


Abstract
α7 nicotinic acetylcholine receptors (nAChRs) have been a puzzle since their discovery in brain and non-neuronal tissues. Maximal transient probability of an α7 nAChR being open with rapid agonist applications is only 0.002. The concentration dependence of α7 responses measured from transfected cells and Xenopus laevis oocytes shows the same disparity in potency estimations for peak currents and net charge, despite being studied at 1000-fold different time scales. In both cases the EC50 was approximately 10-fold lower for net charge than for peak currents. The equivalence of the data obtained at such disparate time scales indicates that desensitization of α7 is nearly instantaneous. At high levels of agonist occupancy, the receptor is preferentially converted to a ligand-bound nonconducting state, which can be destabilized by the positive allosteric modulator N-(5-chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea (PNU-120596). Such currents can be sufficiently large to be cytotoxic to the α7-expressing cells. Both the potentiating effect of PNU-120596 and the associated cytotoxicity have a high temperature dependence that can be compensated for by serum factors. Therefore, despite reduced potentiation at body temperatures, use of type II positive allosteric modulators may put cells that naturally express high levels of α7 nAChRs, such as neurons in the hippocampus and hypothalamus, at risk. With a low intrinsic open probability and high propensity toward the induction of nonconducting ligand-bound states, it is likely that the well documented regulation of signal transduction pathways by α7 nAChRs in cells such as those that regulate inflammation may be independent of ion channel activation and associated with the nonconducting conformational states.
Introduction
Nicotinic α7 receptors have become one of the most interesting and promising new therapeutic targets since they were first discovered to be the sites of α-bungarotoxin (α-btx) binding in the brain approximately 20 years ago. Data suggest that activation of α7 nicotinic acetylcholine receptors (nAChRs), a subtype with uniquely high calcium permeability, can provide cytoprotection, enhance performance in behavioral tasks related to cognitive function, reduce auditory gating deficits seen in schizophrenia (for review, see Haydar and Dunlop, 2010; Thomsen et al., 2010), and modulate inflammation (de Jonge and Ulloa, 2007). Although structurally related to the high-affinity nicotine-binding sites in the brain and the α-btx-sensitive nAChR of the neuromuscular junction, α7 receptors are in many ways unique from other ligand-gated ion channels and confound our conventional approaches and usual assumptions. In several ways, the α7 nAChR represents a primordial type of receptor. They are functional without being coassembled with specialized accessory subunits required by other nAChR subtypes (Drisdel and Green, 2000). They are activatable by the ubiquitous acetylcholine (ACh) precursor, choline (Papke et al., 1996), and are found in many types of nonexcitable, non-neuronal cells (de Jonge and Ulloa, 2007). They open rather inefficiently, and, although they rapidly desensitize in the presence of high concentrations of agonist, once desensitized they do not convert to a high-affinity state, as do other nAChRs (Williams et al., 2011c). These are the features that one might imagine would be present in an ion channel of an organism lacking an evolved nervous system with rapid chemical synaptic transmission. They make α7 nAChRs invisible to agonist-binding experiments, and for many years they were almost impossible to characterize electrophysiologically.
Both academic and industrial laboratories have discovered numerous agonists with selectivity for α7 receptors (Horenstein et al., 2008), and in recent years an alternative therapeutic approach based on allosteric modulation has gained momentum because of the discovery of many structurally diverse positive allosteric modulators (PAMs) that are selective for α7 receptors (Williams et al., 2011c). To date, the α7 PAMs are classified into one of two categories based on their properties of modulation. The type I PAMs increase the magnitude of α7-mediated responses without large effects on response kinetics. The type II PAMs not only increase the magnitude of responses but also greatly slow or reverse the decay of macroscopic currents, resulting in very prolonged responses and enormous increases in net charge, which could translate into large changes in intracellular calcium. In addition, the type II PAMs can evoke currents from receptors desensitized by previous agonist applications (Grønlien et al., 2007).
In cases in which activation of the α7 ion channel is necessary for a desired therapeutic effect (Briggs et al., 2009), a PAM-based therapeutic approach might offer several potential advantages over agonist-based strategies. However, a PAM-based strategy may also be subject to some potentially important limitations, especially if the potentiation of the calcium-permeable α7 receptor currents becomes so large that calcium homeostasis is disrupted, causing cell death (Orr-Urtreger et al., 2000; Lukas et al., 2001). Nonetheless, in vivo studies with α7 PAMs have reported no major concerns regarding toxicity (Williams et al., 2011c), which might suggest that specific factors may prevent overactivation of α7 receptors in the physiological context. We have previously reported that with strong stimulation by the very efficacious PAM N-(5–2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea (PNU-120596), the majority of the receptors revert to a PAM-insensitive desensitized state (Di) (Williams et al., 2011b). In addition, it has been reported that the ability of α7 PAMs to potentiate α7 currents may be significantly reduced at physiological temperature (Sitzia et al., 2011), a factor that would reduce both potential toxicity and therapeutic utility.
To improve our understanding of the unique properties of the α7 nAChR and potential complications that may rise from the use of ostensibly strong PAMs such as PNU-120596, we developed a HEK 293 cell line stably expressing both the human α7 nAChR and the chaperone protein RIC-3 (Treinin, 2008) (A7R3HC10). These cells were used for whole-cell patch-clamp studies, which validate previously published models of α7 activation and desensitization that were based on data from the Xenopus oocyte expression system. We also characterize the degree to which effects of type I and type II α7 PAMs may be temperature-dependent. Whereas temperature can affect the in vitro cytotoxicity profile of an α7 PAM, in vivo factors may compensate for the effect of temperature.
Materials and Methods
cDNA Clones Used for Stable Expression of Human α7 and Human RIC-3 in HEK 293 Cells.
The human α7 receptor clone was obtained from Dr. Jon Lindstrom (University of Pennsylvania, Philadelphia, PA). The human RIC-3 clone was obtained from Dr. Millet Treinin (Hebrew University, Jerusalem, Israel) and was cotransfected with α7 to improve receptor expression (Halevi et al., 2002).
Chemicals.
Solvents and reagents were purchased from Sigma-Aldrich (St. Louis, MO). BSA (fraction V) was from Thermo Fisher Scientific (Waltham, MA). PNU-120596 was synthesized by Dr. Jingyi Wang as described in Williams et al. (2011b). Unlabeled α-btx was purchased from Biotoxins, Inc. (Saint Cloud, FL). 125I-labeledα-btx was prepared and generously provided by Dr. Ralph Loring (Northeastern University, Boston, MA). Cell culture supplies were purchased from Life Technologies (Grand Island, NY). The Hanks' balanced saline solution (HBSS) (Life Technologies) contained 1.26 mM CaCl2, 0.493 mM MgCl2, 0.407 mM MgSO4, 5.33 mM KCl, 0.441 mM KH2PO4, 4.17 mM NaHCO3, 137.93 mM NaCl, 0.338 mM Na2HPO4, and 5.56 mM d-glucose. Fresh ACh stock solutions were made on each day of experimentation. PNU-120596 stock solutions were prepared in DMSO, stored at −20°C, and used for up to 30 days. PNU-120596 solutions were prepared fresh each day at the desired concentration from the stored stock. 5-Hydroxyindole (5-HI) was purchased from Sigma-Aldrich. N-(5-Chloro-2-hydroxyphenyl)-N′-[2-chloro-5-(trifluoromethyl)phenyl]urea (NS-1738) was purchased from R&D Systems (Minneapolis, MN). TQS was generously supplied by Institut De Recherches Internationales Servier (Suresnes, France).
Equilibrium Radioligand Binding Assay.
Two to 3 days before binding assays, cells were plated in poly-d-lysine-treated 24-well dishes. Experiments were performed when the cells were ∼60 to 80% confluent. The growth medium was removed, and cells were washed one time with Dulbecco's phosphate-buffered saline (Life Technologies). 125I-α-btx-containing solutions (0.05–7 nM) were added to the cells and incubated at room temperature for 3 h. Nonspecific binding was determined with the addition of 1 μM unlabeled α-btx in separate wells. After the 3-h incubation, the radioligand was removed, and the cells were washed three times with ice-cold Dulbecco's phosphate-buffered saline. The cells were then solubilized with 0.1 M NaOH-0.1% SDS, and samples were counted in a gamma counter (Beckman Coulter, Brea, CA). Saturation binding curves were fit to the equation Bmax [ligand]/(EC50 + [ligand]) with Kaleidagraph 3.0.2 (Abelbeck Software, Reading, PA). Each condition was tested in triplicate for each experiment, and each experiment was repeated three independent times.
Cytotoxicity Experiments.
The cells were maintained in normal growth medium, and experiments were completed in HBSS. One day before the toxicity studies were performed, two sets of A7R3HC10 cells from the same passage and two sets of untransfected HEK 293 cells from the same passage were plated in 96-well plates at a density of 15,000 cells/well in normal growth medium [Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS)] and incubated at 37°C with 5% CO2. Experimental solutions containing the desired concentrations and chemicals of sufficient volume to treat both sets of untransfected HEK 293 cells and both sets of A7R3HC10 cells were prepared in HBSS and then applied to the cells after the normal growth medium was removed. One set of A7R3HC10 cells and one set of untransfected HEK 293 cells were placed in an incubator set to 28°C with 5% CO2. The other sets of A7R3HC10 cells and untransfected HEK 293 cells were placed in an incubator set to 37°C with 5% CO2. Incubations with experimental treatments were 2 h because maximal toxicity occurred within 2 h, as determined from separate experiments evaluating the onset of toxicity at various time points during 24 h after treatments (data not shown). After the 2-h treatment period, the experimental solutions were replaced with 100 μl of HBSS and 20 μl/well of CellTiter 96 Solution (Promega, Madison, WI) and incubated for 2 to 4 h at 37°C with 5% CO2 after which absorbance readings were made with a microplate spectrophotometer at 490 nm (BioTek Instruments, Winooski, VT). Each condition in an experiment was tested in triplicate. The triplicate values were averaged to obtain a mean value for each treatment condition in an individual experiment, and experiments were repeated on at least three independent occasions. Background absorbance was measured from cell-free wells and subtracted from all control and experimental test conditions. Absorbance readings from experimental test conditions were normalized to the absorbance values of untreated/DMSO vehicle controls, which were defined as 100% cell viability. The DMSO was used to dissolve PNU-120596, which is insoluble in water. The highest concentration of DMSO applied with PNU-120596 was limited to 0.3%, and this occurred in the 30 μM PNU-120596 condition. The 20% DMSO and 30 μM thapsigargin conditions were used as positive controls for toxicity. The statistical significance of PNU-120596 treatments was assessed by comparing the viability values obtained with a given concentration of choline with or without PNU-120596 via two-tailed Student's t-tests. The comparison was made between choline versus choline and PNU-120596 cotreatment groups, rather than between untreated controls versus choline and PNU-120596 cotreatments, because addition of choline alone tended to increase the apparent cell viabilities relative to those for the untreated/vehicle controls. Cells with fewer than 30 passages after stable transfection were used in all toxicity experiments.
Whole-Cell Patch-Clamp Electrophysiology.
Whole-cell responses were recorded using an Axopatch 200 amplifier (Molecular Devices, Union City, CA). In the experiments that involved temperature adjustments, the temperature was controlled with a TC-324B temperature controller (Warner Instruments, Hamden, CT). Cells were bathed in an external solution containing 165 mM NaCl, 5 mM KCl, 2 mM CaCl2, 10 mM glucose, 5 mM HEPES, and 0.001 mM atropine, with the pH adjusted to 7.3 with NaOH. Patch pipettes (Sutter Instruments, Novato, CA) were pulled to a tip diameter of ∼2 μm, fire-polished to approximately 5 MΩ and filled with an internal solution containing 147 mM CsCl, 2 mM MgCl2, 1 mM CaCl2, 10 mM EGTA, 10 mM HEPES, and 5 mM Mg-ATP, pH adjusted to 7.3 with CsOH. Cells were held at −70 mV. Recordings were low-pass-filtered to 5 kHz and digitized at 50 kHz with a DigiData 1440 or 20 kHz with a DigiData 1322A (Molecular Devices) using Clampex data acquisition software (Molecular Devices). A 10-ms test pulse of −10 mV was used to determine access and input resistances before each response. For experiments performed at room temperature, whole-cell recordings were analyzed if access resistances were <40 MΩ and membrane resistances were >200 MΩ. On average, the access resistance, membrane resistance, and cell capacitance values were 15.8 ± 0.6 MΩ, 1.55 ± 0.15 GΩ, and 55.2 ± 6.6 pF, respectively. For experiments evaluating the temperature dependence of PNU-120596, these basic criteria were necessarily relaxed at 37°C. Sweeps with access resistance <40 MΩ, input resistance >100 MΩ, and holding current <700 pA were included in the analysis. No attempt was made to compensate for series resistance or for the liquid junction potential, although the liquid junction potential was calculated to be 4.7 mV. The whole-cell recordings were analyzed with Clampfit 10 (Molecular Devices).
For the ACh concentration response and methyllycaconitine (MLA) inhibition curves, rapid drug application to whole cells was performed with a Burleigh piezoelectric stepper (EXFO, Concord, ON, Canada) as described previously (Williams et al., 2011a). Theta glass (Sutter Instruments, Novato, CA) was pulled, scored, and then broken by hand to create a drug application pipette with a diameter of 350 μm, which was mounted to the stepper. The voltage signal used to control the piezoelectric stepper was conditioned by an RC circuit (τ = 2 ms) to reduce oscillations and avoid damage to the crystal. Solution exchange times were 2.6 ± 0.8 ms as determined by measuring holding current shifts (10–90% rise times) upon moving diluted external solution over an open recording pipette on each day of experimentation. The open recording pipette was positioned just above the surface of the recording chamber and in the same arrangement relative to the drug application pipette during a whole-cell recording. It should be stated that the exchange time estimate measured in this manner is probably only a lower-limit (i.e., fastest time possible) of the solution exchange time achieved for a whole cell during a recording. Three applications of 300 μM ACh were applied initially followed by 3 test applications of 1 μM to 3 mM ACh. Interstimulus intervals were 60 s. The responses from the initial three responses to 300 μM ACh were averaged, as were the three responses from the test concentrations of ACh. The averaged response of each cell to the test ACh concentration was normalized to the averaged response to 300 μM ACh to compensate for varying levels of receptor expression among individual cells. The normalized responses were subsequently adjusted to reflect the response relative to the maximal normalized ACh-evoked response, which was defined as 1. Both peak current and net charge responses were measured; net charge responses were measured as the area under the activation curve during 1 s of ACh application. Means and S.E.M.s were calculated from the responses of four to eight cells at each test concentration. Experiments for the MLA inhibition curve were performed in a manner similar to that for the ACh concentration-response curve, with the initial concentration of ACh being 170 μM and test concentrations consisting of 170 μM coapplication with 3 nM to 100 μM MLA. No preapplications of MLA were made. Each data point represents the mean and S.E.M. of four to six cells at each test concentration. For concentration-response relationships, the data were plotted using Kaleidagraph 3.0.2, and curves were generated as the best fit of the average values to the Hill equation, response = (Imax[agonist]n)/([agonist]n + (EC50)n), where Imax denotes the maximal normalized response and n represents the Hill coefficient. Imax values of the curve fits were constrained to equal 1. In the case of the MLA inhibition concentration-response relationship, negative Hill slopes were used. Error estimates of the EC50 values are the S.E.M.s of the parameters based on the Levenberg-Marquardt algorithm used for the generation of the fits (Press, 1988).
In all other whole-cell electrophysiology experiments, local applications of drug were made using single-barrel glass pipettes attached to a Picospritzer (General Valve, Fairfield, NJ) with Teflon tubing (11.5–14.5 psi). The application pipette was placed within 10 to 15 μm of the cell. Drug applications were 3 s in duration and were made every 60 s. Drugs applied with this method were determined to be diluted 1.5-fold by the time they reached the cell surface (see supplemental data). In the temperature experiments, three baseline responses were recorded at room temperature (23.5°C), after which responses were recorded as the temperature was increased to 37°C. Three responses were recorded at 37°C, and then the temperature was returned to 23.5°C. Cells with responses that failed to recover to 50% of the average baseline response upon temperature reduction from 37 to 23.5°C were excluded from analysis; 6 of 70 total cells failed to meet this criterion. Responses were measured as peak currents. In most cases, the currents recorded at 37°C were normalized to the average peak amplitude of the three initial baseline responses recorded at 23.5°C. It is important to note that at 37°C the quality of the whole-cell seals usually decreased. Because of this, the parameters used to define an acceptable whole-cell recording at 37°C were more relaxed than they would be for a typical whole-cell recording made at room temperature. Whole-cell seals with access resistance <40 MΩ, input resistance >100 MΩ, and holding current <700 pA at 37°C were included in the analysis. Before the increase in temperature, access resistances were <15 MΩ, input resistances were >1 GΩ, and the holding current was between −50 and 0 pA. In most cases, if the patch survived the time at 37°C, the whole-cell parameters improved as temperatures returned to room temperature.
In the comparison of the effect of BSA on potentiation by PNU-120596 at 37°C, responses are shown as both normalized currents and the absolute magnitude of evoked currents. To test for the statistical significance of BSA on PNU-120596 potentiation at 23.5 or 37°C, the sweeps obtained from each cell at 23.5 or 37°C were averaged to obtain one mean response from each cell at 23.5 or 37°C. The mean responses from cells recorded at 23.5 or 37°C either in the absence or presence of 30 μM BSA were then compared with a two-tailed Student's t test using Microsoft Excel (Microsoft, Redmond, WA).
Results
Concentration Dependence of ACh-Evoked Peak Current and Net Charge Responses from A7R3HC10 Cells.
The ACh concentration-response relationship for ACh-evoked whole-cell currents using a rapid solution switching onto A7R3HC10 cells resulted in nonsuperimposable curves for peak current and net charge measurements, as expected for α7 receptors (Fig. 1). The EC50 values determined from peak currents and net charge were 167 ± 20 and 26 ± 6 μM, respectively. Note that these values are not significantly different from those previously reported for human α7 nAChRs expressed in Xenopus laevis oocytes, 173 ± 8 and 21 ± 3 for peak currents and net charge, respectively (Papke and Porter Papke, 2002), even though the time scale of the responses was 1000-fold faster than that of oocyte recording.

A, representative currents illustrating the varying shapes of responses at different concentrations of ACh contrasted with responses to 300 μM ACh. B, the concentration-response relationship for whole-cell peak currents and net charge responses evoked by ACh. The curve for net charge was fit between 1 and 300 μM ACh (■). Each point represents the mean ± S.E.M. from four to eight cells. Net charge responses were calculated for a period of 1 s after ACh application.
The curve for net charge was fit between the 1 and 300 μM ACh points because the net charge was reduced from the maximum at ACh concentrations greater than 300 μM. A unique feature of the α7 receptor is the concentration-dependent desensitization that is rapidly induced with applications of high agonist concentrations. The reductions in net charge at high concentrations seen in this experiment were more pronounced than normally occurs in oocyte experiments. This was probably because of differences in the presentation of agonist; drug applications were made here with a system that provided solution exchanges on the order of several milliseconds versus the drug delivery that occurs on a time scale of 3 to 4 s in a typical oocyte experiment. The rapid application of high agonist concentrations produced synchronous activation and desensitization of the α7 receptor population, resulting in extremely sharp macroscopic responses with minimal area (Fig. 1A). This is also demonstrated by comparing the rise times and rise slopes with increasing concentrations of ACh: the 10 to 90% rise times became shorter and the 10 to 90% rise slopes became steeper as ACh concentrations increased (Table 1). The peak in the responses to the applications of 1 or 3 mM ACh occurred faster than the solution exchange, indicating that the maximal synchronous channel opening occurs at ACh concentrations substantially lower than these high concentrations, which would probably saturate the agonist binding sites. This finding is consistent with channel activation occurring with the highest probability when there is only partial occupancy of the multiple binding sites (Williams et al., 2011a,b).
Ten to 90% rise times and rise slopes with increasing concentrations of ACh
Calibration of ACh-Evoked Currents Based on Specific Binding of 125I-α-btx to Intact A7R3HC10 Cells.
We wished to obtain an estimate of the total number of receptors contributing to the ACh-evoked responses of A7R3HC10 cells. Specific labeling with 3 nM 125I-α-btx was observed in intact A7R3HC10 cells, whereas no specific labeling was detected in untransfected HEK 293 cells treated in parallel with the same solutions (Fig. 2A). From three saturation binding experiments to intact cells, the Kd of α-btx was 991 ± 67 pM and the Bmax was 7.7 × 10−8 ± 1.3 × 10−8 pmol/cell (Fig. 2, B and C). This Bmax translates to an average of 9300 ± 1500 receptors expressed/cell, assuming that 5 molecules of radiolabeled α-btx were bound to each α7 receptor (Palma et al., 1996).

Saturation binding of 125I-α-btx binding to intact A7R3HC10 cells. A, no specific binding was detected with untransfected HEK 293 cells. Values are the mean ± S.E.M. of five to six replicates. B, a saturation binding curve from one representative experiment. C, Scatchard transformation of the same data. The average Kd and Bmax values from three independent saturation binding experiments are 1000 ± 70 pM and 7.7 × 10−8 ± 1.3 × 10−8 pmol/cell, respectively. Specific binding is defined as the difference between total binding and nonspecific binding. Nonspecific binding was determined using 1 μM unlabeled α-btx.
The average peak current and net charge evoked by 300 μM ACh from A7R3HC10 cells (n = 33) were 163 ± 26 pA and 9800 ± 1500 pA × ms, respectively. The single-channel amplitude of α7 channels (potentiated by PNU-120596) was determined to be approximately 7.8 pA (Williams et al., 2011b); this means that ∼20 α7 channels were open at the peak of an average current. If an average cell expresses 9300 α7 ion channels, and all of those ion channels are equally activatable just before the agonist stimulation, the probability of an α7 receptor being open (Popen) at the peak of the current evoked by 300 μM ACh is approximately 0.002. Assuming an average single-channel open lifetime of 0.1 ms (Williams et al., 2011b), an average net charge response to a 1-s application of 300 μM ACh contains ∼12,000 channel openings. On the basis of these numbers, individual channels opened on average only 1.3 times during the 1 s of 300 μM ACh application. These numbers are certainly rough estimates, but they suggest that the instantaneous Popen of α7 is never high, not even immediately after the presentation of a strong agonist stimulus and that an average individual α7 channel opens less than 2 times before becoming desensitized in response to 300 μM ACh. The reduced net charge observed with the application of ACh concentrations higher than 300 μM is also indicative of lower Popen with higher levels of agonist occupancy. Under steady-state conditions in which ACh is presented for prolonged periods of time, the α7 Popen will also be substantially less than the estimated maximum of 0.002 due to desensitization. The peak current of PNU-120596-potentiated responses from outside-out patches was previously used to obtain a lower limit of the number of channels in a patch. This lower limit estimate of channel number was used to estimate that the upper limit of the α7 Popen during a 12-s application of 60 μM ACh is 7.4 × 10−6 ± 3.0 × 10−6 (Williams et al., 2011b).
Evaluation of the In Vitro Cytotoxicity Profile of PNU-120596 in A7R3HC10 Cells.
We have previously characterized the potentiating activity of PNU-120596 on single α7 receptors in outside-out patches from transiently transfected HEK 293 cells (Williams et al., 2011b). We confirmed that the α7-mediated whole-cell currents of A7R3HC10 cells were similarly sensitive to this type II PAM (Fig. 3). Similar to what we have reported for α7 currents in oocytes, with these responses activated by pressure application of ACh or ACh plus PNU-120596, there was a modest increase in peak current and a 100-fold larger increase in net charge. The potentiated currents increased throughout the pressure application, and, as expected, the current evoked by the relatively high concentration of ACh alone decayed to baseline rapidly while the agonist pulse was still occurring.

Representative whole-cell currents from A7R3HC10 cells evoked by 3-s applications of either 1 mM ACh or 100 μM ACh in combination with 10 μM PNU-120596.
Neuronal α7 receptors have a high relative permeability to calcium (Séguéla et al., 1993), and the generation of large calcium-rich currents such as those promoted by coapplications of PNU-120596 with agonists might be likely to perturb the calcium homeostasis of the α7-expressing cells past the “set point” that most effectively promotes cell survival (Johnson et al., 1992). Contradictory results have been published regarding the in vitro cytotoxic effects of PNU-120596 (for review, see Williams et al., 2011c). In contrast to previous studies, which tested limited agonist and/or PAM concentrations, we wished to assess the potential toxicity profile of PNU-120596 over a range of agonist and PNU-120596 concentrations with the A7R3HC10 cell line. On the basis of the recent finding that high concentrations of agonist and PNU-120596 promote nonconducting Di states (Williams et al., 2011b), maximal cytotoxicity was hypothesized to occur upon treatment with relatively low concentrations of both agonist and PNU-120596 because this condition produces the greatest degree of ion channel activation over time. In addition, the existence of Di states was hypothesized to account for the discrepancy in the literature regarding the toxicity of PNU-120596, given that the two studies (Ng et al., 2007; Dinklo et al., 2011) reporting PNU-120596 toxicity used 100 μM choline as the agonist, whereas the one study (Hu et al., 2009) that reported a lack of PNU-120596 toxicity used a very strong agonist stimulus that potentially stabilized Di states.
Note that in these experiments we chose to use choline, rather than ACh, as the stimulating agonist to avoid issues that may accompany the labile nature of ACh. Choline has been shown to selectively activate α7 nAChRs with efficacy similar to that of ACh, although with approximately 10-fold lower potency (Papke et al., 1996). We based our measurements of “cytotoxicity” or “reductions in cell viability” on the reduction of a tetrazolium salt (CellTiter 96 solution) by dehydrogenase enzymes in metabolically active cells to a soluble formazan dye, the quantity of which is directly related to absorbance at 490 nm.
Initial cytotoxicity experiments were performed in full rich DMEM containing 10% FBS with treatments incubated at 37°C. A range of choline concentrations between 0 and 3 mM were coapplied with 10 μM PNU-120596, and in these experiments cell viabilities were reduced to approximately 15 to 25% of the controls whenever PNU-120596 was applied (data not shown), even without additional choline. Because PNU-120596 applied alone without agonist caused the same degree of toxicity as when applied with agonist, we conducted all further experiments under more defined conditions, which would allow us to better determine whether the cytotoxic effects of PNU-120596 were indeed dependent on α7 receptor activation.
When experiments were performed in HBSS, treatments with PNU-120596 alone failed to produce cytotoxicity, and all future experiments were performed in HBSS. However, when choline was coapplied with PNU-120596 and cells were incubated at 37°C in HBSS, no significant degree of toxicity was observed for any of the choline and PNU-120596 combinations (Fig. 4).
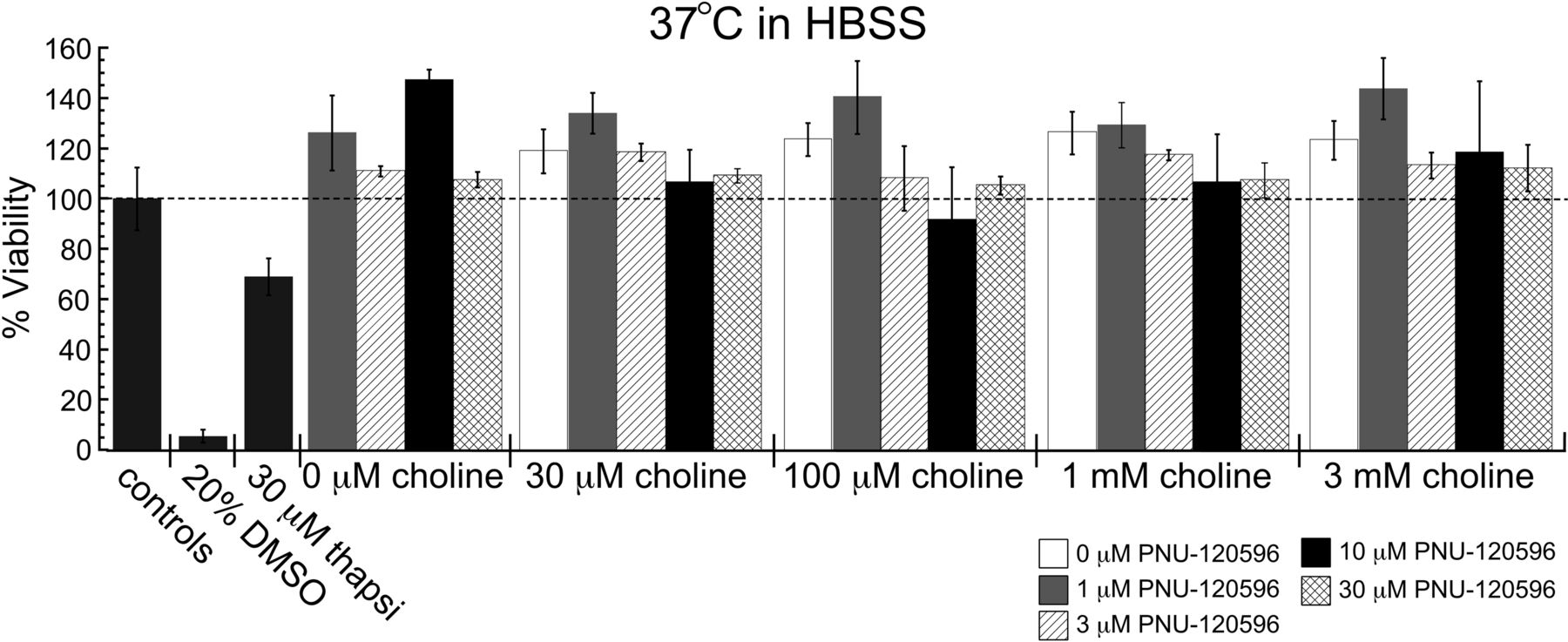
Effects of PNU-120596 and choline on the survival of A7R3HC10 cells. The cytotoxicity profile of 0 to 30 μM PNU-120596 with 0 to 3 mM choline when treatments were incubated at 37°C with 5% CO2. *, two-tailed p < 0.05. Each value is the average ± S.E.M. of three to five independent experiments. Two sets of cells were plated from the same passage 1 day before experiments. Untransfected HEK 293 cells were treated in parallel and were not affected by any choline and PNU-120596 treatments (data not shown). thapsi, thapsigargin.
Temperature Dependence of PNU-120596 on Potentiation of α7-Mediated Responses.
It has recently been reported that the potentiation of responses by the α7 PAMs PNU-120596 and 3,5-dihydro-5-methyl-N-3-pyridinylbenzo[1,2-b:4,5-b']dipyrrole-1(2H)-carboxamide hydrochloride (SB-206553) may have a dependence on the temperature, with potentiation being drastically reduced near physiological temperatures (Sitzia et al., 2011). To investigate the effect of temperature on α7 PAM efficacy, we tested the activities of type I and type II PAMs at 37°C with whole-cell recordings from the A7R3HC10 cell line. Control experiments (Supplemental Fig. 5) were conducted to determine the response stability at a fixed temperature and determine the effects of temperature on responses to ACh alone. In rundown control experiments performed without temperature adjustments, the amplitude of the responses at the end of the experiment were ∼70% of the responses at the beginning.
To determine the temperature effects on the α7-mediated currents, we obtained three responses at room temperature (23.5°C), progressively increased the temperature, recorded three responses at 37°C, and then reduced the temperature back down to 23.5°C. When 1 mM ACh was applied alone, peak currents at 37°C were 47 ± 3% of the initial baseline currents recorded at 23.5°C, and they recovered to ∼80% upon temperature reduction back to 23.5°C, a full recovery relative to the rundown control (Supplemental Fig. 5, C and D; Table 2).
Summary of the observed temperature effects on α7 PAM activity from whole-cell electrophysiology experiments
When 10 μM PNU-120596 was coapplied with 100 μM ACh, potentiated responses at 37°C were reduced to a much greater extent relative to those at 23.5°C than the response reduction that occurred when 1 mM ACh was applied alone (Fig. 5, A and B; Table 2). At 37°C PNU-120596-potentiated responses were only 12 ± 3% (88% reduction) of the baseline responses obtained initially at 23.5°C but recovered when the temperature was returned to 23.5°C. This result is consistent with recent findings (Sitzia et al., 2011) and predicts that there may be temperature dependence of the PNU-120596 cytotoxicity. However, because recent studies of the concentration dependence of PNU-120596 potentiation (Williams et al., 2011b) suggested that potentiation may be reduced with high concentrations of the agent owing to the induction of resistant forms of desensitization, producing an inverted U concentration-response relationship, we also wished to test whether temperature might have the effect of shifting this concentration-response relationship so that potentiation by a lower concentration of PNU-120596 might show less temperature sensitivity. We tested this hypothesis by repeating the temperature experiment with a 10-fold lower concentration of PNU-120596. The result with 1 μM PNU-120596 was similar to that with 10 μM PNU-120596 (Fig. 5, C and D; Table 2). When 1 μM PNU-120596 was coapplied with 100 μM ACh, potentiated responses at 37°C were 13 ± 3% (87% reduction) of the initial responses at 23.5°C and then recovered when the temperature was returned to room temperature.

Temperature dependence of PNU-120596 on potentiation of α7-mediated responses. Whole-cell currents from A7R3HC10 cells evoked by 3-s coapplication of 100 μM ACh and 10 or 1 μM PNU-120596 were recorded every 60 s with various temperatures between 23.5 and 37°C. A, time course for 100 μM ACh and 10 μM PNU-120596-evoked peak responses (○, n = 8). Temperature is indicated by □. B, representative traces of 100 μM ACh and 10 μM PNU-120596-evoked responses recorded at the indicated temperature. C, time course for 100 μM ACh and 1 μM PNU-120596-evoked peak responses (○, n = 9). Temperature is indicated by □. D, representative traces of 100 μM ACh and 1 μM PNU-120596-evoked responses recorded at the indicated temperature. Responses were normalized to the average peak amplitude of the three initial responses obtained at 23.5°C. Each data point is represented as the average normalized value ± S.E.M.
To determine whether the apparent temperature sensitivity of PNU-120596 is applicable to other PAMs, the protocol was repeated with the type I PAMs 5-HI and NS-1738 and the alternative type II PAM TQS (Grønlien et al., 2007) (Supplemental Fig. 6). The effects of temperature on potentiation of responses evoked by 100 μM ACh with either 1 mM 5-HI or 10 μM NS-1738 were similar (Table 2). Relative to baseline responses at room temperature, at 37°C 1 mM 5-HI-potentiated responses were 33 ± 2% (67% reduction) and 10 μM NS-1738-potentiated responses were 34 ± 5% (66% reduction). Neither of these effects was significantly larger than the reduction seen with ACh alone (p < 0.05). In both cases the potentiated responses recovered when temperatures returned to 23.5°C to an extent expected (∼70–80%) on the basis of the rundown control.
Likewise, the potentiation of 100 μM ACh-evoked responses by 10 μM TQS was not reduced any more at 37°C than the reduction in response amplitude that occurred when ACh was applied alone (Supplemental Fig. 6; Table 2). At 37°C, the responses potentiated by TQS were 46 ± 3% (54% reduction) of the baseline responses and recovered to the level of the rundown controls when the temperature was returned to 23.5°C.
Effect of Temperature on PNU-120596 Cytotoxicity.
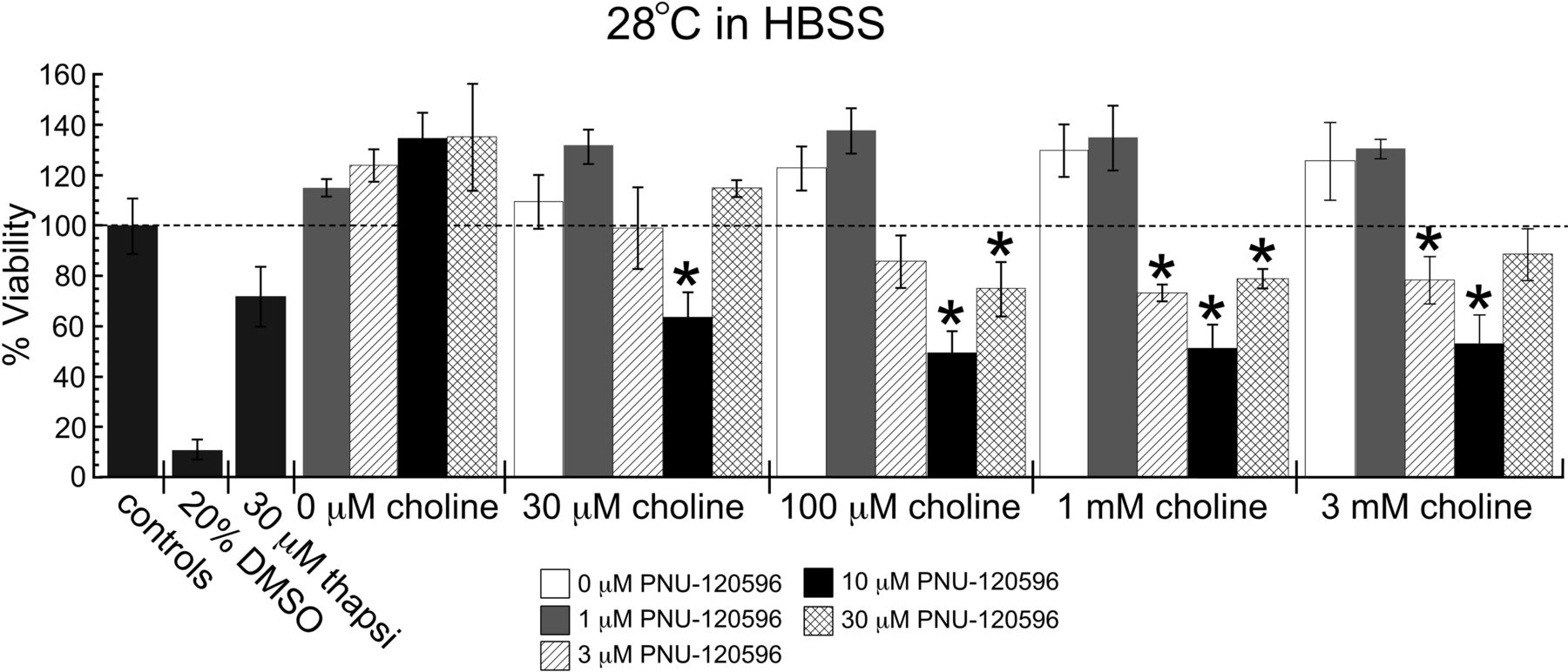
Our data indicate that among the agents tested, temperature had a uniquely large effect on potentiation by PNU-120596. Therefore, we reevaluated PNU-120596 for potential toxic effects at 28°C. Significant toxicity was observed when treatments were incubated at 28°C in HBSS in a manner that was dependent on the concentration of PNU-120596 and, to a lesser extent, on the concentration of choline (Fig. 6). Applications of choline alone or PNU-120596 alone did not reduce cell viabilities, although choline had a tendency to increase the viability relative to that of the control cells. No significant toxicity was produced with 1 μM PNU-120596 over the range of choline concentrations tested; however, at 3 μM PNU-120596 statistically significant toxicity was observed with 1 and 3 mM choline coapplications. The greatest degree of toxicity was observed when choline was applied with 10 μM PNU-120596, for all concentrations of choline tested, resulting in approximately 50% cell viabilities relative to those of the controls. Of interest, consistent with an inverted U concentration response, with 30 μM PNU-120596, significant reductions in cell viability were only observed when coapplications were made with 100 μM and 1 mM choline, and the magnitude of toxicity in this case was less than what was observed when treatments included 10 μM PNU-120596. None of the choline and/or PNU-120596 treatments had toxic effects on untransfected HEK 293 cells (data not shown).

PNU-120596 cytotoxicity measured at 28°C. Cytotoxicity profile of 0 to 30 μM PNU-120596 with 0 to 3 mM choline when treatments were incubated at 28°C with 5% CO2. *, two-tailed p < 0.05. Each value is the average ± S.E.M. of three to five independent experiments. Sets of cells were plated from the same passage 1 day before experiments as for the cells illustrated in Fig. 4. Untransfected HEK 293 cells were treated in parallel and were unaffected by all choline and PNU-120596 treatments at 28 and 37°C (data not shown). thapsi, thapsigargin.
Cytotoxic Effects of PNU-120596 Are MLA-Sensitive.
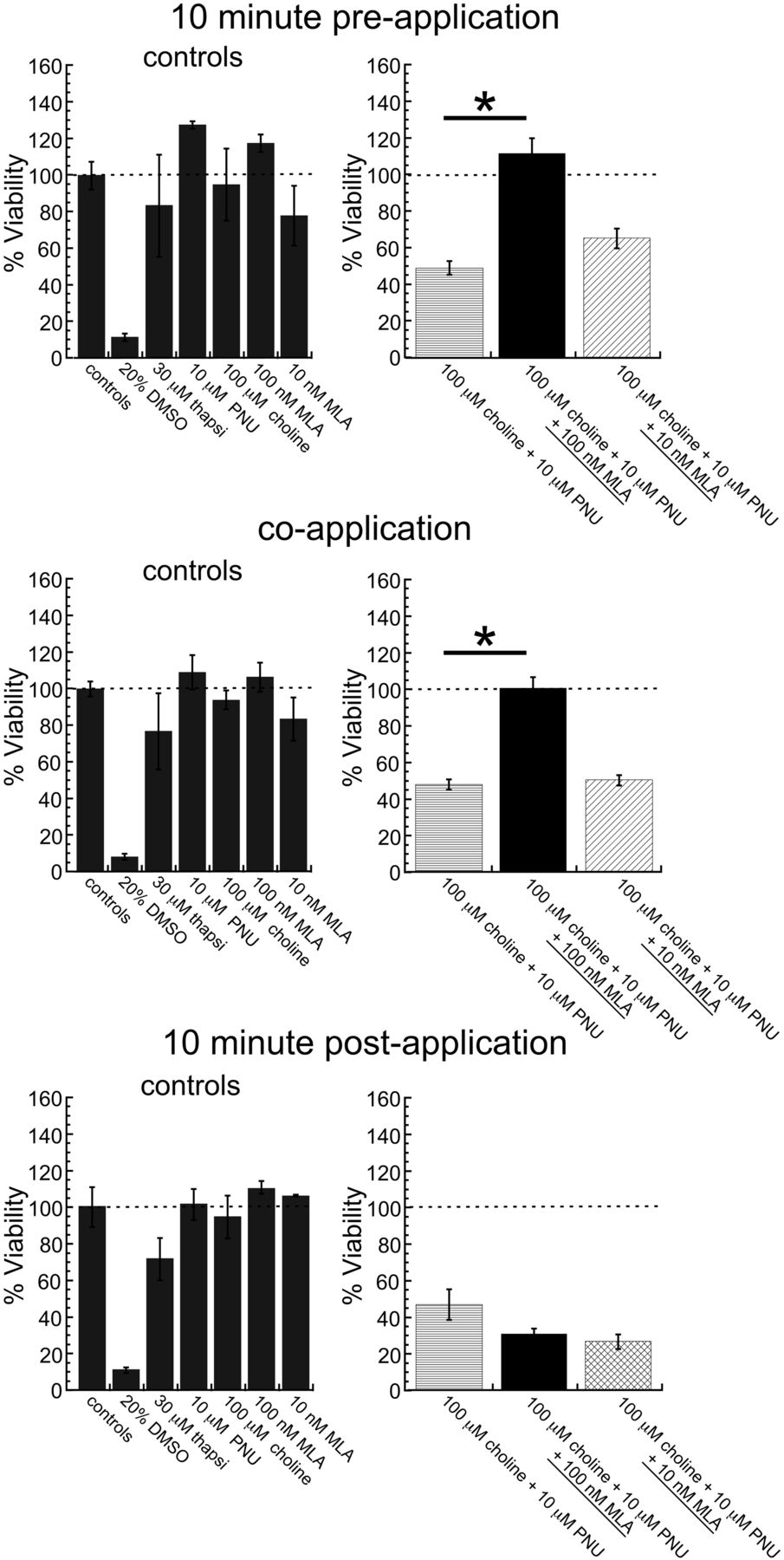
To confirm that the observed toxicity occurred via α7 nAChRs, the competitive antagonist MLA was given at various time points relative to toxic 100 μM choline and 10 μM PNU-120596 cotreatments incubated at 28°C. As shown in Fig. 7, neither a 10-min preapplication nor coapplication of 10 nM MLA was able to block the toxicity produced by the choline and PNU-120596 coapplication. However, this finding is consistent with the recent observation that 10 nM applications of MLA on top of steady-state currents elicited by choline and PNU-120596 actually transiently increase current rather than inhibit current, suggesting that low concentrations of MLA may alter the equilibrium between Ds and Di toward Ds (Williams et al., 2011b). Application of a 10-fold higher MLA concentration either 10 min before or with 100 μM choline and 10 μM PNU-120596 cotreatment was able to completely block the toxic effect, confirming that the toxicity is mediated by α7 receptors. In contrast, 100 nM MLA was unable to block the effect if it was applied 10 min after the toxic 100 μM choline and 10 μM PNU-120596 cotreatment. This result suggests that the onset of PNU-120596-induced toxicity in these cells occurs rapidly, in less than 10 min.

Sensitivity of the cytotoxic effect of 100 μM choline and 10 μM PNU-120596 cotreatment in HBSS at 28°C to the competitive antagonist MLA. Either 10 or 100 nM MLA was added at the time indicated, relative to the toxic 100 μM choline and 10 μM PNU-120596 cotreatment. *, two-tailed p < 0.05. Values are averages ± S.E.M. from 3 independent experiments. thapsi, thapsigargin; PNU, PNU-120596.
Bovine Serum Albumin Modulation of PNU-120596 Toxicity and Potentiation Activity.
As stated above, we noted in preliminary experiments that treatments with 10 μM PNU-120596 alone were toxic at 37°C when the solutions were prepared in DMEM with 10% FBS. Because both the medium and FBS are choline-containing, it is not too surprising that the PNU-120596 effects did not require added agonist; however, it was not clear why the activity was present at 37°C, given the effects of temperature on PNU-120596 potentiation and toxicity in HBSS. We therefore tested the hypothesis that the FBS supplied a factor that augmented the potentiating effects of PNU-120596 or reversed the temperature sensitivity of PNU-120596 potentiation.
Solutions were therefore prepared with choline and 10 μM PNU-120596 in HBSS supplemented with 10% FBS. As seen in Fig. 8, A and B, the presence of FBS eliminated the temperature-dependence of the PNU-120596 toxicity. Bovine serum albumin is a primary constituent of FBS and select serum albumins were previously shown to potentiate α7 nAChR-mediated responses (Conroy et al., 2003). Therefore, the experiments were repeated in HBSS containing 30 μM BSA, the approximate concentration of BSA found in solutions containing 10% FBS (Giles and Czuprynski, 2003; Granato et al., 2003). Again, the temperature dependence of cytotoxicity induced by PNU-120596 was eliminated (Fig. 8, C and D), suggesting that BSA is the constituent of FBS primarily responsible for the effect. Given that BSA is a nonspecific carrier of hormones and fatty acids in blood plasma, it is possible that a substance bound to the BSA is responsible for the effect, rather than BSA itself. Nonetheless, this observation suggests that although the ability of PNU-120596 to potentiate α7-mediated current at physiological temperature is reduced, some factors are likely to exist in vivo that maintain the activity to PNU-120596 at physiological temperatures.

Elimination of the temperature dependence of PNU-120596 cytotoxicity. A, cytotoxicity of 0 to 3 mM choline and 10 μM PNU-120596 treatments at 28°C with 5% CO2 in HBSS with 10% FBS. B, cytotoxicity of 0 to 3 mM choline and 10 μM PNU-120596 treatments at 37°C with 5% CO2 in HBSS with 10% FBS. Of note, a factor in FBS appears to remove the apparent temperature-dependent cytotoxicity in Fig. 9. C, cytotoxicity of 0 to 3 mM choline and 10 μM PNU-120596 treatments at 28°C with 5% CO2 in HBSS with 30 μM BSA. D, cytotoxicity of 0 to 3 μM choline and 10 μM PNU-120596 treatments at 37°C with 5% CO2 in HBSS with 30 μM BSA. The effect of 30 μM BSA appears to be very similar to that of FBS. *, two-tailed p < 0.05. Values are averages ± S.E.M. from three independent experiments. thapsi, thapsigargin; PNU, PNU-120596.
To confirm that the PNU-120596 toxic effects in the BSA-containing HBSS were α7-receptor activation-dependent, we tested for sensitivity to the α7-selective competitive antagonist MLA. In addition, to determine whether it required α7 ion channel currents, we also tested the noncompetitive antagonist mecamylamine (Fig. 9). As before, 10 nM MLA was unable to completely block the toxic effect of the treatment. Ten-minute preapplications and coapplications of 100 nM MLA completely reversed the toxicity, whereas applications of MLA 10 min or more after the choline and PNU-120596 treatment had no apparent effect. In addition, 10-min pretreatment and cotreatment with 100 μM mecamylamine were able to partially block the toxicity of choline and PNU-120596 treatment, whereas mecamylamine treatments 10 min or more after the treatment had no effect. The IC50 for mecamylamine on α7 nAChRs was approximately 10 μM in experiments performed in oocytes in which mecamylamine was coapplied with 300 μM ACh (Papke et al., 2001). In addition, 100 μM mecamylamine appeared to fully block steady-state currents generated by choline and PNU-120596 coapplication in recent experiments (Williams et al., 2011b). Although the 100 μM concentration of mecamylamine was unable to completely block the toxic effect in these studies, the partial block that was observed combined with the rapid onset of the toxicity in less than 10 min is consistent with a requirement for direct ion channel activity.
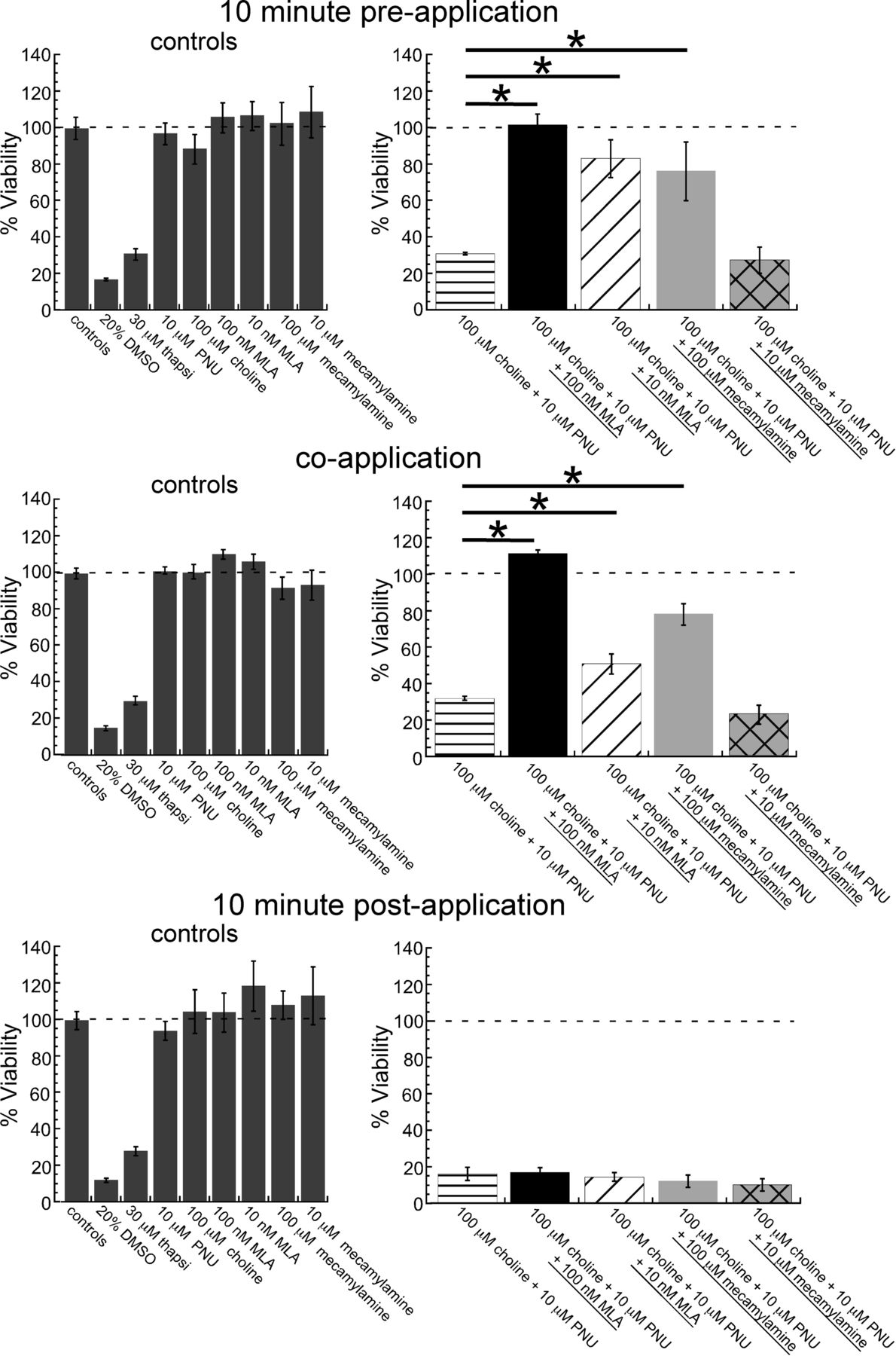
Sensitivity of the cytotoxic effect of 100 μM choline and 10 μM PNU-120596 cotreatment in HBSS with 30 μM BSA at 37°C to the competitive antagonist MLA and the noncompetitive antagonist mecamylamine. At the time indicated, 10 and 100 nM MLA or 10 and 100 μM mecamylamine was added, relative to the toxic 100 μM choline and 10 μM PNU-120596 cotreatment. *, two-tailed p < 0.05. Values are averages ± S.E.M. from 3 independent experiments. thapsi, thapsigargin; PNU, PNU-120596.
Effects of BSA on the Temperature Dependence of PNU-120596 Potentiation.
Because 30 μM BSA appeared to eliminate the temperature dependence of PNU-120596 cytotoxicity in a manner that was dependent on α7 nAChR signaling, whole-cell electrophysiology experiments were performed in the presence of 30 μM BSA. When the responses recorded with 30 μM BSA at 37°C were expressed relative to the initial responses obtained at 23.5°C, the effect of BSA on the normalized peak currents was not statistically significant (Fig. 10A; Table 2) (p > 0.05). However, when the same data were plotted on the basis of the absolute magnitude of the recorded peak currents, the responses recorded in the presence of 30 μM BSA at 23.5 and 37°C were significantly larger than responses recorded in the absence of BSA (p < 0.05) (Fig. 10B). On average, the peak currents evoked by 100 μM ACh and 10 μM PNU-120596 coapplication at 23.5°C in the absence and presence of 30 μM BSA were 1300 ± 400 and 2800 ± 300 pA, respectively. The average peak currents recorded at 37°C in the absence and presence of 30 μM BSA were 100 ± 30 and 390 ± 100 pA, respectively. Our data suggest that 30 μM BSA potentiates α7-mediated responses in an additive manner with PNU-120596 that is not temperature-sensitive.

Partial preservation of PNU-120596 potentiation at 37°C in external solution containing 30 μM BSA. A, whole-cell recordings from A7R3HC10 cells evoked by a 3-s coapplication of 100 μM ACh and 10 μM PNU-120596 in the absence or presence of 30 μM BSA with various temperatures between 23.5 and 37°C. Data in the absence of BSA are the same as those shown in Fig. 5A. Responses were normalized to the average peak amplitude of the three initial responses obtained at 23.5°C. Data in the presence of BSA are represented as the average normalized value ± S.E.M. of 11 cells. B, data from the same cells as in A are shown as the average absolute peak amplitude ± S.E.M. C, representative traces of 100 μM ACh and 10 μM PNU-120596-evoked responses in the presence of 30 μM BSA recorded at the indicated temperature.
Discussion
Our studies of the α7-mediated currents evoked by rapid ACh applications provide valuable insights into the unique activation and desensitization properties of this receptor and allow us to provide, for the first time, an estimate of the maximal nonstationary Popen of this receptor. Our estimate of 0.002 is in remarkable contrast to the values of 0.7 to 0.8 for heteromeric receptors such as α4β2 (Li and Steinbach, 2010) and muscle-type receptors (Land et al., 1981).
The decrease in net charge in the responses evoked during concentration ramps that approached final ACh concentrations more than 300 μM is consistent with the hypothesis that the transient α7 peak Popen is further reduced at high levels of agonist occupancy, supporting a model we have proposed previously (Papke et al., 2000a; Williams et al., 2011b) based in part on oocyte experiments. However, conclusions based on slow macroscopic responses may sometimes be questionable. For example, the peak currents of heteromeric receptor macroscopic oocyte currents are more likely to represent a condition incorporating a large degree of steady-state desensitization than conditions of maximal Popen (Papke, 2010). However, this is not the case with the α7 responses, which appear to reflect instantaneous agonist concentration (Papke and Thinschmidt, 1998), regardless of how slowly or rapidly agonist concentration changes (Papke et al., 2000b; Uteshev et al., 2002). The data from the present study, obtained on a millisecond time scale, showed concentration-response relationships for peak current and net charge that were essentially identical to those reported for the oocyte responses, despite a 1000-fold difference in the time scales of the recordings. This result supports the hypothesis that the kinetics of the responses in both systems are following the instantaneous changes in agonist concentration and, by inference, the levels in agonist occupancy. The primary difference between the A7R3HC10 ACh concentration-response data and those from oocytes (Papke and Papke, 2002) was that, with the very rapid high concentration ramps applied to the cells, receptors were not at the optimal range of agonist occupancy long enough for maximal net charge responses to occur.
The therapeutic utility of PAMs for ligand-gated ion channels of the Cys-loop receptor family has been amply proven by the development of benzodiazepines, GABAA receptor PAMs. Benzodiazepines were preceded in clinical development by barbiturates, which have a larger spectrum of effects than benzodiazepines and subsequently a significantly smaller therapeutic index. With the recognition of α7 as a potentially important therapeutic target, α7 PAMs also appear to be an attractive approach for new drug development. Just as benzodiazepines can be distinguished from barbiturates, we can distinguish type I and type II α7 PAMs, with the type II agents appearing to have such high efficacy that they might lead to unregulated activation.
With a type II PAM, the tonic presence of the α7 agonist choline could disrupt native signaling dynamics and, if the potentiation of the calcium permeable receptor currents becomes sufficiently large to disrupt calcium homeostasis, cause cell death (Orr-Urtreger et al., 2000; Lukas et al., 2001). This could be especially true in cases of trauma or stroke, when choline concentrations in the brain increase to as high as 100 μM (Jope and Gu, 1991; Scremin and Jenden, 1991).
Of the PAMs tested, only PNU-120596 showed strong temperature-dependent potentiation, despite the facts that TQS is also a type II PAM and that PNU-120596 and NS-1738 share the common structural feature of a central urea group. The data suggest that, although PNU-120596 and TQS have similar potentiating properties, they work through distinct mechanisms. Although PNU-120596 and TQS appear to bind at similar sites in the intrasubunit cavity formed by the four membrane-spanning helices (Young et al., 2008; Gill et al., 2011), there may be alternate protein-PAM interactions, with increased differences in the response to membrane fluidity changes that occur at higher temperature. Although increased temperature had only a modest effect on receptor activation by the allosteric agonist 4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (Jindrichova et al., 2012), Sitzia et al., 2011 reported that the potentiating activity of SB-206553, a PAM with properties intermediate to those of the type I and type II PAM classes (Dunlop et al., 2009), was also reduced at 37°C. Although the published data agree that type I PAMs appear to lack in vitro cytotoxicity (at the concentrations of agonists and modulators tested), the data are contradictory regarding the in vitro toxicity of the type II PAM PNU-120596 (Ng et al., 2007; Hu et al., 2009; Dinklo et al., 2011). In vivo studies with α7 PAMs have reported no major concerns regarding toxicity, which might suggest that specific factors can prevent overactivation of α7 receptors in a physiological context. Even for the temperature-sensitive agent PNU-120596, endogenous potentiating factors may provide a narrow margin of safety for cells expressing high levels of α7 receptors, although it should be noted that the peak current of the A7R3HC10 cells is approximately 2- to 3-fold higher than what we have previously reported for α7-expressing cells in hippocampal (Lopez-Hernandez et al., 2007) or hypothalamic brain slices (Uteshev et al., 2003).
A major concern for considering PAM-based therapeutics is whether a PAM will allow α7 receptors to perform their usual functions more effectively or whether it will force α7 receptors into playing alternative roles. To approach the answer to that question, the first step must be to consider exactly what the usual functions of α7 may be and how effectively those functions will be amplified by PAMs that work through altering the Popen of the receptor's ion channel. As we confirm in these experiments, α7 receptor ion channels are intrinsically rather inefficient compared with other nAChRs, a fact that has led to the proposal that α7 receptors are most effective at integrating low-level signals over long periods of time. They desensitize rapidly but also resensitize rapidly (Mansvelder and McGehee, 2000; Wooltorton et al., 2003). Low concentrations of the α7-selective partial agonist 3-(2,4-dimethoxybenzylidene)anabaseine (GTS-21) can promote the survival of nerve growth factor-differentiated PC12 cells after trophic factor withdrawal, provided that the cells are maintained in GTS-21 for several hours. In contrast, treatment of PC12 cells with a high concentration of GTS-21, sufficient to produce a large synchronous current followed by profound desensitization, was toxic to the cells, and the toxic effect was nearly instantaneous (Li et al., 1999). These two treatment paradigms had different effects on intracellular signaling pathways, highlighting a potential dichotomy in functional roles for α7 receptors.
Our understanding of α7 receptors has arguably been encumbered by the usual assumption that α7 function relies entirely on ion channel activation. Nonetheless, α7 receptors have been shown to modulate numerous intracellular signal transduction pathways, in many cases under conditions in which it has not been possible to demonstrate a requirement for ion channel activation (de Jonge et al., 2005; Arredondo et al., 2006) (Parrish et al., 2008; Marrero and Bencherif, 2009). In many cases, although clearly dependent on the presence of α7 and stimulation of α7 by putative agonists, the activation of the signal transduction mechanisms appears to be independent of α7 ion channel activation (Suzuki et al., 2006; de Jonge and Ulloa, 2007). These observations support the hypotheses that α7 receptors may function in multiple ways and that various ligands differ in their ability to stimulate ion channel activation and/or signal transduction. Ligands such as GTS-21 and (1,4-diazabicyclo[3.2.2]nonan-4-yl(5-(3-(trifluoromethyl)phenyl)furan-2-yl)methanone (NS-6740) (Briggs et al., 2009), which are poor ion channel activators but are very effective for PNU-120596-induced activation of desensitized receptors (Papke et al., 2009), may also be effective for ion channel-independent signal transduction. Much of the α7-mediated signal transduction data have come from studies of α7-mediated suppression of inflammation, and the low efficacy, strongly desensitizing agent GTS-21 has been shown to be very effective in several of these models (van Westerloo et al., 2006; Giebelen et al., 2007a,b; Pavlov et al., 2007; Kageyama-Yahara et al., 2008). This finding suggests that there may be one or more ligand-bound states in which the ion channel activation mechanism is “desensitized,” but the receptor is otherwise activated, generating a ligand-bound nonconducting activated state. These states may include the state that the type II PAMs convert into conducting states.
If α7 receptors are capable of both channel-mediated and channel-independent signaling, then it may be that PAMs will augment one type of function but not the other. Although our data caution against the therapeutic development of strong type II PAMs, in cases in which activation of the α7 ion channel is necessary for a desired therapeutic effect (Briggs et al., 2009), a type I PAM-based therapeutic approach might offer several potential advantages over agonist-based strategies. The temporal firing dynamics of native cholinergic signaling should be conserved because PAMs would theoretically only augment the response provided by endogenous agonists.
Clearly there is still much more to be learned about the α7 nAChR. One challenge that now exists is to define experimental models that might be able to discriminate between channel-dependent and channel-independent α7 functions. The α7 PAMs are therapeutic leads that may also serve as probative tools for the study of these two different forms of signaling, as will ligands such as GTS-21 and NS-6740 that may selectively activate specific signaling modes.
Authorship Contributions
Participated in research design: Williams, Peng, and Papke.
Conducted experiments: Williams, Peng, and Kimbrell.
Performed data analysis: Williams and Peng.
Wrote or contributed to the writing of the manuscript: Williams and Papke.
Acknowledgments
We thank Dr. Ralph Loring (Northeastern University, Boston, MA) for providing the 125I-α-btx used in the binding assay. We thank Dr. Cecilia Gotti (University of Milan, Milan, Italy) for providing the α7 antibodies used in the Western blot. We thank Dr. Stephen Baker and Debbie Otero for assistance in conducting the binding assays, Monica Santisteban for conducting the immunoprecipitation and western blot, Institut De Recherches Internationales Servier for supplying TQS and NS-1738, and Drs. Jingyi Wang and Nicole Horenstein for supplying PNU-120596 and for many helpful discussions.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the National Institutes of Health National Institute of General Medical Science [Grant R01-GM57481]; National Institutes of Health National Institute on Aging [Grant T32-AG000196]; and Florida Biomedical James and Esther King Research [Grant KG12].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
ABBREVIATIONS:
- α-btx
- α-bungarotoxin
- nAChR
- nicotinic acetylcholine receptor
- ACh
- acetylcholine
- PAM
- positive allosteric modulator
- PNU-120596
- N-(5-chloro-2,4-dimethoxyphenyl)-N'-(5-methyl-3-isoxazolyl)-urea
- HEK
- human embryonic kidney
- BSA
- bovine serum albumin
- HBSS
- Hanks' balanced saline solution
- DMSO
- dimethyl sulfoxide
- 5-HI
- 5-hydroxyindole
- NS-1738
- N-(5-chloro-2-hydroxyphenyl)-N′-[2-chloro-5-(trifluoromethyl)phenyl]urea
- TQS
- 3a,4,5,9b-tetrahydro-4-(1-naphthalenyl)-3H-cyclopentan[c]quinoline-8-sulfonamide
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- MLA
- methyllycaconitine
- GTS-21
- 3-(2,4-dimethoxybenzylidene)anabaseine
- NS-6740
- (1,4-diazabicyclo[3.2.2]nonan-4-yl(5-(3-(trifluoromethyl)phenyl)furan-2-yl)methanone.

- Received May 30, 2012.
- Accepted July 24, 2012.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}