


Abstract
The ability of dopamine receptors to interact with other receptor subtypes may provide mechanisms for modulating dopamine-related functions and behaviors. In particular, there is evidence suggesting that the trace amine-associated receptor 1 (TAAR1) affects the dopaminergic system by regulating the firing rate of dopaminergic neurons or by altering dopamine D2 receptor (D2R) responsiveness to ligands. TAAR1 is a Gαs protein-coupled receptor that is activated by biogenic amines, “trace amines,” such as β-phenylethylamine (β-PEA) and tyramine that are normally found at low concentrations in the mammalian brain. In the present study, we investigated the biochemical mechanism of interaction between TAAR1 and D2R and the role this interaction plays in D2R-related signaling and behaviors. Using a bioluminescence resonance energy transfer biosensor for cAMP, we demonstrated that the D2R antagonists haloperidol, raclopride, and amisulpride were able to enhance selectively a TAAR1-mediated β-PEA increase of cAMP. Moreover, TAAR1 and D2R were able to form heterodimers when coexpressed in human embryonic kidney 293 cells, and this direct interaction was disrupted in the presence of haloperidol. In addition, in mice lacking TAAR1, haloperidol-induced striatal c-Fos expression and catalepsy were significantly reduced. Taken together, these data suggest that TAAR1 and D2R have functional and physical interactions that could be critical for the modulation of the dopaminergic system by TAAR1 in vivo.
Introduction
The trace amines (TAs) β-phenylethylamine (β-PEA), p-tyramine, octopamine, tryptamine, and synephrine are endogenous biogenic amines that are present in mammalian brain at very low concentrations compared with classic monoamines (Baldessarini, 1978; Grandy, 2007). For many years, TAs were believed to have a minor role in neurotransmission, and they were traditionally referred to as side products of the synthesis of classic monoamines. Their function as sympathomimetic compounds has been known since the last century, and in humans, TA activity is particularly evident in subjects treated with monoamine oxidase inhibitors or who consume food containing TAs in high concentrations (McCabe and Tsuang, 1982). The “amphetamine-like” effect of TAs is believed to occur at high, nonphysiological concentrations and has been explained by their interaction with the plasma membrane monoamine transporters, particularly the dopamine transporter (DAT) (Berry, 2004). In normal mice, β-PEA administration induces hyperactivity and an increase in dopamine release, and these effects are disrupted in mice lacking the DAT (Sotnikova et al., 2004). The dysregulation of TAs has been linked to different neurological and psychiatric disorders, such as schizophrenia, depression, Parkinson's disease, attention-deficit/hyperactivity disorder, and migraine (Grandy, 2007; Sotnikova et al., 2009). Altered levels of β-PEA have been found in patients with depression and psychotic episodes (Sabelli and Mosnaim, 1974; Davis and Boulton, 1994).
The recent discovery of a class of G protein-coupled receptors (GPCRs) that can be activated by TAs, trace amine-associated receptors (TAARs), has further increased interest in these amines and their roles in both physiology and pathology (Borowsky et al., 2001; Bunzow et al., 2001). Six TAAR genes and three TAAR pseudogenes exist in humans, whereas rodents express an even greater number of TAAR genes. Only TAAR1 and TAAR4, however, possess any demonstrable TA-responsiveness.
TAAR1 signals through the stimulatory G protein (Gαs) and is localized in several brain areas, including the limbic regions and in the nuclei containing monoaminergic cell bodies (Lindemann et al., 2008). This expression pattern makes TAAR1 a potential therapeutic target to modulate critical behaviors related to monoamine systems (Lindemann and Hoener, 2005; Sotnikova et al., 2009). In animal studies, TAAR1-deficient mice (TAAR1-KO mice) display an increased sensitivity to the neurochemical and locomotor effects of amphetamine (Wolinsky et al., 2007; Lindemann et al., 2008). Whereas in vitro studies have suggested that TAAR1 may directly alter DAT function (Miller et al., 2005), there is substantial evidence that TAAR1 is able to modulate firing activity of dopaminergic neurons in the ventral tegmental area (Lindemann et al., 2008) via potential interaction with dopamine D2 receptor (D2R) signaling. In particular, the D2R agonist quinpirole seems to be more potent, and the D2R-mediated electrophysiological responses desensitize less well in TAAR1-KO mice (Bradaia et al., 2009).
The D2R serves as the main target of antipsychotic drugs such as haloperidol (Strange, 2001), and we sought to determine whether a functional interaction occurs between TAAR1 and D2R that modulates their signaling. First, we measured TAAR1 cAMP production in the presence of D2R/antagonist complexes using a cAMP-responsive bioluminescence resonance energy transfer (BRET) biosensor (Barak et al., 2008). We found that the D2R antagonists haloperidol, raclopride, and amisulpride enhanced β-PEA/TAAR1-mediated production of cAMP in a Gi-dependent and D2R-selective manner. Furthermore, we discovered in cells that plasma membrane TAAR1 and D2R form constitutive heterodimers that can be disrupted in the presence of haloperidol, and in mice lacking TAAR1 that haloperidol-induced catalepsy and striatal c-Fos expression are reduced. These data suggest that the in vivo effects of TAAR1 ligands and antipsychotic drugs may depend on the mutual regulation of signaling that occurs between TAAR1 and D2R.
Materials and Methods
Animals and Reagents.
TAAR1 knockout (TAAR1-KO) mice of mixed C57BL/6J × 129Sv/J background were generated as described previously (Wolinsky et al., 2007; Sotnikova et al., 2010). Three- to six-month-old mice of both genders were used in these experiments.
All cell culture reagents and buffers were from Invitrogen (Carlsbad, CA) and Sigma-Aldrich (St. Louis, MO), and FBS was from JRH Biosciences (Lenexa, KS). Coelenterazine h was purchased from Promega (Madison, WI). Anti-hemagglutinin (HA) antibody was from Roche Applied Sciences (Indianapolis, IN), and anti-FLAG was from Sigma-Aldrich. Plasmids containing the cDNA for the human trace amine receptor were obtained from the cDNA resource center at the University of Missouri-Rolla and the American Type Culture Collection (Manassas, VA). All compounds used in this study were obtained from Sigma-Aldrich.
Construction of Expression Vectors.
A modified version of human TAAR1 was used as described previously to enable plasma membrane expression of the mature receptor. This construct, described below, is hereafter referred to in this article as TAAR1 (Barak et al., 2008). In brief, full-length human TAAR1 cDNA without a stop codon was amplified by PCR with 5′ and 3′ in-frame restriction enzyme sites of EcoRI and KpnI, respectively, and PCR product was cloned into a pcDNA3.1 vector with N-terminal triple HA tag. The cDNA corresponding to the first nine amino acids of the β2-adrenergic receptor was inserted in-frame between the triple HA and TAAR1 to generate the triple HA-β2N9-TAAR1-green fluorescent protein. The Rluc version was generated amplifying by PCR using specific primers with 5′ and 3′ in-frame restriction enzyme sites of XhoI and KpnI, respectively, and subcloned into a phRluc N3 vector (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Mouse D2R long dopamine receptor tagged on the C terminus with Rluc or YFP was modified as described previously (Salahpour and Masri, 2007; Barak et al., 2008). In brief, it was amplified by PCR using a 5′-primer containing an EcoRV restriction site and a 3′-primer containing a NotI restriction site. PCR product was cloned into a pcDNA3 vector downstream three HA tags or FLAG tag, which generated amino-terminally HA-tagged or FLAG tagged D2RLR. For the D2R-YFP version, the receptor was directly cloned into pEYFP N1 vector.
The BRET sensor for cAMP (Barak et al., 2008) was constructed by the modification of an existing FRET-based intramolecular biosensor EPAC (Ponsioen et al., 2004), in which residues 148 to 881 of EPAC1 were surrounded by enhanced cyan fluorescent protein upstream and citrine downstream (DiPilato et al., 2004; Violin et al., 2008). Using the restriction enzymes BamHI and KpnI, the enhanced cyan fluorescent protein was removed and replaced by a humanized Renilla reniformis luciferase gene that was PCR-amplified from phRluc-C1 (PerkinElmer Life and Analytical Sciences) and cloned using the same restriction sites to preserve the frame of translation (Barak et al., 2008).
Cell Culture and Transfection of Cell Lines.
Human embryonic kidney (HEK) 293 (HEK-293T) cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) FBS, 2 mM l-glutamine, and 0.05 mg/ml gentamicin at 37°C in a humidified atmosphere at 95% air and 5% CO2. Transient transfections were performed 24 h after cells seeding using calcium phosphate protocol. TAAR1 (5 μg) and 2 μg of D2R for each milliliter of transfection solution were used for the experiments. For the BRET experiments, 24 h after transfection, the cells were plated in poly (d-lysine)-coated 96-well microplates (well assay plate with clear bottom; Thermo Fisher Scientific, Waltham, MA) at a density of 80,000 cells per well in phenol red free minimum essential medium containing 2% FBS, 10 mM HEPES, and 2 mM l-glutamine. The cells were then cultured for an additional 24 h.
Bioluminescence Resonance Energy Transfer Measurement.
For BRET assays, at the day of the experiment the phenol red free medium was removed from HEK-293T cells and replaced by phosphate-buffered saline containing calcium and magnesium and 0.003% (w/v) ascorbic acid. The assay was started by adding 10 μl of the cell-permeant substrate specific for R. reniformis luciferase, coelenterazine h, to the well to yield a final concentration of 5 μM. The antagonist compounds were added 5 min before the agonist. For time course experiments, the plate was read immediately after the addition of the agonist and for approximately 30 min. For dose-response experiments, the plate was read 10 min after agonist addition. All of the experiments were conducted in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (Sigma-Aldrich) at 200 μM final concentration as described previously (Barak et al., 2008). BRET readings were collected using a Mithras LB940 instrument that allows the sequential integration of the signals detected in the 465 to 505 nm and 515 to 555 nm windows using filters with the appropriate band pass and by using MikroWin 2000 software (Berthold Technologies USA, Oak Ridge, TN). For titration experiments, constant concentration of TAAR1-Rluc was used with increasing amount of D2R-YFP. The acceptor/donor ratio was calculated as described previously (Salahpour and Masri, 2007). Curve was fitted using a nonlinear regression and one site-specific binding with Prism 5 (GraphPad Software Inc., San Diego, CA). For evaluation of the basal cAMP level, data were expressed as a percentage of vehicle-treated cells.
Cellular Fractionation.
Cells were lysed in ice-cold hypotonic buffer (20 mM HEPES, pH 7.4, 2 mM EDTA, 2 mM EGTA, 6 mM MgCl2, and protease inhibitor cocktail) using a Dounce homogenizer. Cellular debris was removed by centrifugation (1000g for 5 min at 4°C). Sucrose was added to the supernatant to a final concentration of 0.2 M, and then lysates were layered to a discontinuous sucrose gradient (0.5, 0.9, 1.2, 1.35, 1.5, and 2.0 M). Samples were centrifuged at 28,000 rpm for 16 h at 4°C using a rotor (SW32Ti; Beckman Coulter, Fullerton, CA) Thirty-two 1-ml fractions were collected, and BRET was measured from 100-μl aliquots of each fraction. As endoplasmic reticulum (ER) and plasma membrane (PM) markers, anti-KDEL (Abcam Inc., Cambridge, MA) and anti-Na+/K+-ATPase (Millipore Corporation, Billerica, MA) antibodies were used.
Quantitative Measurement of Cell Surface Receptors.
Cell-surface expression of HA-GPCRs or FLAG-GPCRs constructs was determined by ELISA using monoclonal anti-HA or anti-FLAG antibody and the horseradish peroxidase-conjugated secondary anti-mouse antibody. The peroxidase activity was detected by specific reagent (SigmaFast; Sigma-Aldrich), and the colorimetric reaction was measured using a spectrophotometer (Beckman Coulter) using a 492-nm filter. Total receptor level was assessed in the same sample by measuring the Rluc activity as in the BRET experiments (Salahpour et al., 2004).
Immunofluorescence.
One hour after intraperitoneal administration of haloperidol (0.5 mg/kg) or saline, mice were rapidly anesthetized with ketamine-xylazine and perfused transcardially with 4% (w/v) paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4. Brains were postfixed overnight in the same solution and stored at 4°C. Thirty-micrometer thick sections were cut with a Cryostat (Leica, Wetzlar, Germany) and stored at −20°C in a solution containing 30% (v/v) ethylene glycol, 30% (v/v) glycerol, and 0.1 M sodium phosphate buffer until they were processed for immunofluorescence. Striatum was identified using a mouse brain atlas, and sections were processed as follows.
On Day 1, free-floating sections were rinsed in Tris-buffered saline (TBS; 0.25 M Tris and 0.5 M NaCl, pH 7.5) three times for 10 min each. After a 20-min incubation in 0.2% Triton X-100 in TBS, sections were rinsed three times in TBS again. Finally, they were incubated overnight at 4°C with the c-Fos primary antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA). On day 2, sections were rinsed three times for 10 min in TBS and incubated for 45 min with AlexaFluor 488 (Invitrogen). Sections were rinsed for 10 min twice in TBS and twice in Tris buffer (0.25 M Tris) before mounting in Vectashield (Vector Laboratories, Burlingame, CA).
Confocal microscopy and images from each region of interest were obtained bilaterally using sequential laser-scanning confocal microscopy (SP2; Leica). Neuronal quantification was performed in 375 × 375-μm images by counting c-Fos-positive nucleus.
Haloperidol-Induced Catalepsy.
Wild-type (WT) and TAAR1 heterozygous (HET) and TAAR1-KO mice were treated with different doses of haloperidol or vehicle, and catalepsy was measured 3 h later in the bar test as described previously (Sotnikova et al., 2005). In brief, the presence of catalepsy was determined and measured by placing the animal's forepaws on a horizontal wooden bar (0.7 cm in diameter) 4 cm above the tabletop. The time until the mouse removed both forepaws from the bar was recorded, with a maximum cutoff time of 180 s.
Statistical Analysis.
Data were analyzed by two-tailed Student's t test, one-way ANOVA, or two-way ANOVA with Bonferroni post hoc test. Values in graphs are expressed as mean ± S.E.M.
Results
Haloperidol Enhances β-PEA-Induced Stimulation of TAAR1.
We measured the accumulation of cAMP induced by the activation of TAAR1 and evaluated the capacity of D2R to modulate this process. Gαs-coupled TAAR1 signals through cAMP (Borowsky et al., 2001; Bunzow et al., 2001), whereas agonists of D2R decrease adenylyl cyclase activity by coupling to an inhibitory Gαi protein (Masri et al., 2008). To measure cAMP, we used an EPAC biosensor validated for TAAR1 and D2R (Barak et al., 2008; Masri et al., 2008; Violin et al., 2008). With TAAR1 expressed alone, β-PEA at 1 μM induced a robust increase in cAMP levels as measured by a reduction in BRET signal (Fig. 1A). In the cells expressing TAAR1 alone, haloperidol at 1 μM was not able to alter cAMP concentrations either under basal conditions or in β-PEA-stimulated cells. When we cotransfected TAAR1 with D2R, we observed a reduced (by approximately 25%) β-PEA stimulation to cells expressing TAAR1 alone, and in this case, haloperidol enhanced the β-PEA induced cAMP production without affecting the basal level of cAMP (Fig. 1B). This enhancement lasted for the duration of the 30-min experiment. It is also evident that the basal level of cAMP did not differ between TAAR1- and TAAR1-D2R-expressing cells, indicating a lack of effect of D2R on basal cAMP levels (TAAR1 = 1.213 ± 0.005 versus TAAR1 + D2R = 1.217 ± 0.002, p ≥ 0.05). However, β-PEA responses were significantly lower in cells coexpressing TAAR1 and D2R (β-PEA in TAAR1 cells: 1.124 ± 0.002 versus TAAR1 + D2R 1.151 ± 0.002, p < 0.05, n = 7).

D2R antagonists enhance TAAR1-mediated stimulation of cAMP by β-PEA in HEK-293 cells. A, time course effects of haloperidol and β-PEA in cells transiently transfected with EPAC and TAAR1. BRET ratio is measured as YFP/Rluc ratio, and the readings were started right after β-PEA addition. Cells were exposed to 1 μM β-PEA or control medium in presence or absence of 1 μM haloperidol. The decrease in BRET ratio indicates an increase in cAMP concentration. β-PEA induced a robust increase in cAMP level, whereas haloperidol alters neither the basal nor the stimulated response. B, the same time course experiment was performed in cells coexpressing TAAR1 and D2R. In this case, 1 μM haloperidol enhanced β-PEA stimulation without altering basal cAMP. C and D, an analogous time course experiment with raclopride, another D2R antagonist. In cells expressing only EPAC biosensor and TAAR1, raclopride at 1 μM had no effect on β-PEA stimulation (C) while demonstrating the ability to increase this effect in cells coexpressing D2R (D). E and F, similar results were obtained with D2R antagonist amisulpride. At 1 μM, amisulpride induced long-lasting enhancement in cAMP levels only in cells expressing D2R with TAAR1 (F) but not in the cells expressing TAAR1 alone (E). Results shown are representative of three to four independent experiments.
D2R Dopamine Receptor Mediates Haloperidol Effects on TAAR1 Signaling.
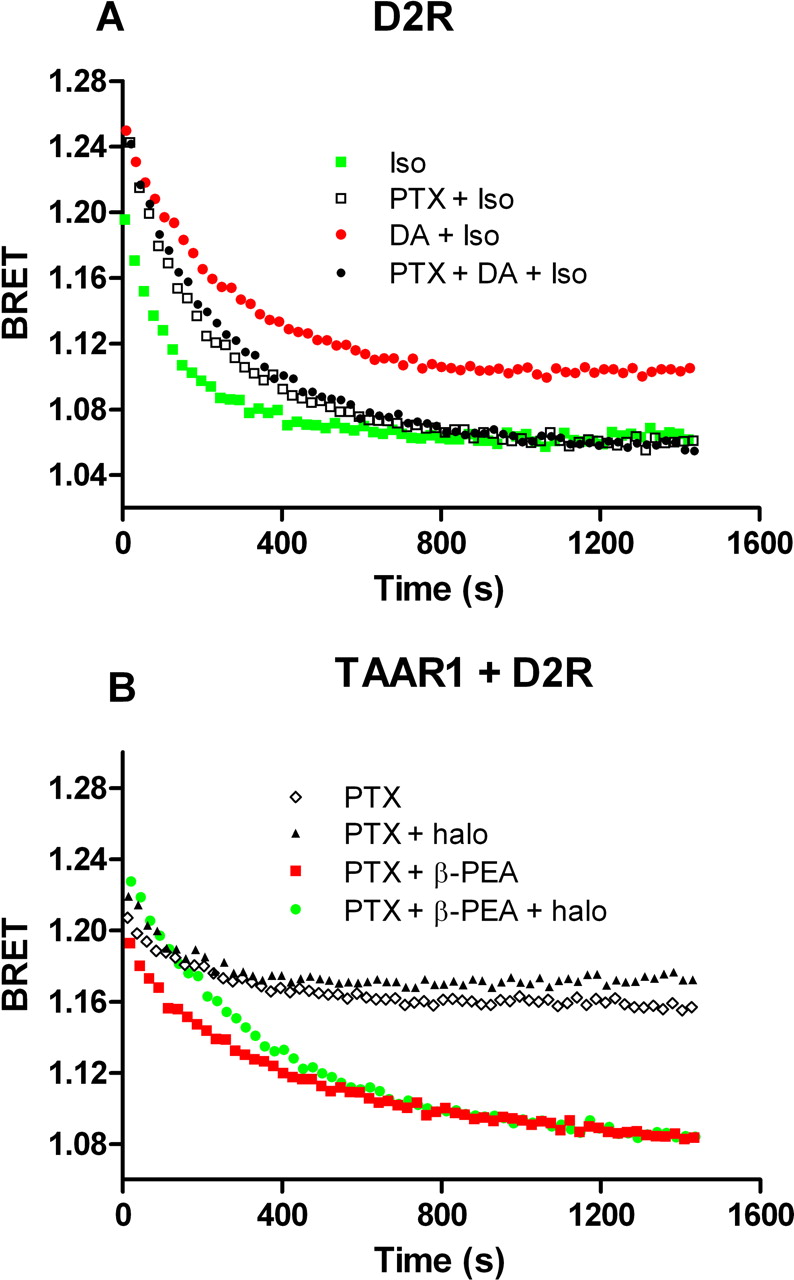
It has been reported that haloperidol does not interact with TAAR1 (Bunzow et al., 2001; Barak et al., 2008) and, accordingly, we have observed that haloperidol does not directly stimulate TAAR1 in our assay (Fig. 1, A and B). To confirm that an enhancement in TAAR1 signaling by haloperidol is mediated through D2R we investigated two other D2R antagonists, raclopride and amisulpride. The first is a classic selective D2R antagonist, and the second is an atypical antipsychotic with good D2R selectivity (Möller, 2003). In cells expressing TAAR1 and D2R, each of these compounds at 1 μM was able to enhance β-PEA stimulation without affecting basal cAMP (Fig. 1, C–F). Because D2R receptor is coupled to Gαi protein, we verified whether this protein was the mediator of the observed enhancement. We therefore pretreated cells overnight with pertussis toxin (PTX) to prevent the coupling between Gαi protein and D2R receptor. In control experiments, pretreatment with PTX prevented the inhibitory effect of dopamine on the isoproterenol stimulation of cAMP production mediated by β2-AR (DA + iso = 1.10 ± 0.004, PTX + DA + iso = 1.06 ± 0.001, p < 0.05; Fig. 2A). Likewise, PTX treatment prevented the ability of haloperidol to potentiate TAAR1 stimulation of cAMP production (PTX + β-PEA = 1.09 ± 0.005, PTX + β-PEA + halo = 1.09 ± 0.006, p ≥ 0.05; Fig. 2B). A loss of the stimulatory effects of raclopride and amisulpride on TAAR1-mediated cAMP signaling was also observed in PTX-treated cells (data not shown).

Pertussis toxin disrupts D2R-mediated effect of haloperidol on TAAR1 signaling. A, HEK-293 cells transfected with EPAC biosensor and D2R are treated with PTX overnight to prevent coupling of Gi protein to D2R. These cells were exposed to 1 μM isoproterenol to stimulate β2-AR endogenously expressed and were compared with PTX-untreated cells. Isoproterenol readily stimulates cAMP production, and this effect is partly inhibited by dopamine at 0.1 μM as a consequence of D2R activation. This inhibition is abolished in cells pretreated with PTX, indicating the efficacy of this toxin to disrupt Gαi-mediated effect. B, time course experiment was conducted to evaluate PTX effect on haloperidol enhancement of β-PEA response. Cells transfected with EPAC, TAAR1, and D2R were pretreated with PTX, and the day after, the cells were stimulated with β-PEA that induce an increase in cAMP level. In PTX-treated cells, haloperidol lost its modulatory action on the β-PEA effect. Results shown are representative of three to four independent experiments.
D2R Blockade Selectively Enhances TAAR1 Signaling.
The above data indicate that the D2R is able to modulate TAAR1-induced cAMP production. To validate the selectivity of this effect for TAAR1, we performed a similar experiment using 1 μM isoproterenol activation of endogenous β2-AR. Cells expressing only biosensor or biosensor and D2R showed robust response to isoproterenol, and this response was not affected by haloperidol (1 μM) treatment (data not shown). Likewise, isoproterenol produced a robust increase of cAMP in cells transfected with only TAAR1 or in cells coexpressing TAAR1 and D2R (Fig. 3, A and B). Haloperidol at 1 μM had no effect on basal cAMP concentration and did not augment isoproterenol cAMP increases, thus confirming specificity of the TAAR1/D2R interaction for eliciting this phenomenon (iso = 1.025 ± 0.003, iso + halo = 1.027 ± 0.005, p ≥ 0.05 in TAAR1-expressing cells; iso = 1.016 ± 0.003, iso + halo = 1.012 ± 0.005, p ≥ 0.05 in TAAR1-D2R-expressing cells). Isoproterenol tested at lower concentrations (10 and 100 nM) with haloperidol also showed no potentiation in cAMP production (data not shown).
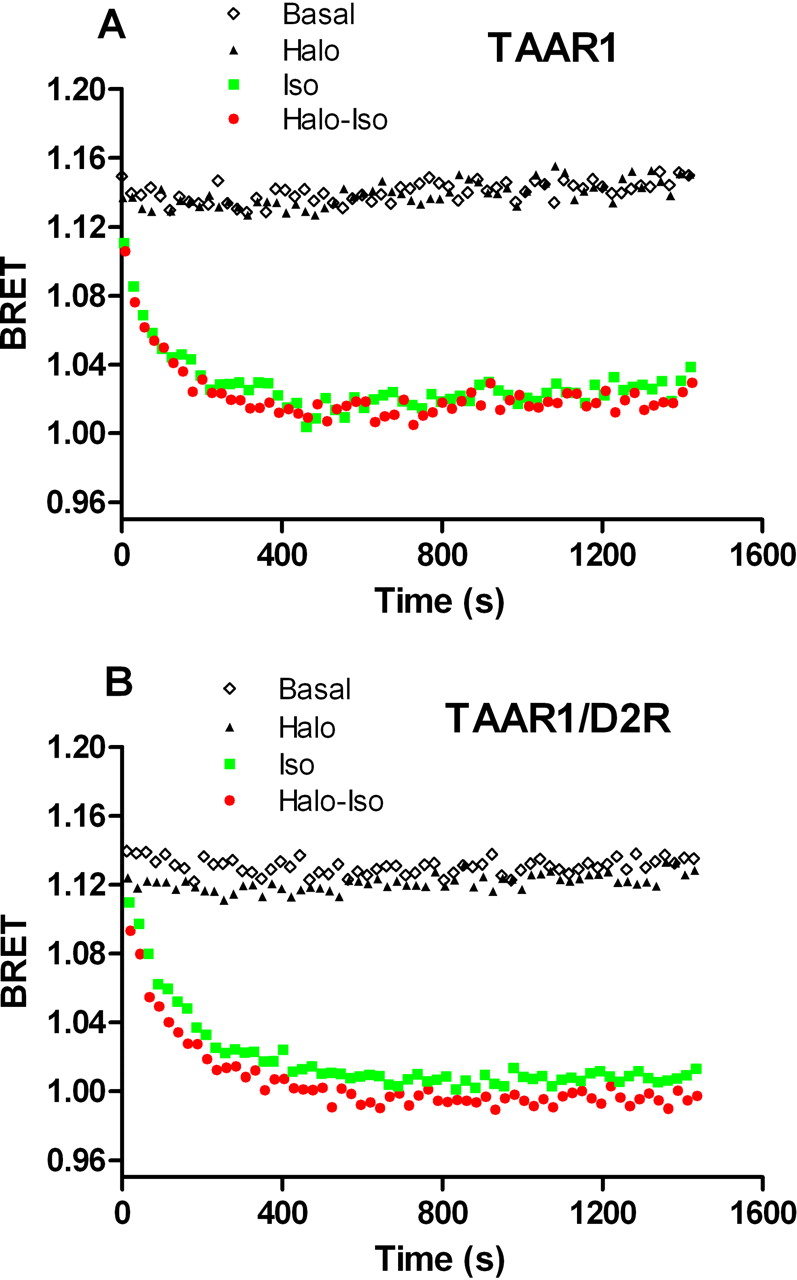
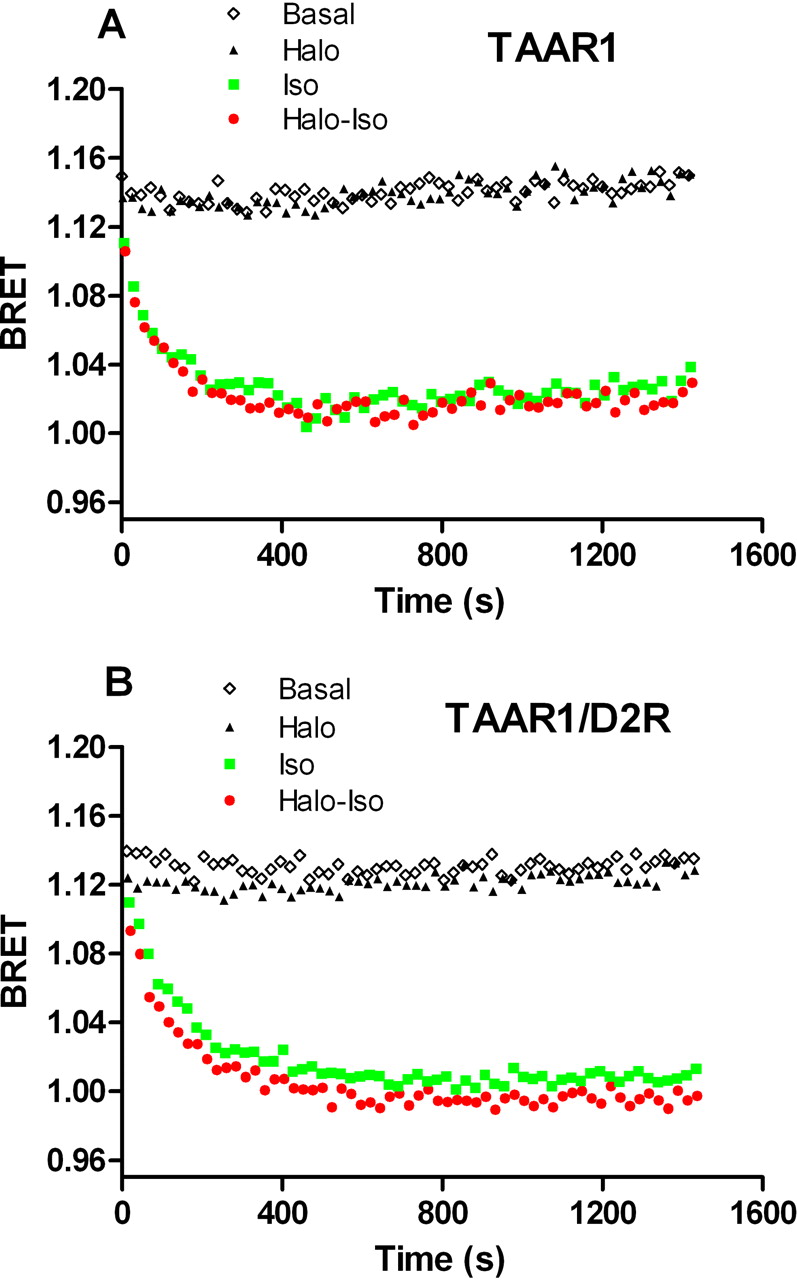
Lack of effect of haloperidol on isoproterenol-induced stimulation of β2-AR. A, the selectivity of haloperidol ability to enhance TAAR1 response was tested by assessing the effects of haloperidol on β2-AR stimulation. Cells expressing TAAR1 and the EPAC biosensor were exposed to isoproterenol (1 μM) or control medium. The activation of endogenously expressed β2-AR induced a robust increase in cAMP levels that were not modulated by haloperidol coadministration. B, when D2R was coexpressed with TAAR1 in the same cells, haloperidol was still not able to increase cAMP production induced by isoproterenol. Results shown are representative of three to four independent experiments.
D2R Blockade Increases Maximal Response but Not Potency of TAAR1 Coexpressed with D2R.
We examined a range of β-PEA concentrations from 10−11 to 10−4 M in cells expressing exclusively TAAR1 and in cells expressing both TAAR1 and D2R with and without haloperidol. As expected, haloperidol had no effect in cells expressing TAAR1 alone; in contrast, in cells expressing both TAAR1 and D2R, haloperidol doubled the maximum effect of β-PEA (Emax = 209 ± 13%, p < 0.001) with little change in the EC50 (basal EC50 = 28 ± 69 nM; haloperidol-treated EC50 = 125 ± 123 nM) (Fig. 4, A and B). Raclopride and amisulpride had similar effects, increasing the efficacy of β-PEA for TAAR1 stimulation (raclopride-treated Emax = 195 ± 8%, p < 0.001; amisulpride-treated Emax = 252 ± 24%, p < 0.05) with minor changes in EC50 values (basal EC50 = 48 ± 119 nM, and raclopride-treated EC50 = 151 ± 115 nM; basal EC50 = 111 ± 213 nM and amisulpride-treated EC50 = 215 ± 326 nM) (Fig. 4, C–F).

Dose-response of β-PEA effect on TAAR1-dependent cAMP accumulation under conditions of D2R blockade. A, various concentrations of β-PEA were applied to cells expressing TAAR1, and cAMP levels were detected using the BRET cAMP biosensor. BRET signal was measured 10 min after the addition of β-PEA. Effects of 10−11 to 10−4 M TAAR1 agonist β-PEA and 1 μM haloperidol were assessed. β-PEA induced an increase in cAMP level that was not modulated by an addition of haloperidol in cells expressing TAAR1 alone. B, the same experiment was conducted in cells coexpressing TAAR1 and D2R. In this case, haloperidol enhanced TAAR1 response with a 2-fold increase in maximum effect (p < 0.001) and no change in EC50. C and D, modulatory effect of raclopride at 1 μM on the ability of different β-PEA concentrations to stimulate cAMP via TAAR1. Like haloperidol, raclopride doubled the β-PEA maximum effect (p < 0.001) only in cells coexpressing D2R (D) with no change in EC50. E and F, analogous experiment with amisulpride at 1 μM reveals an increase in maximum effect of β-PEA (p < 0.05) in cells coexpressing D2R without alteration in EC50. Three to four independent experiments were performed for each compound and condition. Data were analyzed using a two-way ANOVA with repeated measures and Bonferroni post hoc test.
D2R Coexpression Modulates TAAR1 Levels.
Next, we analyzed the membrane expression of co- and singularly expressed HA-TAAR1 and FLAG-D2R by ELISA in nonpermeabilized cells (Salahpour et al., 2004). When TAAR1 and D2R were present in the same cells, TAAR1 membrane expression was reduced by approximately 50% compared with cells without D2R or in cells coexpressing D1R (Fig. 5A). D2R membrane expression was not influenced by TAAR1 presence (Fig. 5C). We performed an additional experiment to exclude the possibility that this modulation could be a nonspecific effect by measuring membrane expression of D1R in the presence of D2R. Coexpression of D2R did not affect D1R membrane immunoreactivity (Fig. 5B). Because we used a TAAR1 that was HA-tagged on the N terminus and tagged with Rluc on the C terminus, we were able to simultaneously measure its membrane and total expression. The same cells that were used for ELISA experiments were detached from their dishes, and Rluc counts were measured as an indicator of total presence of TAAR1. The total amount of TAAR1 was reduced in the presence of D2R but not in the presence of D1R (Fig. 5D). Finally, we investigated whether haloperidol could modulate TAAR1 membrane expression. Cells were treated with haloperidol at 1 μM for 15 min and, as shown in Fig. 5E, haloperidol did not modulate TAAR1 surface expression in cells coexpressing TAAR1 and D2R.
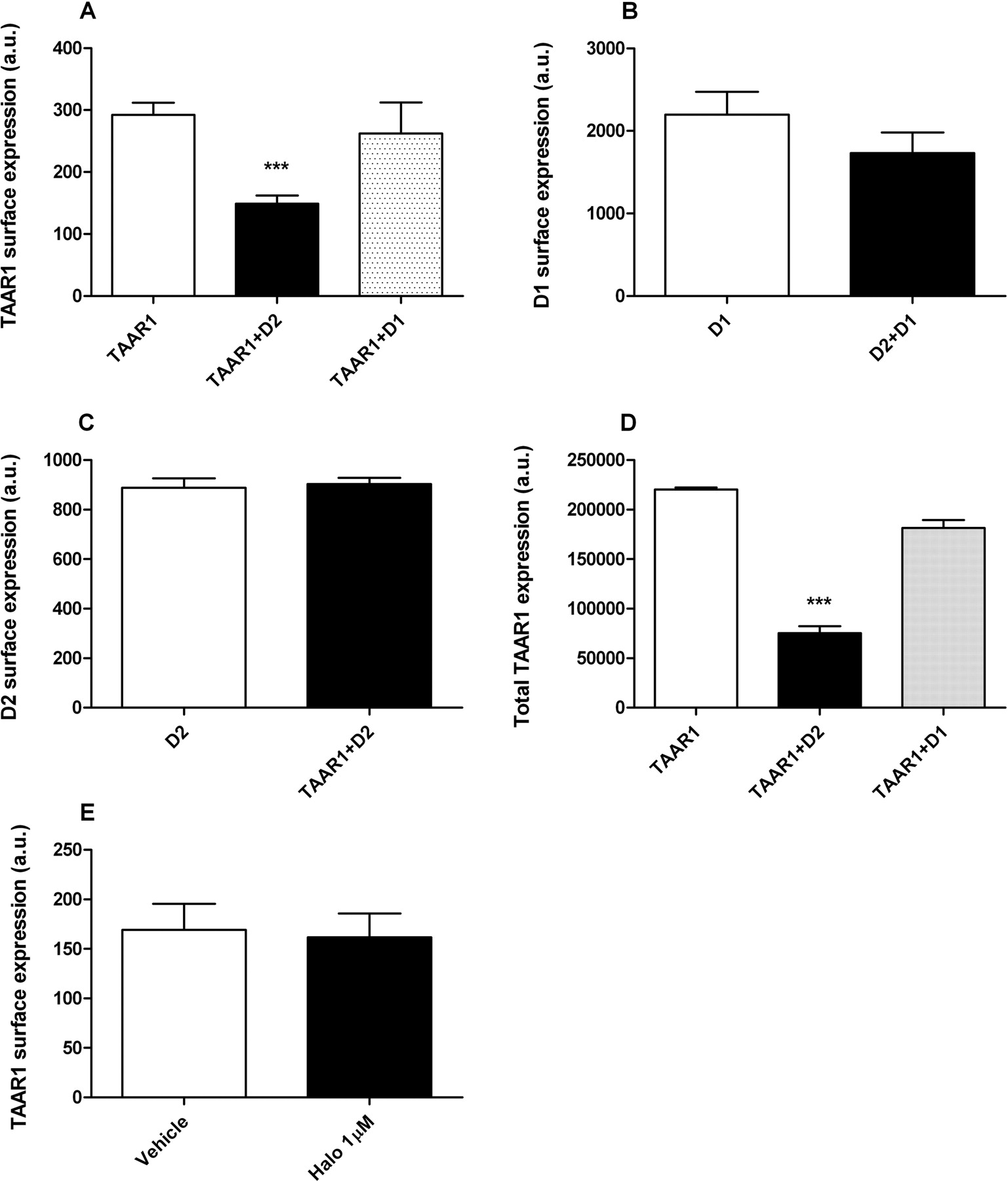
Surface and total expression of TAAR1 is modulated by D2R coexpression. A, the analysis of surface expression of TAAR1 alone or when coexpressed with D2R or D1R was performed in HEK-293 cells. ELISA assay was performed in nonpermeabilized cells by using HA-tagged TAAR1 and FLAG-tagged D2R and D1R and antibodies specific for these two tags. Using an appropriate substrate for the peroxidase (see Materials and Methods) linked to secondary antibody, the absorbance of the supernatant was correlated with the presence of the receptors in the membrane. When TAAR1 is coexpressed with D2R, its presence on the membrane is significantly reduced by approximately 50% (p < 0.001) compared with cells in which this receptor is expressed alone. The reduction in TAAR1 levels was not observed in cells expressing TAAR1 and D1R. B, D2R coexpression does not affect membrane levels of D1R. By using a HA-D1R, we measured the effect of D2R on D1R expression. D2R coexpression did not cause significant changes in D1R membrane levels. C, D2R membrane expression is not modulated by coexpression with TAAR1. In cells expressing both TAAR1 and D2R receptors, D2R surface immunoreactivity was not changed compared with cells only expressing D2R. D, D2R coexpression, but not D1R, significantly reduced TAAR1 total expression. TAAR1 tagged with R. reniformis luciferase in the C terminus was used to monitor the total expression of the receptor by using the cell-permeable Rluc substrate coelenterazine h. The cells were detached from their dishes, and approximately 90,000 cells were placed for each well in a 96-well plate. Coelenterazine h was added at a final concentration of 5 μM, and 10 min later, the luminescence was measured. Similar to surface expression, the total amount of TAAR1 was also reduced in the presence of D2R by approximately 60% (p < 0.01). E, TAAR1 membrane expression is not modulated by haloperidol (1 μM). All values are expressed as means ± S.E.M. (n = 3–5 independent experiments for each experimental condition).
TAAR1 and D2R Form a Heterodimer in Living Cells.
GPCR dimerization occurs for many receptors and this process has important functional consequences (Milligan, 2009). For instance, the dimerization of the two subunits of the metabotropic GABAB receptor is a prerequisite for the correct cell-surface expression and activation of the receptor (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998). BRET is a versatile and validated method to study protein-protein interactions in living cells (Bouvier, 2001), and we used this technique to study the dimerization of TAAR1 tagged with Rluc and D2R tagged with YFP. We performed a titration curve between the two receptors using a fixed TAAR1-Rluc (BRET donor) expression and an increasing amount of D2R-YFP (BRET acceptor). The hyperbolic nature of the curve indicates an association between TAAR1 and D2R (Fig. 6) and heterodimer formation (Salahpour and Masri, 2007). Pretreatment with haloperidol at 1 μM reduced the BRET signal and resulted in a linear titration curve, suggesting the disassembling of the dimer. As a negative control, we cotransfected TAAR1-Rluc and increasing amount of D1-YFP, and we observed no detectable BRET signal between these two receptors (Fig. 6A). Moreover, by cotransfecting the cells with an excess of untagged D2R, we saw a significant reduction of BRET between TAAR1-Rluc and D2-YFP (−53.53% ± 17.66, p < 0.005; Fig. 6B), indicating the specificity of the dimer formation. In addition, we also investigated possibility of cointernalization of the receptors. Because agonist pretreatment may induce a physiological desensitization of a receptor by its internalization, we verified whether TAAR1 stimulation could trigger D2R internalization. Using the same ELISA approach described above, we used HA-TAAR1 and FLAG-D2R to monitor receptors membrane expression. Quinpirole at 1 μM was able to decrease D2R membrane expression (−23.97% ± 0.03, p < 0.05; Fig. 6C). It is noteworthy that TAAR1 stimulation by β-PEA at 1 μM induced a weak but significant decrease in D2R membrane expression (−6.16% ± 0.01, p < 0.05). The observed cointernalization of the receptors after selective stimulation of only one of them further strengthens the evidence of TAAR1-D2R heterodimer formation.

BRET titration curve of physical interaction between TAAR1-Rluc and D2R-YFP. A, fixed amount of TAAR1-Rluc (donor) and increasing amount of D2R-YFP (acceptor) were coexpressed in the same cells. BRET was measured 10 min after the addition of the substrate coelenterazine h in presence or absence of 1 μM haloperidol. To test the specificity of BRET signal between TAAR1 and D2R, BRET was also measured between TAAR1-Rluc and increasing amount of D1R-YFP. The hyperbolic shape of the curve indicates that TAAR1-Rluc and D2R-YFP form a constitutive heterodimer when coexpressed in the same cells. Haloperidol (1 μM), on the contrary, abolished the BRET signal between the two receptors, suggesting the disassembling of the dimer. A linear increase in the BRET signal is observed between TAAR1-Rluc and D1-YFP indicating a nonspecific, bystander BRET between these receptors. B, fixed amounts of TAAR1-Rluc and D2R-YFP were transfected, and BRET was measured. To evaluate the specificity of the complex, an untagged D2R was also transfected. The cotransfection on the untagged-D2R reduced the complex formation between TAAR1-Rluc and D2R-YFP as measured by BRET. C, cointernalization of TAAR1 and D2R was measured by an ELISA approach (see Materials and Methods). HA-TAAR1 and D2R-FLAG were expressed in cells. Upon stimulation by quinpirole (1 μM) or β-PEA (1 μM), D2R-FLAG surface expression decreased. All values are expressed as means ± S.E.M. (n = 3–5 independent experiments for each experimental condition).
TAAR1 and D2R Heterodimer Is Formed Mainly at the Plasma Membrane.
We further investigated whether the heterodimer is formed at the level of the ER or at the PM. To study dimer localization, we transfected the cells with TAAR1-Rluc and D2-YFP or with only TAAR1-Rluc, and we separated ER and PM by centrifugation of the cell lysates using a discontinuous sucrose gradient (Salahpour et al., 2004). We obtained ER in fractions 2 to 6 and PM in 6 to 14 using Na+/K+ ATPase and KDEL as PM and ER markers, respectively (data not shown). Under these conditions, we could detect significant Rluc activity in the first 11 fractions, but as shown in Fig. 7, TAAR1-D2R heterodimer was found mainly in the plasma membrane fractions.

Subcellular distribution of TAAR1-D2R heterodimer. HEK-293T cells expressing TAAR1-Rluc and D2R-YFP or with only TAAR1-Rluc were lysed, and ER and PM were fractionated on a discontinuous sucrose gradients as described under Materials and Methods. TAAR1-D2R heterodimer was found mainly in the PM fractions and was determined by measuring BRET in every fraction.
Haloperidol-Induced c-Fos Expression Is Reduced in TAAR1-KO Mice.
Haloperidol treatment can induce the expression of several marker proteins in the brain indicative of neuronal activity, in particular c-Fos expression in the dorsal striatum of mice (Nguyen et al., 1992). We injected TAAR1-KO mice with saline or haloperidol at a dose of 0.5 mg/kg i.p. and evaluated by immunofluorescence the c-Fos expression in dorsal striatum at 1 h after injection. As shown in Fig. 8A, vehicle did not induce c-Fos expression in any of the genotypes, whereas haloperidol induced a marked fluorescence in neurons of both WT and TAAR1-KO mice, indicating expression of the c-Fos protein. It is noteworthy that the number of neurons activated by haloperidol was significantly reduced in TAAR1-KO animals by approximately 30% (p < 0.05) (Fig. 8B).

Disrupted effects of haloperidol in TAAR1-deficient mice. A, haloperidol-induced c-Fos expression is reduced in the striatum of TAAR1-KO mice. WT and TAAR1-KO mice were treated with haloperidol 0.5 mg/kg i.p. or saline for 1 h. c-Fos expression was evaluated by immunofluorescence staining with specific antibody. Confocal microscopy and images from each region of interest were obtained bilaterally using sequential laser-scanning confocal microscopy. B, quantification of haloperidol-induced c-Fos-positive neurons in WT and TAAR1-KO mice. Neuronal quantification was performed in 375 × 375-μm images by counting c-Fos-positive nucleus. Please note that no c-Fos-positive neurons were noted in vehicle-treated WT or TAAR1-KO mice. Positive neurons in striatal slices from haloperidol-treated TAAR1-KO mice were significantly reduced compared with WT control (p < 0.05). C, haloperidol-induced catalepsy in WT, TAAR1-HET, and TAAR1-KO mice measured 3 h after the treatment. Two-way ANOVA with Bonferroni post hoc test revealed significant differences in all doses of haloperidol tested in both TAAR1-HET and TAAR1-KO mice in comparison with WT mice (***, p < 0.001; **, p < 0.01).
Haloperidol-Induced Catalepsy Is Reduced in TAAR1-KO Mice.
To further evaluate the consequences of a TAAR1-D2R interaction in vivo we tested the ability of haloperidol to induce classic D2R mediated cataleptic behaviors in mice deficient in TAAR1. As shown in Fig. 8C, haloperidol caused a dose-dependent increase in cataleptic behaviors in WT, TAAR1-HET, and TAAR1-KO mice; however, the responses to haloperidol were significantly reduced in TAAR1-HET and TAAR1-KO mice (two-way ANOVA revealed significant effect of dose, p < 0.001; genotype, p < 0.001 and interaction dose × genotype, p < 0.05) These observations taken together with the c-Fos data demonstrate that the interaction between TAAR1 and D2R observed in vitro may have important physiological consequences in vivo.
Discussion
In this study, we demonstrate that TAAR1 is able to physically and functionally interact with the D2R both in vitro and in vivo. D2R coexpression with TAAR1 results in formation of haloperidol-sensitive constitutive heterodimers in plasma membranes of cells. In addition, the inhibition of D2R signaling by specific D2R antagonists enhances β-PEA-induced TAAR1 signaling. This D2R-dependent potentiation of β-PEA stimulation is Gαi protein-dependent, and the stimulatory effect seems limited specifically to TAAR1. Furthermore, we observe in vivo in TAAR1-KO mice a role for TAAR1 in D2R-related signaling and behavior; a reduction in haloperidol induced c-Fos expression that parallels a reduction in haloperidol-induced catalepsy.
The clinical efficacies of antipsychotics are related to their abilities to antagonize dopamine action at D2Rs (Creese et al., 1976; Strange, 2001) and even the newer serotonin receptor active, atypical antipsychotics antagonize D2R signaling through G-protein and β-arrestin2-dependent pathways (Masri et al., 2008; Beaulieu and Gainetdinov, 2011). TAAR1 is also well positioned to modulate brain dopaminergic activity. For example, TAAR1-KO mice show increased sensitivity to amphetamine (Wolinsky et al., 2007; Lindemann et al., 2008) and dopaminergic drugs (Sotnikova et al., 2008; Bradaia et al., 2009). These mutants also demonstrate a deficit in prepulse inhibition and have a larger proportion of striatal D2Rs in a high-affinity state (Wolinsky et al., 2007). Dopamine supersensitivity and increased activity of D2R have been observed in patients with schizophrenia (Breier et al., 1997), and as a consequence, TAAR1-KO mice have been suggested as an animal model of this disorder (Wolinsky et al., 2007).
In the in vitro model system, we observed that our reference compound and prototypical antipsychotic drug haloperidol significantly enhanced TAAR1-dependent β-PEA signaling in cells coexpressing TAAR1 and D2R, whereas it is known that haloperidol does not act on TAAR1 directly (Bunzow et al., 2001; Barak et al., 2008). The ability of other D2R antagonists, raclopride and amisulpride, to mimic the characteristic effects of haloperidol further indicated that those effects are due to modulation by D2R. It is noteworthy that β-PEA-stimulated but not basal cAMP levels were decreased in cells coexpressing TAAR1 and D2R, whereas D2R blockade with antagonists enhanced the efficacy of β-PEA TAAR1 signaling. An analogous response to D2R expression was not observed with isoproterenol acting on endogenously expressed Gαs-coupled β2-AR, indicating that the result for TAAR1 is not simply due to cross-talk between the agonist of Gαs-coupled TAAR1 and the antagonist of Gαi-coupled D2R. Although several mechanisms may be responsible for the potentiation in TAAR1 signaling, including enhancement of TAAR1 G-protein coupling, it is certain that the D2R and Gαi subunits are critical, because overnight pretreatment of cells with PTX prevents the increase in TAAR1 signaling caused by D2R antagonist.
GPCRs may interact at multiple subcellular locations, such as those that occur during cell trafficking (Dong et al., 2007). We therefore studied receptor expression levels in cells coexpressing both receptors. Our results show that D2R coexpression decreases both the surface and total levels of TAAR1 by approximately 50%, whereas the coexpression of D1R had no effect on TAAR1 membrane levels. In addition, D2R coexpression did not affect surface levels of D1R. It is noteworthy that whereas haloperidol treatment did not affect the level of the surface expression of TAAR1, TAAR1-mediated signaling is markedly increased when the antagonist blocks D2R. Thus, the observed alterations in TAAR1 expression found in cells coexpressing D2R cannot be a basis of the increase in TAAR1 signaling caused by haloperidol under these conditions. Rather, it is possible that the pharmacological properties of TAAR1 are changed when it is coexpressed with D2R.
A simple hypothesis to explain our findings is receptor heterodimerization (Angers et al., 2002). GPCR homo- and heterodimerization has been demonstrated for many receptors (Dalrymple et al., 2008), and direct interaction between receptors could lead to a modulation of their function, such as differential trafficking and/or changes in their pharmacological profile (Salahpour et al., 2004; Milligan, 2009). We tested this hypothesis by using a BRET approach and performed a titration curve analysis of heterodimerization between TAAR1-Rluc and D2R-YFP. This method is commonly used to study homo- and heterodimerization between different GPCRs (Pfleger and Eidne, 2006). Using this approach, we showed that TAAR1 and D2R formed constitutive heterodimers and that haloperidol treatment abolished the BRET signal resulting from the formation of the heterodimer. We further demonstrated that heterodimer formation was specific to D2R and that heterodimers formed mainly on the plasma membrane. Although further detailed studies are necessary to understand the molecular mechanisms of altered pharmacological properties of these heterodimers, it is likely that the disruption of the heterodimer formation or conformational change in the complex structure caused by haloperidol contributes to the enhanced TAAR1 signaling.
To directly investigate whether this interaction of D2R and TAAR1 has physiological consequences at the in vivo level, we first studied the effect of haloperidol treatment on c-Fos expression. c-Fos is an immediate early gene that has been widely used as an indicator of neuronal activation. It has been demonstrated that haloperidol and many other antipsychotics can induce c-Fos expression in several brain regions, and particularly, typical antipsychotics can activate neurons located in dorsal striatum (Nguyen et al., 1992). We observed that c-Fos expression in dorsolateral striatum, after haloperidol treatment, was decreased in TAAR1-KO mice, suggesting that D2R-mediated signaling is affected. Haloperidol and other D2R antagonists induce cataleptic behaviors in animals, and these extrapyramidal side effects are commonly used in pharmacological modeling of Parkinson's disease (Sotnikova et al., 2005). Using a bar test to evaluate catalepsy, we have shown that haloperidol is less potent in inducing catalepsy in TAAR1 full and partial (heterozygous) knockout mice, indicating that TAAR1 modulates D2R-related physiology in vivo.
In conclusion, we report that antagonism of D2R enhances TAAR1 cAMP signaling. The enhancement seems unique to this pair of receptors and possibly is a result of the disruption of TAAR1-D2R complex. Moreover, TAAR1 modulates in vivo D2R antagonism-related signaling and behaviors. The observations that both TAAR1 antagonism and a constitutive genetic lack of TAAR1 result in increased dopamine potency at D2R in dopaminergic neurons (Bradaia et al., 2009), combined with our data showing enhancement of TAAR1 signaling under conditions of D2R blockade indicate that the TAAR1 and D2R can potently modulate each other's activity. Thus, the observed functional D2R/TAAR1 interaction may have important consequences for current and future therapeutic strategies based on the application of dopaminergic drugs and TAAR1 ligands in the treatment of dopamine-related disorders (Revel et al., 2011).
Authorship Contributions
Participated in research design: Espinoza, Salahpour, Masri, Sotnikova, Barak, Caron, and Gainetdinov.
Conducted experiments: Espinoza, Salahpour, Masri, Sotnikova, and Messa.
Contributed new reagents or analytic tools: Espinoza, Salahpour, Masri, Barak, Caron, and Gainetdinov.
Performed data analysis: Espinoza, Salahpour, Masri, Sotnikova, Messa, and Gainetdinov.
Wrote or contributed to the writing of the manuscript: Espinoza, Salahpour, Masri, Barak, Caron, and Gainetdinov.
Acknowledgments
We thank Katherine Clark and Yushi Bai for excellent technical assistance in performing these experiments. We acknowledge Lundbeck A/G and Lundbeck USA for generously providing TAAR1 mutant mice.
Footnotes
This work was supported in part by F. Hoffmann-La Roche Ltd. (Basel, Switzerland); Fondazione Compagnia di San Paolo (Torino, Italy); the National Institutes of Health National Institute on Drug Abuse [Grants U01-DA022950, P30-DA029925]; the National Institutes of Health National Institute of Mental Health [Grant R01-MH073853]; and a European Marie-Curie Outgoing International Fellowship [FP6–2005-Mobility-6].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.073304.
-
ABBREVIATIONS:
- TA
- trace amine
- β-PEA
- β-phenylethylamine
- D2R
- dopamine D2R receptor
- BRET
- bioluminescence resonance energy transfer
- EPAC
- exchange protein directly activated by cAMP
- β2-AR
- β2-adrenergic receptor
- DA
- dopamine
- TAAR
- trace amine-associated receptor
- TAAR1-KO
- trace amine-associated receptor 1 knockout
- DAT
- dopamine transporter
- Rluc
- Renilla reniformis luciferase
- GPCR
- G protein-coupled receptor
- PTX
- pertussis toxin
- FBS
- fetal bovine serum
- HA
- hemagglutinin
- PCR
- polymerase chain reaction
- YFP
- yellow fluorescent protein
- ER
- endoplasmic reticulum
- PM
- plasma membrane
- ELISA
- enzyme-linked immunosorbent assay
- TBS
- Tris-buffered saline
- WT
- wild type
- HET
- heterozygous
- iso
- isoproterenol
- halo
- haloperidol
- ANOVA
- analysis of variance
- HEK
- human embryonic kidney.
- Received April 28, 2011.
- Accepted June 10, 2011.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}