 were determined using liquid chromatography coupled to tandem mass spectrometry.
Detection of MRP4 Proteins by Immunoblotting
Homogenates prepared from tissues harvested from C57BL/6J WT and KO adult female mice (Leggas et al., 2004) were analyzed by immunoblotting as previously described (Takenaka et al., 2007). Antibodies used were M4I10 (MRP4; Abcam, Cambridge, MA), MRP1 (Abcam), P-glycoprotein (J.D.S., St. Jude Children's Research Hospital, Memphis, TN), MRP5 (a gift from Dr. George Scheffer, VU Medical Center, Amsterdam, The Netherlands), BXP53 (ABCG2; Kamiya, Seattle, WA), and β-actin (Sigma-Aldrich, St. Louis, MO).
PMEA Efflux in the Presence of NFV
PMEA efflux was measured over the indicated time in WT or Abcc4/Mrp4 KO MEFs as described previously using a trace amount of [3H]Bis(POM)-PMEA in the absence or presence of 50 μM NFV (Nagai et al., 2011) (Supplemental Methods).
MRP4 Pharmacophore Models
PI MRP4 Pharmacophore.
Computational molecular modeling studies were performed using Discovery Studio 2.5.5. and 3.5.5. (Acers, San Diego, CA). Common feature pharmacophore models describe the arrangement of key features important for biologic activity (Clement and Mehl, 2000; Ekins et al., 2007). A common features pharmacophore was developed for PIs using nelfinavir as the most active molecule, followed by ritonavir and then amprenavir, indinavir, and saquinavir as inactive (Supplemental Table 1). Up to 255 molecule conformations were generated with the FAST conformer generation method, with the maximum energy threshold of 20 kcal/mol.
This common features pharmacophore was applied to screen several databases—including the US Food and Drug Administration Collaborative Drug Discovery database (http://www.collaborativedrug.com) (Hohman et al., 2009), Human Metabolome Database (Wishart et al., 2009), SCUT (Ekins et al., 2005), MicroSource US Drug Collection (http://msdiscovery.com/), and the Kyoto Encyclopedia of Genes and Genomes (Kanehisa and Goto, 2000)—using the FAST search method as previously described (Ekins et al., 2005). The quality of the molecule mapping to the pharmacophore was determined by the fit value, which is dependent on the proximity of a compound to the pharmacophore feature centroids and the weights assigned to each centroid, where a higher fit value represents a better fit.
MRP4 Inhibitor Pharmacophore.
A more diverse training set of 10 MRP4 inhibitors were selected from the literature (Russel et al., 2008) and used for both a common features model and a quantitative pharmacophore (Supplemental Table 2). The common features model was described as above but instead used the CAESAR conformer generation algorithm. Dipyridamole and quercetin were used as the most active molecules with 10 μM as the cutoff for activity. The quantitative MRP4 pharmacophore used the IC50 data associated with each molecule. Both pharmacophores were then used to search the SCUT database.
Mapping Prostaglandin E2 and Quercetin to the PI MRP4 Substrate Pharmacophore.
The common features pharmacophore developed with the five PIs was used to map prostaglandin E2 (PGE2) and quercetin using the ligand pharmacophore mapping protocol with rigid fitting.
Statistical Analysis
For proliferation assays, cytotoxicity for each drug was expressed as IC50 values calculated with ADAPT II modeling software (Biomedical Simulations Resource, Los Angeles, CA). Nonlinear and linear regression analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA).
Results
MRP4 ATP Hydrolysis and HIV PIs.
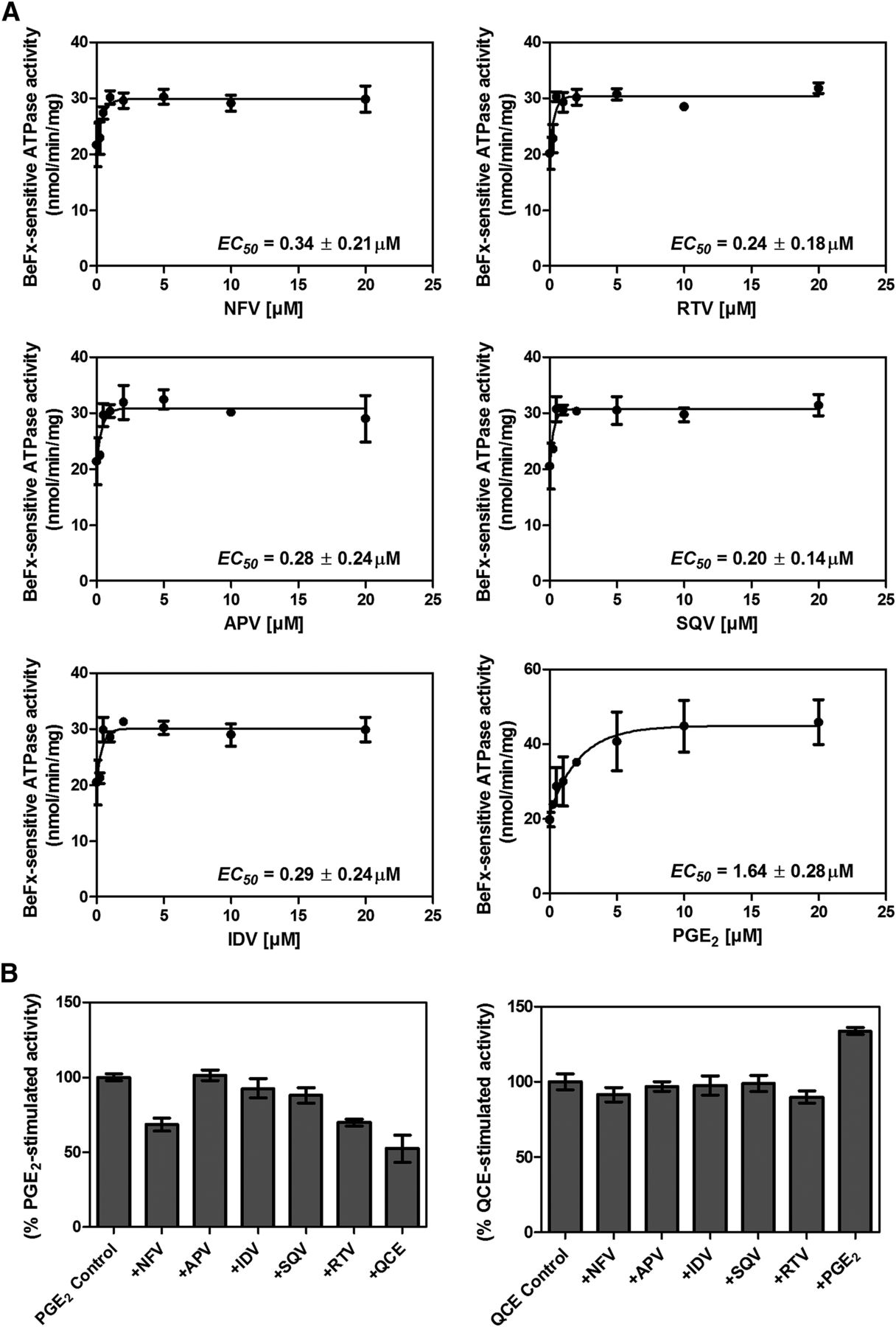
Previous studies classified MRP4 substrates into the following categories: 1) cyclic nucleotide or nucleoside phosphate analogs, 2) anticancer agents, 3) steroids, and 4) prostaglandins (Russel et al., 2008). Previously, we demonstrated that some substrates (e.g., PGE2) stimulate ATPase activity, whereas others show concentration-dependent biphasic kinetics (stimulation and inhibition) (Sauna et al., 2004). ATP hydrolysis mediated by ABC transporters is a useful surrogate assay to identify potential transport substrates based on the premise that the transport of substrates is powered by ATP binding and hydrolysis; however, not all transport substrates stimulate ATP hydrolysis (Sauna et al., 2004). We evaluated a panel of HIV PIs (amprenavir, indinavir, saquinavir, ritonavir, and nelfinavir; Supplemental Fig. 1) for their ability to affect ABCC4/MRP4 ATPase activity. Each of the PIs tested produced some stimulation (approximately 50%) of ATP hydrolysis by MRP4 (Fig. 1A) with the positive control, PGE2, showing >100% stimulation. A notable feature is the inability of these PIs to inhibit ATP hydrolysis, unlike cGMP, which stimulates and inhibits ATPase (Sauna et al., 2004). To monitor whether these PIs compete for the same binding site as known MRP4 substrates (Sauna et al., 2004; Wu et al., 2005), PGE2 and quercetin were used to stimulate ATP hydrolysis. In Fig. 1B, we show that, among the PIs, only NFV and ritonavir inhibit the PGE2-stimulated ATP hydrolysis (P < 0.0005). We extended these studies to determine whether these PI affected quercetin-stimulated activity. None of the PI inhibited quercetin-stimulated activity, suggesting that NFV and ritonavir share a common binding site with PGE2, but not quercetin.

Nelfinavir and ritonavir modulate MRP4 ATPase activity. (A) The beryllium fluoride (BeFx)–sensitive ATPase activity of ABCC4/MRP4 was determined using the Pi release assay in the presence of various concentrations of NFV, ritonavir (RTV), amprenavir (APV), saquinavir (SQV), or indinavir (IDV). PGE2, a known MRP4 substrate (Reid et al., 2003) that stimulates ATPase activity (Sauna et al., 2004), was used as a positive control. (B) The effect of indicated compounds on PGE2-stimulated ATPase activity (left) and quercetin (QCE)-stimulated ATPase activity (right) was evaluated.
PI Modulation of MRP4 Transport of PMEA.
To directly determine whether the PIs inhibit MRP4-mediated transport, we used the PMEA prodrug, [3H]Bis(POM)-PMEA. Bis(POM)-PMEA bypasses the PMEA uptake carrier organic anion transporter 1 (Hatse et al., 1998; Schuetz et al., 1999; Adachi et al., 2002) and is hydrolyzed to the MRP4 substrate, PMEA, by intracellular esterases. Two different cell lines of different histotypes, Saos-2 (osteosarcoma) and Hek293 (kidney), were engineered to express MRP4 because each cell line has different endogenous levels of transporters (Fig. 2A). Each cell type was coincubated with 10 μM Bis(POM)-PMEA and either the positive control MRP4 inhibitor, indomethacin (100 μM), or a PI (at 50 μM) for 6 hours followed by determination of total intracellular radioactivity. Neither amprenavir, indinavir, nor saquinavir consistently increased PMEA accumulation in both cell types. In contrast, whereas ritonavir modestly increased PMEA concentration, NFV strongly increased intracellular PMEA in HEK293 with a more modest effect in Saos2 (Fig. 2B). These studies suggest that NFV is a good inhibitor of MRP4.

Among common HIV PIs, only NFV is an inhibitor. (A) Immunoblot analysis of MRP4, P-glycoprotein (P-gp), or ABCG2 expression in either HEK293 or Saos2 cells programmed with either empty vector of an MRP4 expression vector. (B) Bis(POM)-PMEA uptake by Saos2 or HEK293 cells containing either empty vector or an MRP4 expression vector was determined in the presence of the indicated PIs.
Nelfinavir Is an MRP4 Substrate.
To determine whether NFV inhibits MRP4 at concentrations that are achievable clinically (7–10 μM) (Markowitz et al., 1998), we incubated both HEK293 and Saos-2 cells with various concentrations of NFV (Fig. 3A) before adding Bis(POM)-PMEA. NFV dose-dependently increased PMEA accumulation, with an estimated IC50 of approximately 24 and 15.8 μM for Saos2 and Hek293, respectively. We extended these studies to determine whether NFV (15 μM) was capable of reversing MRP4-mediated resistance to PMEA (Fig. 3B). NFV reduced the PMEA IC50 over 3-fold from 3.5 μM to 1.1 μM in MRP4-expressing cells. In contrast, only a modest shift in IC50 was observed for the empty vector cells (from 0.8 to 0.5 μM). These studies suggest that NFV increases PMEA accumulation by inhibiting MRP4, producing greater cytotoxicity.

NFV is a substrate and inhibitor of MRP4. (A) Bis(POM)-PMEA uptake was determined in Saos2 or HEK293 cells containing either empty vector or an MRP4 expression vector in the presence of various NFV concentrations. (B) NFV strongly increased Bis(POM)-PMEA cytotoxicity in MRP4-expressing cells (left). NFV uptake was strongly reduced in MRP4-expressing cells (middle). MRP4 expression reduced NFV cytotoxicity (right). (C) The presence of 50 μM NFV (right) increased MTX cytotoxicity in MRP4-expressing Saos2 cells compared with MTX alone (left).
NFV impacts MRP4 ATPase activity (Fig. 1) and inhibits its function to enhance accumulation of a well-known substrate, PMEA (Figs. 2B and 3A). To test whether NFV is an MRP4 substrate, either vector or MRP4-expressing cells were incubated with NFV followed by determination of intracellular concentrations of NFV by liquid chromatography coupled to tandem mass spectrometry (Fig. 3B, middle). The uptake of NFV was lower in MRP4 cells with an estimated 40% lower steady-state accumulation of NFV.
NFV is capable of killing cells by multiple mechanisms (Gills et al., 2007; Xie et al., 2011). To extend these studies, we evaluated whether MRP4 impacted NFV cytotoxicity. We cultured cells in various concentrations of NFV for 4 hours. Subsequently, cell survival was determined 72 hours after NFV treatment. Cells expressing MRP4 had a 3-fold shift in NFV IC50 from 28.6 to 84.6 µM relative to vector cells (Fig. 3B, right).
NFV Reduces MRP4-Mediated Resistance to MTX.
In total, these studies support our proposition that NFV is an MRP4 substrate. On the basis of these studies and the potential for HIV patients to develop cancers that are typical of the non-HIV-infected population (Rudek et al., 2011; Deeken et al., 2012), we tested whether NFV would affect MTX cytotoxicity, because MTX is an MRP4 substrate (Chen et al., 2002) that is widely used in combination therapy to treat multiple cancers from acute lymphoblastic leukemia to breast cancer (Bonadonna et al., 1995; Pui and Evans, 2006). Like NFV, MTX inhibits PGE2-stimulated MRP4 ATP hydrolysis (Sauna et al., 2004), suggesting that these compounds occupy a similar or identical substrate binding site. On the basis of these findings, we tested whether NFV modulated sensitivity of MRP4-overexpressing cells to MTX (Fig. 3C, left). Cells were incubated with various concentrations of MTX for 4 hours in the absence and presence of NFV followed by incubation in drug-free medium for 72 hours. As shown in Fig. 3C, the IC50 for Saos2 empty vector cells was 1.0 μM and overexpression of MRP4 shifts the IC50 to 1.8 μM. The addition of NFV during the MTX incubation had minimal effect on the IC50 for the empty vector cells (1.0 versus 1.1), whereas the IC50 for MRP4 is shifted to 1.5-fold (1.2 μM; Fig. 3C, right), which indicates that NFV enhances MTX cytotoxicity by MRP4 inhibition.
Constitutive MRP4 Protects against NFV.
To assess whether constitutive levels of Abcc4/Mrp4 were sufficient to protect cells from NFV cytotoxicity and affect Abcc4/Mrp4-mediated export, we developed cell lines from Abcc4/Mrp4 KO and WT MEFs (Fig. 4). Immunoblot analysis of three independent WT and Abcc4/Mrp4 KO MEF lines shows comparable amounts of Abcc4/Mrp4 among WT MEF cell lines (Fig. 4A, left). Unlike our previous studies showing compensation for Abcc4/Mrp4 absence by upregulation of Abcg2 in some tissues (e.g., spleen and brain), the KO MEFs do not display upregulation of Bcrp/Abcg2, nor do they exhibit upregulation of Mrp5, a transporter also capable of exporting PMEA (Fukuda and Schuetz, 2012). The level of expression of Mrp4/Abcc4 is comparable with spleen but less than kidney (Fig. 4A, middle). Our previous studies have demonstrated MRP4 absence impacts accumulation of its substrates in spleen (Takenaka et al., 2007). We next determined Mrp4 function in the WT MEFs by evaluating PMEA accumulation as described above (Figs. 2 and 3). In WT MEFs, incubation with the ABCC4/MRP4 inhibitor MK571 (25 μM) produced a strong increase in PMEA accumulation in WT MEFs (2.1-fold) with a small effect in KO MEFs, demonstrating that MRP4 is functional in WT MEFs (Fig. 4A, right). In addition, although Bcrp/Abcg2 transports PMEA (Takenaka et al., 2007), inhibition of Bcrp/Abcg2 with the specific inhibitor fumitremorgin C (Rabindran et al., 2000) revealed only a small increase in PMEA accumulation, indicating that Abcg2 levels in MEFs are insufficient to impact PMEA accumulation. To confirm that NFV blocks Abcc4/Mrp4-mediated export of PMEA, KO and WT MEFs were preloaded with Bis(POM)-PMEA in media containing deoxyglucose (to inhibit glycolysis), but also lacking glucose to block regeneration of ATP, thereby depriving MRP4 the energy to fuel export as previously described (Schuetz et al., 1999). Subsequently, cells were washed in ice-cold media to stop PMEA accumulation. Export was restored by readdition of warmed media (37°C) containing glucose (5 mM) with or without NFV (50 μM). The rate of PMEA export from WT MEFs was 21 pmol/mg per minute versus 5.6 pmol/mg per minute for KO MEFs (a 3.8-fold difference in rate), confirming a highly functional Mrp4 in WT MEFs. Further support for NFV blocking Abcc4/Mrp4-mediated export is demonstrated by NFV suppression of PMEA export from WT MEFs, which is essentially complete after 30-minute exposure to NFV (Fig. 4B, left). The total amount of PMEA exported from WT MEFs (over a 2-hour interval) was 60% of the total PMEA. In contrast, KO MEFs exported <20% of total PMEA (Fig. 4B, right). Notably, NFV strongly reduced export of PMEA from WT MEFs to a level comparable with KO MEFs.
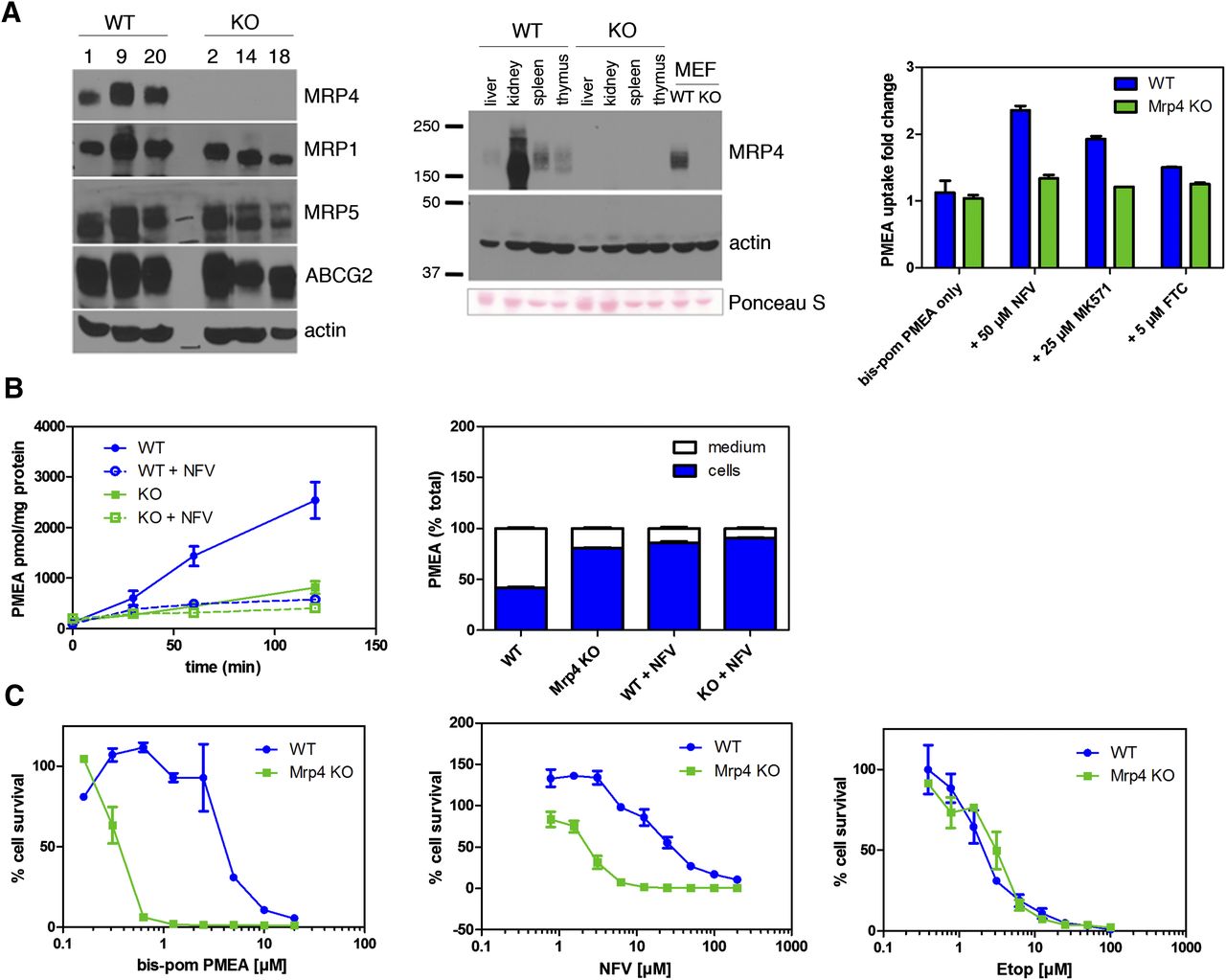
Mrp4/Abcc4 absence reveals nelfinavir is a substrate and inhibitor. (A) Immunoblot analysis of three independent clones for each genotype revealed no upregulation of Abcc1, Abcc5, and Abcg2 in Mrp4 KO MEFS (left). MEFs expressed Mrp4 at levels comparable with normal tissues containing functional Mrp4 (middle). Bcrp/Abcg2 was only minimally functional in Mrp4 KO MEFs (right). (B) NFV blocks Mrp4/Abcc4-mediated export of PMEA. KO and WT MEFs were incubated with Bis(POM)-PMEA under energy-depleted conditions. Subsequently, energy-containing media were restored and export of PMEA was determined (left). The proportion of PMEA in the media and cells was determined and expressed as the percentage of the of total 120 minutes after PMEA export was initiated (right). (C) Mrp4 KO MEFs were more sensitive to the cytotoxic effects of Bis(POM)-PMEA (left) and NFV (middle) but not to etoposide (right). Etop, etoposide; FTC, fumitremorgin C.
Enhanced MRP4 levels reduce NFV accumulation and cytotoxicity (Fig. 3). We assessed whether Abcc4/Mrp4 absence altered NFV cytotoxicity in KO MEFs (Fig. 4C). Prior to conducting these studies, we assessed sensitivity to Bis(POM)-PMEA in WT and KO MEFs by determining viability 3 days after 4-hour Bis(POM)-PMEA treatment. The absence of MRP4/ABCC4 dramatically sensitizes MEFs to Bis(POM)-PMEA, producing a shift in the IC50 from 4 μM in WT to 0.33 μM in KO MEFs (Fig. 4C, left). The NFV IC50 was 2.4 μM for Abcc4/Mrp4 KO MEFs, whereas it was 18.3 μM for WT MEFs, a 7.6-fold increase (Fig. 4C, middle). This indicates that endogenous Mrp4 protects against NFV cytotoxicity. Moreover, KO MEFs are not generally sensitized to cytotoxic agents because WT and KO MEFs are equally sensitive to etoposide (Fig. 4C, right).
Pharmacophore Models for MRP4 Substrates and Inhibitors.
Ten diverse MRP4 inhibitors from the literature were used to generate a common feature (Supplemental Fig. 2A) and a quantitative pharmacophore (Supplemental Fig. 2B). These different approaches have been widely used for infer pharmacophores for multiple transporters such as P-glycoprotein (Ekins et al., 2002a,b), organic anion transporting polypeptide (Chang et al., 2005), and multidrug and toxin extrusion protein 1 (Astorga et al., 2012). Notably, using a method that does not require a rigid alignment of all molecules appears advantageous when substrates and/or inhibitors are structurally diverse (Kortagere and Ekins, 2010). The diverse MRP4 common features model was used to search the SCUT database and resulted in 227 hits, whereas the diverse MRP4 quantitative pharmacophore resulted in 192 hits (selected compounds shown in Supplemental Tables 3 and 4). After ranking by fit score, potentially interesting molecules were highlighted along with known substrates. The diverse MRP4 common features model found nine known MRP4 drug substrates (as well as rediscovered five of the training compounds) and the diverse MRP4 quantitative pharmacophore found seven substrate hits (and rediscovered two of the training compounds). The array of features suggests that MRP4 inhibitors are structurally promiscuous.
We extended the current pharmacophore modeling and database screening to identify other potential Mrp4 substrates because ritonavir also appears to be a MRP4 substrate, albeit a weak one (Supplemental Fig. 3), and HIV patients concurrently take other medicines, especially during cancer chemotherapy. The five PIs produced a common feature pharmacophore model exhibiting four hydrogen bond acceptors (green), one hydrogen bond donor (purple), and three hydrophobes (cyan) (Fig. 5). Because NFV inhibits PGE2-stimulated MRP4/ABCC4 ATPase, we hypothesized that the pharmacophore of NFV and PGE2 would share similar features. The common pharmacophore of NFV was used to map PGE2 using the ligand pharmacophore mapping protocol. PGE2 was allowed to miss two features and had a fit value of 1.63. NFV (in yellow) is shown fitting to the pharmacophore, and indicates that PGE2 and NFV, by sharing most features, share a binding site in MRP4 (Fig. 5B, left). In contrast, quercetin was difficult to map and even after allowing for three missing features, the fit value was only 0.056 (Fig. 5B, middle). Moreover, quercetin does not appear to map to the same central hydrogen bond donor as PGE2 (Fig. 5, right). The strong similarity of the NFV and PGE2 pharmacophore suggests either a similar binding mode or site on MRP4. In contrast, quercetin’s markedly different pharmacophore suggests either a different binding mode or distinct binding site.
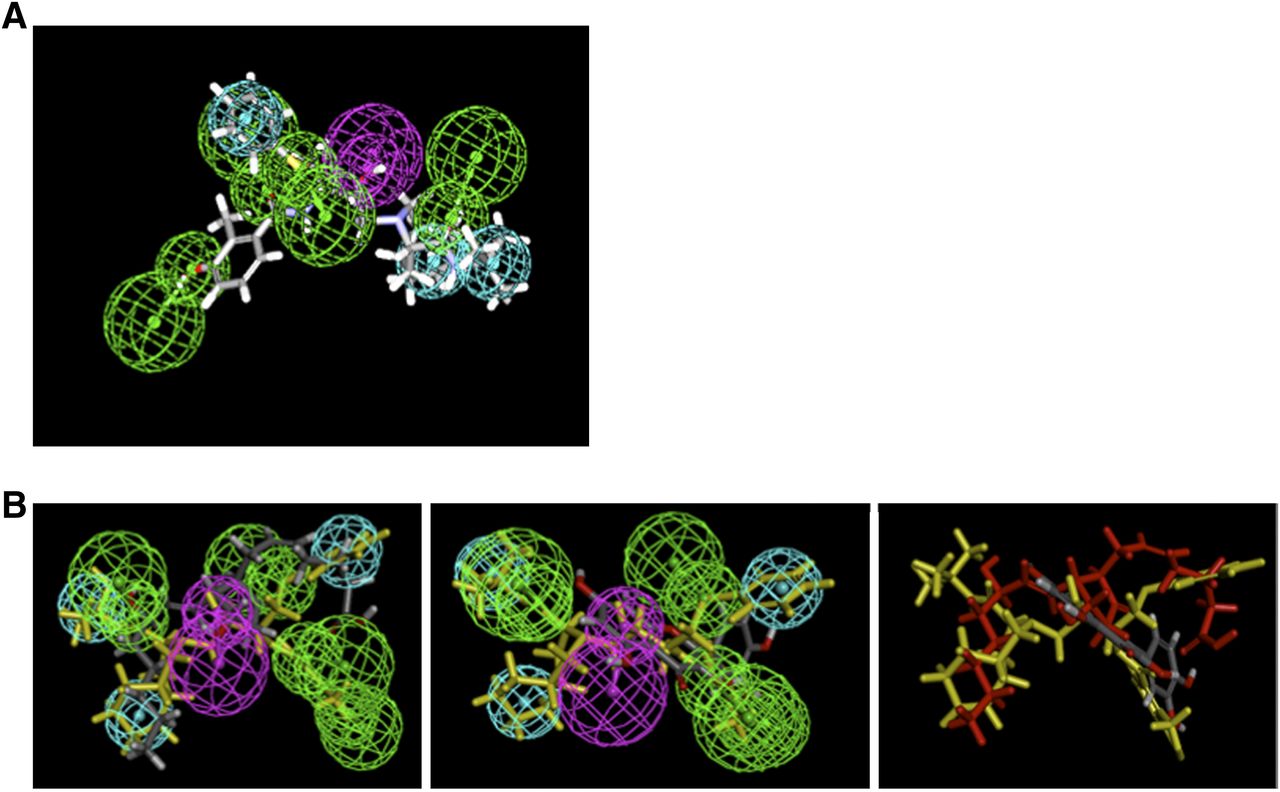
A pharmacophore model of MRP4 reveals distinct substrate properties. (A) PI MRP4 substrate common features pharmacophore showing nelfinavir mapped. Features include hydrogen bond acceptors (green) with vectors, hydrogen bond donor (purple), and vectors and hydrophobic features (cyan). (B) Mapping PGE2 (left) and quercetin (middle) onto the common pharmacophore with NFV shown in yellow. (Right) NFV (yellow), PGE2 (red), and quercetin (gray) are mapped to show that quercetin does not overlap with other two compounds.
Discussion
The success of HAART therapy in the treatment of HIV has increased the life expectancy among those infected with HIV. Their prolonged survival increases the likelihood of non-HIV, but age associated cancers (e.g., lung and breast cancer). These HAART maintenance regimens could enhance the efficacy of cancer therapy because these agents (e.g., tenofovir, adefovir) (Gallant and Deresinski, 2003) also exhibit antitumor activity (De Clercq et al., 1999). Thus, their enhanced accumulation might increase cytotoxicity in cancers in which ABCC4/MRP4 is the major route of egress. We hypothesized that some PIs (another class of drugs in HAART) might interact with ABCC4/MRP4 as either substrates or inhibitors. Among those tested (ritonavir, NFV, amprenavir, indinavir, and saquinavir), only NFV was highly effective. By screening with ATPase assays on ABCC4/MRP4 programmed membrane vesicles as well as multiple cell lines overexpressing ABCC4/MRP4 and Abcc4/Mrp4 KO MEFs, we show that NFV is an ABCC4/MRP4 substrate. This is further supported by findings demonstrating that ABCC4/MRP4 has a role in protecting against the cytotoxic effects of NFV, a recently reported cytotoxin and potential chemotherapeutic (Gupta et al., 2005; Yang et al., 2005; Shim et al., 2012). MRP4 overexpression and absence both protects from and sensitizes to the cytotoxic effects of NFV, respectively. Because NFV enhances the cytotoxicity of both PMEA and MTX, our studies have strong implications for treating HIV patients on HAART with cancer chemotherapeutic regimens. To further characterize the features of drugs interacting with ABCC4/MRP4, we developed ABCC4/MRP4 pharmacophore models. Such models have been described for other transporters as a technique to assist in identifying salient properties of substrates and inhibitors (Ekins et al., 2012). Our ABCC4/MRP4 pharmacophores included a model based upon NFV and other PIs, as well as models derived from MRP4 inhibitors described in the literature. There was overlap between these pharmacophores because each was dominated by multiple hydrogen bond acceptors (Fig. 5; Supplemental Fig. 2). The PI MRP4 substrate pharmacophore included NFV and is likely useful to predict other drugs, especially anticancer and antiviral, that could provoke enhanced cytotoxicity among HIV-infected cancer patients secondary to MRP4 inhibition.
ABCC4/MRP4 and ABCG2 share a number of common substrates from endogenous compounds (e.g., cyclic GMP) (de Wolf et al., 2008; Russel et al., 2008) to antiretroviral medications (PMEA, tenofovir) (Takenaka et al., 2007) to anticancer chemotherapeutics (MTX and irinotecan) (Chen et al., 2002; Volk and Schneider, 2003). However, with respect to PI, none of those tested were good ABCG2 substrates (Kis et al., 2010). Notably, NFV is only an inhibitor of ABCG2 (Gupta et al., 2004). Considering that ABCG2 and ABCC4/MRP4 are broadly expressed (Takenaka et al., 2007), these findings suggest that in cells (both normal and cancer infected with HIV) that coexpress both transporters, ABCG2 inhibition by NFV might be overridden by ABCC4/MRP4-mediated export of NFV. This suggests that ABCC4/MRP4 has not only the potential to disarm nucleoside-based antiviral inhibitors (e.g., PMEA, tenofovir, etc.), but also PIs like NFV that share a pharmacophore recognized by ABCC4/MRP4 (Fig. 6). It is notable that our PI MRP4 substrate pharmacophore analysis suggests that atazanavir (Supplemental Table 4) is a potential ABCC4/MRP4 substrate. Although not formally tested, this seems to be supported by recent studies of Bierman et al. (2010). Validating this in vitro may represent future work.

Schematic showing two potential drug binding pockets in MRP4 and possible drug–drug interactions. (Left) Quercetin (QCE) and PGE2 have distinct binding sites. (Right) PMEA, MTX, and NFV compete for the same binding pocket as PGE2. The black arrow in the left panel shows the direction of transport. NBD, nucleotide binding domain.
After screening a number of clinically relevant HIV drugs, our studies find that unlike several other common PIs, only NFV strongly interacts with human and murine Abcc4/Mrp4. NFV appears to be both a substrate and an inhibitor of MRP4. The ability of ABC transporter substrates to act as both an inhibitor and substrate is not unique to either NFV or MRP4/ABCC4. Notably, tyrosine kinase inhibitors display both substrate and inhibitor properties in their interactions with ABCB1 or ABCG2 (Brózik et al., 2011).
How does NFV inhibit MRP4/ABCC4? Because NFV was recently shown to interact with multiple kinases (Xie et al., 2011), we might infer that it interacts with the ABCC4/MRP4 nucleotide binding domain producing a reduction in ATPase activity. However, this molecular mechanism seems unlikely for two reasons: first, NFV only stimulates and does not inhibit ABCC4/MRP4 ATPase activity; and second, the predicted ATP binding site that NFV reportedly interacts with on EGFR bears little sequence resemblance to the ABCC4/MRP4 nucleotide binding domains (Kool et al., 1997; Xie et al., 2011).
The divergent substrate-dependent effect of NFV on substrate-stimulated ATPase suggests either distinct binding sites or a different binding mode in the same binding pocket. For example, NFV inhibits PGE2-stimulated MRP4/ABCC4 ATPase. This suggests that NFV and PGE2 share either a common binding site or have a similar biding mode. Because pharmacophore modeling revealed shared properties of both PGE2 and NFV on ABCC4/MRP4, it is likely a similar binding mode. This proposition is supported by a PGE2 fit value of 1.62 with respect to NFV pharmacophore. In contrast, quercetin poorly fits the NFV pharmacophore and just minimally overlaps (fit value of 0.056). This agrees with the inability of NFV to affect quercetin-stimulated ATPase. Because quercetin can stimulate ABCC4/MRP4 ATPase but is not inhibited by NFV (Fig. 1B, right), it is likely that the quercetin binding mode or site is distinct. At this point, we cannot distinguish whether different MRP4/ABCC4 substrates and inhibitors have distinct binding sites or binding modes. However, we note that Van Aubel et al. (2005) showed that ABCC4/MRP4 concurrently transports urate and either cAMP or cGMP. This suggests that MRP4/ABCC4 has a large binding pocket that might allow occupation of multiple substrates/inhibitors that adopt different binding modes. Moreover, the propensity of a substrate to assume different binding modes on MRP4 might increase the likelihood of drug–drug interactions among cytotoxic substrates relying on MRP4 export. This is supported, in part, by our studies showing that NFV increases the cytotoxicity of MTX in MRP4-overexpressing cells a finding consistent with the evidence that, like NFV, MTX shares a binding mode with PGE2 as shown by its inhibition of PGE2-stimulated MRP4/ABCC4 ATPase activity (Sauna et al., 2004).
NFV has shown effectiveness as a potential chemotherapeutic against several different tumor cell lines, possibly by suppressing activity of the protein kinase B/Akt pathway (Gills et al., 2007). Consistent with this, a recent proteome-screen predicted that NFV was capable of binding protein kinase B/Akt as well as other members in the protein kinase superfamily (Xie et al., 2011). Although protein kinase B/Akt activity may impact the sensitivity of tumor cells to NFV, our study reveals MRP4/ABCC4 amounts determine the cellular concentration of NFV. Consequently the accumulation of NFV in target cells, tissues, or organs will be determined by the amount of MRP4/ABCC4. These findings suggest that agents impairing MRP4/ABCC4 export might enhance cytotoxicity by increasing intracellular concentration of NFV. Conversely, we show that NFV, as a new ABCC4/MRP4 inhibitor, reduces export, thereby increasing the toxic effects of known ABCC4/MRP4 substrates (e.g., adefovir and MTX, respectively) by way of drug–drug interactions. A further extension of these findings is that inhibition of MRP4/ABCC4-mediated drug export has the potential to alter metabolism of drugs, especially in the kidney, which has high levels of MRP4/ABCC4 (Leggas et al., 2004; Takenaka et al., 2007). In addition, our computational modeling provides new insights into the pharmacophore of drugs with the potential to interact with ABCC4/MRP4, enabling us to predict which drugs might alter ABCC4/MRP4 function. In summary, these in vitro and computational pharmacophore findings highlight an important therapeutic mechanism that might explain both unexpected enhancements in antitumor efficacy, but also host toxicities that could occur when treating HIV-infected cancer patients receiving HAART regimens.
Acknowledgments
The authors kindly acknowledge Accelrys for providing Discovery Studio.
Authorship Contributions
Participated in research design: Fukuda, Takenaka, Sparreboom, Wu, Ekins, Ambudkar, Schuetz.
Conducted experiments: Fukuda, Takenaka, Wu, Ekins.
Contributed new reagents or analytic tools: Sparreboom, Cheepala.
Performed data analysis: Fukuda, Takenaka, Sparreboom, Wu, Ekins, Ambudkar, Schuetz.
Wrote or contributed to the writing of the manuscript: Fukuda, Sparreboom, Ekins, Ambudkar, Schuetz.
Footnotes
- Received April 22, 2013.
- Accepted June 17, 2013.
This work was supported by ALSAC; the National Institutes of Health National Institute of General Medical Science [Grant 2R01-GM60904]; and the National Institutes of Health National Cancer Institute [Grants P30-CA21745, P30-CA21865, and P30-CA021765]. C.-P.W. and S.V.A. were supported by the Intramural Research Program of the National Institutes of Health [National Cancer Institute Center for Cancer Research]. S.E. is an employee of Collaborations in Chemistry and consults for Collaborative Drug Discovery Inc., as well as companies in the pharmaceutical industry, and has no conflicts of interest with this work.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ABC
- ATP binding cassette
- ABCC4
- ATP binding cassette transporter 4
- HAART
- highly active antiretroviral therapy
- HEK
- human embryonic kidney
- HIV
- human immunodeficiency virus
- KO
- knockout
- MEF
- mouse embryo fibroblast
- MK571
- 3-([{3-(2-[7-chloro-2-quinolinyl]ethenyl)phenyl}-{(3-dimethylamino-3-oxopropyl)-thio-methyl]thio)propanoic acid
- MRP4
- multidrug resistance protein 4
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MTX
- methotrexate
- NFV
- nelfinavir
- PGE2
- prostaglandin E2
- PI
- protease inhibitor
- PMEA
- 9-(2-phosphonylmethoxyethyl) adenine
- POM
- pivaloyloxymethyl
- WT
- wild-type
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}