


Abstract
Two orphan leucine-rich repeat-containing G protein-coupled receptors were recently identified as targets for the relaxin family peptides relaxin and insulin-like peptide (INSL) 3. Human gene 2 relaxin is the cognate ligand for relaxin family peptide receptor (RXFP) 1, whereas INSL3 is the ligand for RXFP2. Constitutively active mutants of both receptors when expressed in human embryonic kidney (HEK) 293T cells signal through Gαs to increase cAMP. However, recent studies using cells that endogenously express the receptors revealed greater complexity: cAMP accumulation after activation of RXFP1 involves a time-dependent biphasic pathway with a delayed phase involving phosphoinositide 3-kinase (PI3K) and protein kinase C (PKC) ζ, whereas the RXFP2 response involves inhibition of adenylate cyclase via pertussis toxin-sensitive G proteins. The aim of this study was to compare and contrast the cAMP signaling pathways used by these two related receptors. In HEK293T cells stably transfected with RXFP1, preliminary studies confirmed the biphasic cAMP response, with an initial Gαs component and a delayed response involving PI3K and PKCζ. This delayed pathway was dependent upon G-βγ subunits derived from Gαi3. An additional inhibitory pathway involving GαoB affecting cAMP accumulation was also identified. In HEK293T cells stably transfected with RXFP2, the cAMP response involved Gαs and was modulated by inhibition mediated by GαoB and release of inhibitory G-βγ subunits. Thus, initially both RXFP1 and RXFP2 couple to Gαs and an inhibitory GαoB pathway. Differences in cAMP accumulation stem from the ability of RXFP1 to recruit coupling to Gαi3, release G-βγ subunits and thus activate a delayed PI3K-PKCζ pathway to further increase cAMP accumulation.
Relaxin is a two-chain peptide hormone that is structurally closely related to insulin. It belongs to the insulin-relaxin peptide family that includes the relaxins: human gene (H)1 (product of a gene duplication event in higher primates), H2 (major circulating form), and H3 (principally a neuropeptide); insulin; insulin-like growth factors I and II; and the relaxin/insulin-like factors INSL3, INSL4, INSL5, and INSL6. Although relaxin was first identified for its role in parturition in guinea pigs (Hisaw, 1926), it is now recognized as a hormone with pleiotropic effects. Relaxin has important functions in the heart, kidney, and brain in addition to roles in the regulation of nitric oxide production and neoangiogenesis (for review, see Bathgate et al., 2006). It is noteworthy that relaxin can also prevent the tissue remodeling observed in fibrosis with a conservation of endogenous tissue structure (Unemori et al., 1996), and herein lies its potential as a therapeutic.
Despite the length of time since the discovery of relaxin, its receptor remained elusive until the recent pairing of relaxin family peptides with two highly similar leucine-rich repeat-containing GPCRs: LGR7 and LGR8 (Hsu et al., 2002). LGR7 was subsequently identified as the relaxin receptor (Hsu et al., 2002) and LGR8 as the receptor for the related peptide INSL3 (Kumagai et al., 2002). More recently, two additional GPCRs were deorphanized using ligands from the insulin-relaxin family: GPCR135 is the receptor for H3 relaxin (Liu et al., 2003), and GPCR142 is the receptor for INSL5 (Liu et al., 2004). These four receptors form the relaxin family peptide (RXFP) receptor family, and, based upon recent International Union of Pharmacology recommendations, they are named RXFP1 (LGR7), RXFP2 (LGR8, Great), RXFP3 (GPCR135, SALPR), and RXFP4 (GPCR142, GPR100) (Bathgate et al., 2006).
Although relaxin mediates many physiological effects, the intracellular signaling pathways involved are still unclear. Despite this, there is evidence for relaxin-mediated activation of a number of signaling pathways, including cAMP. Relaxin has long been shown to increase cAMP levels in numerous target tissues, although the precise mechanism of adenylate cyclase stimulation has not been clearly demonstrated. Studies have shown increases in cAMP in response to relaxin stimulation in THP-1 cells (Parsell et al., 1996), although this cAMP increase was very weak and required amplification by phosphodiesterase inhibition and adenylate cyclase priming by forskolin. In addition, MAPK inhibitors were shown to decrease cAMP in THP-1 cells, suggesting that MAPK-mediated inhibition of phosphodiesterases is required to give sustained increases in cAMP (Bartsch et al., 2001). When taken together with little to no cAMP responses in other tissues (Palejwala et al., 1998; Kompa et al., 2002), this suggested that the involvement of cAMP in relaxin signaling was minimal. However, relaxin stimulation of either RXFP1 or RXFP2 receptors stably expressed in HEK293T cells (HEK-RXFP1 and HEK-RXFP2, respectively) resulted in clear increases in cAMP (Hsu et al., 2002), and constitutively active mutants showed ligand-independent cAMP production (Hsu et al., 2000, 2002). Thus, unlike previous studies, this clearly demonstrated that RXFP1 and RXFP2 could act as Gs-linked GPCRs that signal through adenylate cyclase to increase cAMP.
Recent studies in THP-1 cells (which endogenously express RXFP1) showed a biphasic cAMP response to relaxin stimulation, with an early peak (1–2 min) and a later peak (10–20 min) that was sensitive to PI3K inhibitors (Nguyen et al., 2003). Relaxin was subsequently shown to stimulate PI3K, and its effects upon cAMP levels seemed independent of effects on phosphodiesterases (Nguyen et al., 2003). A general inhibitor of PKC, chelerythrine chloride, also inhibited the later phase of the cAMP response, and relaxin was shown to stimulate the translocation of PKCζ to the plasma membrane (Nguyen and Dessauer, 2005). Thus, the relaxin-stimulated cAMP response at RXFP1 is biphasic, and the second phase of the response occurs through a PI3K-PKCζ-dependent pathway.
Even less is known regarding the signaling events initiated by INSL3 binding to RXFP2. Based upon the identification of RXFP2 as a Gs-coupled receptor (Hsu et al., 2002), signaling studies have thus far focused on cAMP pathways. In HEK-RXFP2 and gubernacular cells (which endogenously express RXFP2), INSL3 stimulation caused increased cAMP levels (Kumagai et al., 2002). In contrast, INSL3 stimulation of testicular germ cells and oocytes caused a decrease in cAMP that was prevented by pretreatment with pertussis toxin (PTX), suggesting that RXFP2 couples to Gi/Go proteins (Kawamura et al., 2004).
Here, we aimed to compare and contrast the cAMP signaling pathways of the RXFP1 and RXFP2 receptors. The study confirms the presence of the delayed PI3K-PKCζ signaling pathway mediated by RXFP1 in HEK-RXFP1 cells and has identified for the first time that G-βγ derived from Gαi3 is the mediator of this pathway. We have also identified an additional novel cAMP signaling pathway that is part of the immediate response that involves GαoB-mediated inhibition of adenylate cyclase. Thus, differences in immediate and delayed cAMP responses are due to involvement of distinct Gi/Go isoforms. In HEK-RXFP2 cells, the delayed PI3K-PKCζ pathway was not involved, showing that RXFP2 is unable to couple to Gαi3. Instead, RXFP2 signaling involves a Gαs stimulation, and we show for the first time involvement of a GαoB and G-βγ-mediated inhibition of cAMP accumulation.
Materials and Methods
Hormones and Reagents. Recombinant H2 relaxin was kindly provided by BAS Medical (San Mateo, CA). Human INSL3 was chemically synthesized by Dr. John Wade (Howard Florey Institute, Melbourne, VIC, Australia). PTX was purchased from Sigma-Aldrich (Sydney, NSW, Australia); LY294002, wortmannin, and bisindolylmalemide I were purchased from Calbiochem (Kilsyth, VIC, Australia); and chelerythrine chloride and PKCζ pseudosubstrate inhibitor were purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA). The anti-Gαi/o/t/z antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); biotin molecular weight marker and anti-biotin secondary antibody were both purchased from Cell Signaling Technology, Inc. (Beverly, MA). Donkey anti-goat/sheep secondary HRP-linked antibody was kindly provided by Dr. Grant Drummond (Department of Pharmacology, Monash University, Melbourne, VIC, Australia).
Constructs. β-Adrenergic receptor kinase I-C terminus (βARK-ct) (Koch et al., 1994) was obtained from Dr. Walter Thomas (Baker Heart Research Institute, Melbourne, VIC, Australia) with the kind permission of Dr. Robert Lefkowitz (Duke University Medical Centre, Durham, NC). The construct produces a segment of the C-terminal end of bovine β-adrenergic receptor kinase, which acts to sequester G-βγ subunits.
PTX-insensitive Gi/Go α subunit mutants were kindly provided by Dr. Patrick M. Sexton (Howard Florey Institute). These G protein α subunits have a Cys351Ile mutation (Bahia et al., 1998), which renders them insensitive to ADP-ribosylation by PTX.
Cell Culture. HEK293T cells (ATCC CRL-1573; American Type Culture Collection, Manassas, VA) stably expressing either the RXFP1 (HEK-RXFP1) or RXFP2 (HEK-RXFP2) receptors were maintained in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (all from Trace Biosciences, Sydney, NSW, Australia). All tissue culture plates were coated with 0.1 mg/ml poly-l-lysine (Sigma-Aldrich) before use. Cells were maintained at 37°C in a CO2 water-jacket incubator (Forma Scientific, Marietta, OH) in 5% CO2 and 85% humidity.
HEK-RXFP1 and HEK-RXFP2 cells were used as described previously (Sudo et al., 2003; Halls et al., 2005b). Transient transfections were performed using Metafectene (Biontex, Munich, Germany) as per manufacturer's instructions (Halls et al., 2005b). Cells transiently expressing stated constructs were seeded into 96-well plates 24 h after transfection and used 48 h after transfection.
cAMP Accumulation Assay. cAMP responses were determined using the AlphaScreen cAMP accumulation assay (PerkinElmer, Rowville, VIC, Australia). Cells were seeded into 96-well plates (5 × 104 cells/well) in 10% FBS (v/v) RPMI 1640 medium for 8 h and then partially serum-starved overnight [0.5% FBS (v/v) RPMI 1640 medium]. Inhibitors were preincubated with the cells for 30 min in 0.5% FBS (v/v) RPMI 1640 medium, except the myristoylated PKCζ pseudosubstrate inhibitor and PTX, which were incubated with the cells for 1 and 16 h, respectively.
cAMP assays were performed in duplicate as described previously (Halls et al., 2005b). In brief, cells were incubated with stimulation buffer containing H2 relaxin or INSL3 (concentrations ranging from 0.1 pM to 1 μM, as stated), 0.1 mM forskolin (Sigma-Aldrich), or blank for stated time periods at 37°C. After removal of stimulation buffer, cells were frozen in lysis buffer at –70°C to terminate the reaction and to lyse cells. Samples were transferred to a 384-well white Optiplate (PerkinElmer) after thawing, and anti-cAMP acceptor beads then donor beads with biotinylated cAMP were added to all wells.
After overnight incubation, plates were read using a Fusion-α microplate reader (PerkinElmer), and the data were analyzed against a standard curve using Prism (GraphPad Software Inc., San Diego, CA). Samples were normalized for cell number by expressing the results as a percentage of the 0.1 mM forskolin response. Each experiment was performed in duplicate, and results are expressed as the mean ± S.E. of the mean of n separate experiments (as stated). All dose-response studies (except 3-min INSL3 stimulation of RXFP2) were best described by a bell-shaped model and were fit to a Gaussian equation. The 3-min INSL3 dose-response curves at RXFP2 were best fit using a GraphPad Prism nonlinear regression sigmoidal dose-response model. Statistical analyses were performed on raw data using a GraphPad Prism unpaired t test, with statistical significance accepted at p < 0.05.
Western Blotting. HEK-RXFP1 and HEK-RXFP2 cells transiently transfected with PTX-insensitive Gαi/o(C351I) mutants were lysed and scraped in radioimmunoprecipitation assay buffer (1× phosphate-buffered saline, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride, 100 μM sodium orthovanadate, and 5 μg/ml aprotinin) on ice, collected, and sonicated before addition of 100 μg/ml phenylmethylsulfonyl fluoride and centrifugation (10,000g; 10 min; 4°C). Concentration of the protein samples was determined according to Lowry et al. (1951). Samples were diluted in radioimmunoprecipitation assay buffer to give 1 mg/lane and supplemented with SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 0.1% bromphenol blue, and 10% β-mercaptoethanol) at a ratio of 1:1. Proteins were separated on a 10% polyacrylamide gel and electrotransferred to a Hybond-P polyvinylidene difluoride membrane (pore size 0.45 μm; GE Healthcare, Little Chalfont, Buckinghamshire, UK) using a semidry electroblotter. After blocking (5% nonfat dry milk and 0.1% Tween 20 in Tris-buffered saline; 1 h at room temperature), the membrane was incubated with primary (anti-Gαi/o/t/z, 1:500 dilution in Tris-buffered saline with 0.1% Tween 20 and 5% bovine serum albumin; overnight at 4°C) and then secondary (donkey anti-sheep/goat conjugated-HRP and anti-biotin conjugated HRP, both 1:2000 dilution in Tris-buffered saline with 0.1% Tween 20 and 5% nonfat dry milk; 1 h at room temperature) antibodies. Peroxidase activity was observed by chemiluminescence using the Lumi-light Western blotting substrate (Roche Diagnostics, Indianapolis, IN) and exposure to film.
Results
RXFP1, but Not RXFP2, Shows a Biphasic cAMP Response Involving PI3K and PKCζ in Stably Transfected HEK293T Cells. H2 relaxin (30 nM) stimulation of RXFP1 over a period of 40 min revealed a biphasic time course of activation (Fig. 1A). No significant difference in cAMP accumulation was observed in the presence of the PI3K inhibitors at times between 0 and 15 min. Addition of the PI3K inhibitors LY294002 (10 μM) or wortmannin (0.1 μM) inhibited the second phase (>15 min) of the response. Conversely, 10 nM INSL3 (Fig. 1B) or 10 nM H2 relaxin stimulation (data not shown) of RXFP2 over a similar period revealed a very different cAMP accumulation profile. There was no effect of the PI3K inhibitors wortmannin (100 nM) or LY294002 (1 μM) on either INSL3 or H2 relaxin stimulation at any of the times tested. It is noteworthy that the magnitude of the cAMP response at RXFP2 was significantly smaller than the response obtained by stimulation of RXFP1, despite similar expression levels of both receptors in the cells (Sudo et al., 2003).

Time course of cAMP accumulation: effect of PI3K inhibitors on the RXFP1 and RXFP2 response. cAMP accumulation in response to H2 relaxin stimulation (30 nM) of RXFP1 (A), and INSL3 stimulation (10 nM) of RXFP2 (B), was measured over a period of 40 min in the presence and absence of the PI3K inhibitors LY294002 (10 μM; 30-min preincubation) and wortmannin (0.1 μM; 30-min preincubation). cAMP levels are expressed as a percentage of the 0.1 mM forskolin response (after 30-min incubation). Symbols represent means, and vertical bars represent S.E.M. of five separate experiments performed in duplicate. *, p < 0.05 versus H2 relaxin alone at RXFP1 (unpaired t test).
In HEK-RXFP1 cells, concentration-response curves were performed measuring cAMP levels at 3 min (pEC50 = 8.66 ± 0.25) and 30 min (pEC50 = 9.42 ± 0.22) based upon time-course data, to further define the characteristics of the responses after stimulation over a short and longer period (Fig. 2; Table 1). Two inhibitors of PI3K were used to ensure that the decreases in cAMP were due to specific inhibition of PI3K. LY294002 and wortmannin inhibit PI3K through two distinct mechanisms, thus ensuring PI3K specificity: wortmannin blocks the catalytic subunits of PI3K (Yano et al., 1993), whereas LY294002 acts at the ATP binding site (Vlahos et al., 1994). Both 0.1 μM wortmannin and 10 μM LY294002 significantly decreased the maximum response to 30-min exposure of H2 relaxin by 70 and 60%, respectively (Fig. 2B), but they had no effect on the response to a 3-min exposure (Fig. 2A). As observed in the time-course studies, the two unrelated PI3K inhibitors had no effect on the concentration-response relationship to INSL3 at RXFP2 after either 3 min (pEC50 = 8.64 ± 0.44; Fig. 2C) or 30 min (pEC50 = 9.24 ± 0.16; Fig. 2D) exposure to the peptide. The same held true for H2 relaxin stimulation of RXFP2 (data not shown). There was no effect of the PI3K inhibitors on either basal or forskolin-stimulated cAMP levels in either cell line (data not shown) or upon any of the pEC50 values.
Effect of PI3K and PKC inhibitors, PTX pretreatment, and βARK-ct transfection on the maximum RXFP1 and RXFP2 cAMP accumulation responses
Effect of 0.1 μM wortmannin (30-min preincubation), 10 μM LY294002 (30-min preincubation), 1 μM chelerythrine chloride (30-min preincubation), 100 ng/ml PTX (16-h preincubation), and βARK-ct transient transfection (114 ng/cm2 culture area) on the 3- and 30-min maximum responses to H2 relaxin at RXFP1 or INSL3 at RXFP2 is shown. Maximum responses were determined for each separate experiment (performed in duplicate), and then the mean and S.E.M. were determined for each maximum response. cAMP is expressed as a percentage of the 0.1 mM forskolin response at each time point. Results are expressed as mean ± S.E.M. of (n) experiments, each performed in duplicate.
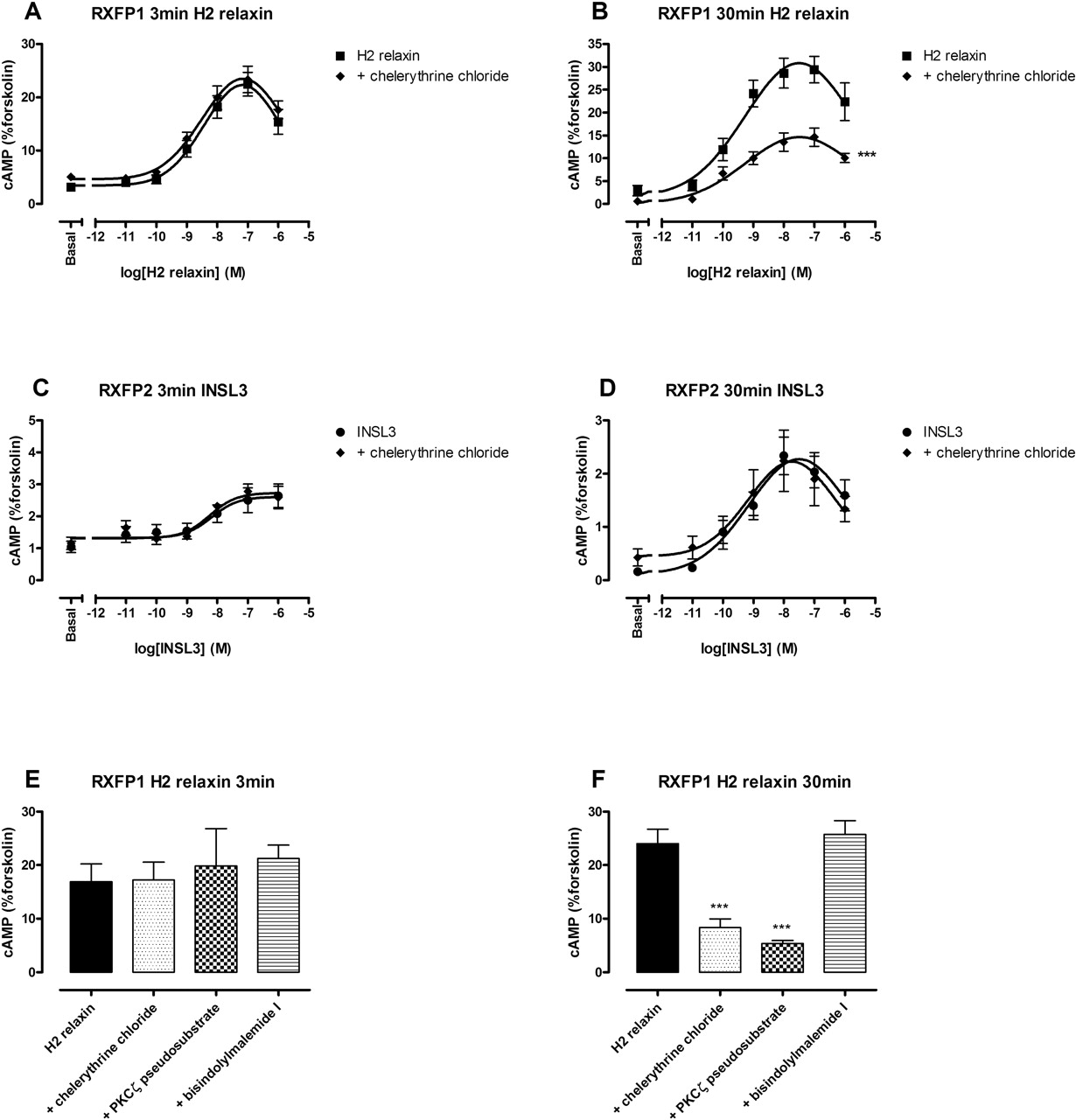
Based upon previous evidence for PI3K activation of PKCζ to increase cAMP after H2 relaxin stimulation (Nguyen and Dessauer, 2005), the effect of a general inhibitor of PKC, chelerythrine chloride, was examined (Fig. 3; Table 1). Chelerythrine chloride (1 μM) significantly inhibited the maximum response to H2 relaxin at RXFP1 at 30 min by 68% (Fig. 3B) but had no effect on the response generated at 3 min (Fig. 3A). The level of inhibition was comparable with that observed for wortmannin and LY294002. To confirm involvement of atypical PKC isoforms in this delayed response, a number of PKC inhibitors were used after stimulation with 30 nM H2 relaxin (Fig. 3, E and F; Table 2). Specificity for atypical PKCs (ζ or ι/λ) was thus confirmed using three PKC inhibitors with different activity spectra across the various PKC isoforms: chelerythrine chloride acts as a general PKC inhibitor by acting upon the catalytic domain (Uberall et al., 1997); bisindolylmaleimide I is also considered a general inhibitor, although does not have any activity at the atypical isoforms (Martiny-Baron et al., 1993; Uberall et al., 1997); and myristoylated PKCζ is a pseudosubstrate inhibitor that is specific for the atypical PKC isoforms because they exhibit identical sequence within their pseudosubstrate domains (Selbie et al., 1993). Thus, the overlap in activity of the inhibitors for the various isoforms of PKC allowed confirmation of the specific involvement of an atypical PKC in this system, probably PKCζ based upon previous studies (Nguyen and Dessauer, 2005). The specific inhibitor of atypical PKCs, myristoylated PKCζ pseudosubstrate inhibitor (10 μM), significantly inhibited the cAMP response at 30 min by 78% (Fig. 3F) but had no effect at 3 min (Fig. 3E). It is noteworthy that the general inhibitor of PKC that does not inhibit the atypical isoforms, bisindolylmaleimide I (1 μM), did not inhibit either the 3-min (Fig. 3E) or 30-min (Fig. 3F) responses.
Effect of PKC inhibitors on the response to H2 relaxin stimulation at RXFP1
Effect of 1 μM chelerythrine chloride (30-min preincubation), 10 μM PKCζ pseudosubstrate inhibitor (1-h preincubation), and 1 μM bisindolylmalemide I (30-min preincubation) on the 3- and 30-min response to 30 nM H2 relaxin at RXFP1. cAMP is expressed as a percentage of the 0.1 mM forskolin response at each time point. Results are expressed as mean ± S.E.M. of (n) experiments, each performed in duplicate.
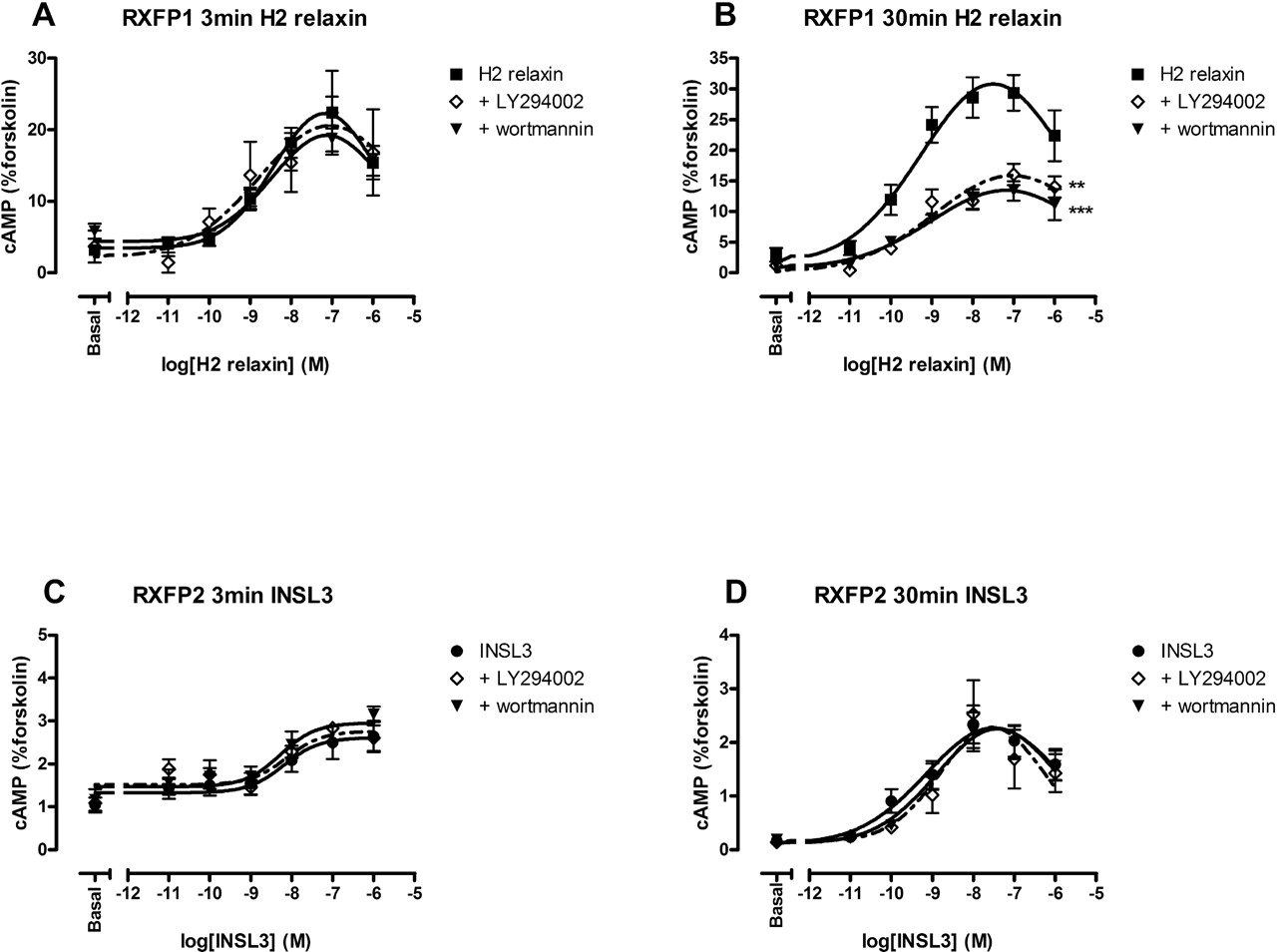
Effect of PI3K inhibition upon the RXFP1 and RXFP2 responses after 3- or 30-min stimulation. Concentration-response curves were performed at either 3 min (A and C) or 30 min (B and D) using H2 relaxin (0.1 pM–1 μM) at RXFP1 (A and B) and INSL3 (0.1 pM–1 μM) at RXFP2 (C and D) in the presence and absence of the PI3K inhibitors wortmannin (0.1 μM; 30-min preincubation) and LY294002 (10 μM; 30-min preincubation). cAMP levels are expressed as a percentage of the 0.1 mM forskolin response at each time point. Symbols represent means, and vertical bars represent S.E.M. of four to eight separate experiments performed in duplicate. **, p < 0.01 and ***, p < 0.001 versus peptide alone (all maximum responses, unpaired t-tests).
As expected, the general PKC inhibitor chelerythrine chloride (1 μM) had no effect on the INSL3 concentration-response curve at RXFP2 at either the 3-min (Fig. 3C) or 30-min (Fig. 3D) time points. The same was true for the H2 relaxin concentration-response relationship at RXFP2 (data not shown). There was no effect of any of the PKC inhibitors used on basal or forskolin-stimulated cAMP levels in either cell line (data not shown). Chelerythrine chloride did not affect any of the pEC50 values.
PTX-Sensitive G Proteins Are Involved in Signaling through Both RXFP1 and RXFP2. There is evidence to suggest involvement of PTX-sensitive G proteins in the cAMP accumulation response after stimulation of both RXFP1 (Halls et al., 2005a) and RXFP2 (Kawamura et al., 2004). Thus, we examined the effect of 100 ng/ml PTX upon time-course and concentration-response relationships at both receptors (Fig. 4; Table 1).
Preincubation of PTX with HEK-RXFP1 cells revealed different effects upon H2 relaxin stimulation that were time-dependent. This was observed in both time-course (Fig. 4A) and concentration-response studies (Fig. 4, C and D). In time-course studies, from 0 to 20 min there was little effect of PTX pretreatment on cAMP accumulation; however, after 20 min PTX significantly inhibited the RXFP1 response to H2 relaxin (Fig. 4A). The detailed concentration-response studies confirmed an additional effect of PTX pretreatment: a significantly increased cAMP response by 46% after pretreatment of cells with PTX at the 3-min time point (Fig. 4C) and a significant inhibition by 37% at the 30-min time point (Fig. 4D). Thus, PTX-sensitive G proteins seem to mediate distinct pathways leading to cAMP accumulation at early compared with late time points, confirming the biphasic nature of the RXFP1 response.

The role of atypical PKC in the delayed cAMP response mediated through RXFP1 but not RXFP2. Concentration-response curves were performed at either 3 min (A and C) or 30 min (B and D) using H2 relaxin (0.1 pM–1 μM) at RXFP1 (A and B) and INSL3 (0.1 pM–1 μM) at RXFP2 (C and D) in the presence and absence of the general PKC inhibitor chelerythrine chloride (1 μM; 30-min preincubation). Single point experiments [30 nM H2 relaxin for 3 min (E) or 30 min (F)] were also performed in the presence and absence of chelerythrine chloride (1 μM; 30-min preincubation); bisindolylmaleimide I, a general PKC inhibitor that does not inhibit PKCζ (1 μM; 30-min preincubation); and myristoylated PKCζ pseudosubstrate inhibitor, which inhibits the atypical (ζ and ι) PKC isoforms (10 μM; 1-h preincubation). cAMP levels are expressed as a percentage of the 0.1 mM forskolin response at each time point. Symbols represent means, and vertical bars represent S.E.M. of four to eight separate experiments performed in duplicate. ***, p < 0.001 versus peptide alone (all maximum responses, unpaired t tests).
It is noteworthy that pretreatment of HEK-RXFP2 cells with PTX increased the INSL3 response at RXFP2 in a consistent manner over a 40-min period (Fig. 4B). This was also confirmed by concentration-response studies at the two time points, reiterating the monophasic nature of the RXFP2 response. Pretreatment of cells with PTX significantly increased the 3-min response to INSL3 by 43% (Fig. 4E) and the 30-min maximum response by 48% (Fig. 4F). PTX pretreatment of HEK-RXFP2 cells had the same effect upon H2 relaxin stimulation (data not shown). Pretreatment of HEK-RXFP2 cells with PTX caused increased basal levels of cAMP. There was no other effect of PTX on basal or forskolin-stimulated cAMP levels in either stable cell line or parent HEK293T cells (data not shown), suggesting some coupling of PTX-sensitive G proteins to RXFP2 at rest. PTX did not affect any pEC50 values.

The role of PTX-sensitive G proteins in the RXFP1 and RXFP2 cAMP responses. cAMP accumulation in response to H2 relaxin stimulation (30 nM) of RXFP1 (A), and INSL3 stimulation (10 nM) of RXFP2 (B), was measured over a period of 40 min in the presence and absence of the Gi/Go inhibitor PTX (100 ng/ml; 16-h preincubation). Concentration-response studies were then performed at either 3 min (C and E) or 30 min (D and F) using H2 relaxin (0.1 pM–1 μM) at RXFP1 (C and D) and INSL3 (0.1 pM–1 μM) at RXFP2 (E and F) in the presence and absence of the Gi/Go inhibitor PTX (100 ng/ml; 16-h preincubation). cAMP levels are expressed as a percentage of the 0.1 mM forskolin response at each time point, or after 30-min incubation for time-course studies. Symbols represent means, and vertical bars represent S.E.M. of four to eight separate experiments performed in duplicate. *, p < 0.05 and **, p < 0.01 versus peptide alone (all maximum responses, unpaired t tests).
G-βγ Proteins Mediate the cAMP Increases of the Delayed RXFP1 Pathway, but Act to Inhibit cAMP Levels in a Delayed Manner after RXFP2 Stimulation. Because PI3K can be activated by G-βγ subunits (Stephens et al., 1994), we tested the effect of the G-βγ scavenger βARK-ct (Koch et al., 1994) on the cAMP responses mediated by RXFP1 and RXFP2 (Fig. 5; Table 1). Cells were transiently transfected with βARK-ct, which sequesters G-βγ subunits within cells. The βARK-ct construct expresses a 28-amino acid peptide derived from the βARK-1 protein, which, after activation by G-βγ subunits, acts to phosphorylate β-adrenoceptors. βARK-ct contains the G-βγ binding sequence within βARK1 and would be expected to bind and sequester available G-βγ subunits within the cell, inhibiting any resulting signaling pathways. In HEK-RXFP1 cells, as was observed for PI3K and PKCζ inhibition, βARK-ct transfection significantly decreased the maximum response after a 30-min stimulation by 68% (Fig. 5B) but had no effect on the immediate cAMP response (Fig. 5A), suggesting that G-βγ subunits mediate the delayed PI3K-PKCζ pathway.
Again, in direct contrast to RXFP1, βARK-ct-transfected HEK-RXFP2 cells (Fig. 4) resulted in significantly increased cAMP responses after 30-min stimulation, with the maximum increasing by 76% in response to INSL3 (Fig. 5D). It is noteworthy that this potentiation of the cAMP response was not observed after 3-min exposure to INSL3 (Fig. 5C). The same effect was observed after H2 relaxin stimulation of RXFP2 (data not shown). This indicates that G-βγ subunits used by RXFP2 inhibit cAMP accumulation in a delayed manner. There was no effect of transient expression of βARK-ct on basal or forskolin-stimulated cAMP levels in either stable cells or the parent HEK293T cell line (data not shown), suggesting that all the observed effects are receptor-mediated. Transient transfection of βARK-ct did not affect pEC50 values.

Sequestration of G-βγ by βARK-ct affects the cAMP response through both RXFP1 and RXFP2. Concentration-response curves were performed at either 3 min (A and C) or 30 min (B and D) using H2 relaxin (0.1 pM–1 μM) at RXFP1 (A and B) and INSL3 (0.1 pM–1 μM) at RXFP2 (C and D) in the presence and absence of the transiently expressed G-βγ scavenger βARK-ct (114 ng/cm2 culture area). cAMP accumulation is expressed as a percentage of the 0.1 mM forskolin response at each time point. Symbols represent means, and vertical bars represent S.E.M. of four to eight separate experiments performed in duplicate. *, p < 0.05 and ***, p < 0.001 versus peptide alone (all maximum responses, unpaired t tests).
The interaction between the various inhibitors, PTX, and βARK-ct was then examined at both RXFP1 and RXFP2 to determine whether the observed effects upon cAMP were due to single or multiple pathways (Fig. 6; Table 1). Concentration-response curves were generated for each condition, with results expressed as the maximum response obtained in each case, because there was no affect of any of the compounds on pEC50 values. After 3-min stimulation with H2 relaxin in HEK-RXFP1 cells, pretreatment with PTX significantly increased the maximum cAMP response by 46% (Fig. 6A). Transfection with βARK-ct had no effect, but transfection with βARK-ct and pretreatment with PTX significantly increased the maximum cAMP response by 39%. This suggests that the initial RXFP1 response involves a Gs-mediated cAMP accumulation and an inhibition by PTX-sensitive G proteins. There is no involvement of G-βγ subunits in the initial cAMP response. At 30-min in HEK-RXFP1 cells (Fig. 6B), which were pretreated with PTX, the H2 relaxin maximum response was significantly inhibited by 37% compared with the response to H2 relaxin alone. As stated previously, transfection with βARK-ct also significantly decreased the maximum cAMP response, but by 68%. However, transfection of these cells with βARK-ct in addition to PTX pretreatment caused a significant decrease in the maximum cAMP response by only 48%, equivalent to the inhibition caused by pretreatment with PTX alone. Transfection with βARK-ct alone thus caused the greatest degree of inhibition. Because there is no additive inhibitory effect of combined treatment with PTX and βARK-ct transfection, these compounds must inhibit components of the same signaling pathway, thus G-βγ subunits must be derived from PTX-sensitive G proteins. However, PTX pretreatment increased the response compared with the effect of βARK-ct alone, suggesting that PTX-sensitive G proteins still mediate the inhibitory cAMP pathway at 30 min.
The same pattern of behavior was observed for cells pretreated with PTX and wortmannin (Fig. 6E) or for cells pretreated with PTX and chelerythrine chloride (Fig. 6F). Both wortmannin and chelerythrine chloride alone gave the greatest inhibition of the maximum response, and this was unaltered in cells treated with these inhibitors and also transfected with βARK-ct. However, pretreatment with PTX in addition to wortmannin or chelerythrine chloride exposure increased the maximum response back to the level of response observed after pretreatment with PTX alone. Thus, PTX reverses the effect (or decreases the degree of inhibition) of βARK-ct, wortmannin, or chelerythrine chloride, because it not only removes the stimulatory G-βγ-PI3K-PKCζ pathway but also the additional inhibition mediated by PTX-sensitive G proteins.

Effect of coincubation with inhibitors reveals both inhibitory and excitatory cAMP pathways through RXFP1 and RXFP2. Concentration-response curves were performed after 3-min (A and C) or 30-min (B, D, E, and F) stimulation with H2 relaxin (0.1 pM–1 μM) at RXFP1 (A, B, E, and F) and INSL3 (0.1 pM–1 μM) at RXFP2 (C and D), in the presence and absence of the Gi/Go inhibitor PTX (100 ng/ml; 16-h preincubation), transient transfection of the G-βγ scavenger βARK-ct (114 ng/cm2 culture area), the PI3K inhibitor wortmannin (0.1 μM; 30-min preincubation), the general PKC inhibitor chelerythrine chloride (10 μM; 30-min preincubation), or combinations of these inhibitors. Maximum responses were determined for each separate experiment (performed in duplicate), and then the mean and S.E.M. were determined for each maximum response. cAMP levels are expressed as a percentage of the 0.1 mM forskolin response at each time point. Bars represent means, and vertical bars represent S.E.M. of the maximum response of four to eight separate experiments performed in duplicate. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus the response to peptide alone; ^, p < 0.05 versus the response to peptide preincubated with PTX (all maximum responses, unpaired t tests).
In HEK-RXFP2 cells, pretreatment with PTX caused a 43% increase in the maximum response after 3-min stimulation with INSL3 (Fig. 6C). Transfection with βARK-ct had no effect on the maximum response; however transfection with βARK-ct in combination with pretreatment with PTX caused an increase in the maximum response by 54%, equivalent to the increase observed with PTX pretreatment alone. Thus, in a manner similar to the initial RXFP1 response, the initial RXFP2 response involves a Gs-mediated cAMP accumulation and an inhibition of cAMP by PTX-sensitive G proteins. Pretreatment with PTX also caused an increase in the maximum response to INSL3 stimulation after 30 min at RXFP2, this time by 48% (Fig. 6D). It is noteworthy that transfection of βARK-ct caused a greater increase in the cAMP response by 76% at this time point. However, there was no difference in the effect of βARK-ct transfection alone compared with the effect of βARK-ct transfection in combination with PTX pretreatment (74%). The same effect was observed after H2 relaxin stimulation of RXFP2 (data not shown). This indicates that G-βγ subunits also inhibit cAMP accumulation at 30 min and that they are derived from PTX-sensitive G proteins, because there was no additive effect of pretreatment with PTX in combination with transient transfection of βARK-ct.

RXFP1 and RXFP2 both couple to a GαoB inhibitory pathway initially, but RXFP1 then recruits Gαi3 to mediate the delayed G-βγ-PI3K-PKCζ stimulatory pathway. All PTX-insensitive G protein isoforms [Gαi1(C351I), Gαi2(C351I), Gαi3(C351I), GαoA(C351I), or GαoB(C351I); all 114 ng/cm2 culture area] were expressed after transient expression in both HEK-RXFP1 and HEK-RXFP2 cells as determined by Western blot (A). Bands were observed at the correct molecular mass of approximately 40 kDa. mGα denotes PTX-insensitive G protein isoform with Cys 351 Ile mutation. Concentration-response curves were performed at 3 min (B and D) or 30 min (C and E) using H2 relaxin (0.1 pM–1 μM) at RXFP1 (B and C) or INSL3 (0.1 pM–1 μM) at RXFP2 (D and E) in the presence and absence of the Gi/Go inhibitor PTX (100 ng/ml; 16-h preincubation) and one of five transiently transfected PTX-insensitive Gi/Go α-isoforms: Gαi1(C351I), Gαi2(C351I), Gαi3(C351I), GαoA(C351I), or GαoB(C351I) (all 114 ng/cm2 culture area). mGα denotes PTX-insensitive G protein isoform with Cys 351 Ile mutation. Maximum responses were determined for each separate experiment (performed in duplicate), and then the mean and S.E.M. were determined for each maximum response. cAMP is expressed as a percentage of the 0.1 mM forskolin response at each time point. Bars represent means, and vertical bars represent S.E.M. of the maximum response of four to eight separate experiments performed in duplicate. **, p < 0.01 and ***, p < 0.001 versus the response to peptide alone; *, p < 0.01 and **, p < 0.001 versus the response to peptide preincubated with PTX (all maximum responses, unpaired t tests).
The Different cAMP Pathways Activated by RXFP1 and RXFP2 Are Mediated by Different Isoforms of Gi/Go. The delayed cAMP signaling pathways activated upon stimulation of RXFP1 and RXFP2 are clearly different and seem to deviate at the level of PTX-sensitive, or Gi/Go protein coupling: at RXFP1, PTX pretreatment and βARK-ct transfection act to decrease cAMP levels (i.e., Gi/Go-derived G-βγ subunits are involved in a stimulatory cAMP pathway), whereas at RXFP2 PTX pretreatment and βARK-ct transfection act to increase cAMP levels (i.e., Gi/Go-derived G-βγ subunits are involved in an inhibitory cAMP pathway). To address which G proteins were driving the cAMP response, transient transfection of PTX-insensitive Gi/Go isoforms was used (Fig. 7; Table 3). The effective interaction of all G protein α subunits with receptors initially requires the exchange of GDP for GTP. In Gi/Go, this exchange can be prevented by PTX, rendering the G proteins inactive. Gi/Go isoforms are sensitive to ADP-ribosylation by PTX because of a conserved cysteine residue positioned four amino acids from the C terminus. Mutation of this residue therefore causes the isoform to become insensitive to PTX. Transient transfection of one of the Gi/Goα (C351I) isoforms (Bahia et al., 1998) and subsequent treatment of cells with PTX (to inactivate endogenous Gi/Go isoforms) leaves only the transfected PTX-insensitive Gi/Go protein active. Thus, the identity of the Gi/Go isoform involved in the cAMP response can be revealed if the isoform transfected restores signaling in the presence of PTX.
Effect of transiently expressed PTX-insensitive Gi/Go isoforms on the cAMP accumulation response
Effects of transient transfection of PTX-insensitive Gi/Go isoforms on the maximum cAMP accumulation response through RXFP1 (3- and 30-min incubations with H2 relaxin) and RXFP2 (3- and 30-min incubations with INSL3) are shown below. Responses are the maximum response determined from each separate experiment and expressed as mean ± S.E.M. of (n) experiments.
There are three isoforms of Gαi (Gαi1, Gαi2, and Gαi3) and two forms of Gαo (GαoA and GαoB). Therefore, to determine the isoforms of Gi/Go proteins mediating the cAMP response at each receptor, stable cell lines were transiently transfected with PTX-insensitive Gi/Go isoforms: Gαi1(C351I), Gαi2(C351I), Gαi3(C351I), GαoA(C351I), and GαoB(C351I) and treated with PTX (Fig. 7; Table 3). All PTX-insensitive G protein isoforms were expressed after transient transfection in both HEK-RXFP1 and HEK-RXFP2 cells as determined by Western blotting (Fig. 7A). Faint bands were observed for both HEK-RXFP1, HEK-RXFP2, and the same cells pretreated with PTX. Much stronger bands were observed in cells transiently transfected with the PTX-insensitive G protein isoforms. In HEK-RXFP1 cells stimulated with H2 relaxin for 3 min (Fig. 7B), only GαoB(C351I)-transfected cells restored the cAMP response in the presence of PTX to the level of cAMP stimulated by H2 relaxin alone. The maximum responses of H2 relaxin and GαoB(C351I) were significantly decreased compared with PTX-treated cells (36% decrease), whereas transfection of any of the other isoforms [Gαi1(C351I), Gαi2(C351I), Gαi3(C351I), and GαoA(C351I)] gave responses that were similar to PTX pretreatment and significantly increased (by a mean of 39, 31, 51, and 35%, respectively) compared with the effect of either H2 relaxin alone or GαoB(C351I). Thus, initially RXFP1 can couple to Gs, and GαoB, which causes inhibition of cAMP. With 30 min of H2 relaxin stimulation (Fig. 7C), PTX pretreatment with or without Gαi1(C351I), Gαi2(C351I), GαoA(C351I), or GαoB(C351I) transfection gave significantly decreased responses (45, 35, 36, 45, and 52%, respectively) compared with the effect of H2 relaxin alone. Transfection with Gαi3(C351I) gave a similar response to H2 relaxin alone, and the response was significantly increased compared with the effect of either PTX pretreatment or transfection with Gαi1(C351I), Gαi2(C351I), GαoA(C351I), or GαoB(C351I) (by a mean increase of 48%). Thus, at 30-min only Gαi3(C351I) restored the cAMP response in the presence of PTX to levels observed in response to H2 relaxin alone. Therefore, RXFP1 couples to Gαi3 with time, which mediates the G-βγ-PI3K-PKCζ pathway.
The effect of transfection of the five PTX-insensitive Gi/Go isoforms was then assessed in HEK-RXFP2 cells. After INSL3 stimulation for both 3 min (Fig. 7D) and 30 min (Fig. 7E), there was a significant increase in the maximum response to pretreatment with PTX, or transfection with Gαi1(C351I), Gαi2(C351I), Gαi3(C351I), and GαoA(C351I) compared with the effect of peptide alone (29, 33, 23, 26, and 26%, respectively). The response after transfection of GαoB(C351I) was not different from the effect of INSL3 alone, but it was significantly decreased compared with the response to either PTX pretreatment or transfection with Gαi1(C351I), Gαi2(C351I), Gαi3(C351I), or GαoA(C351I) (by an average of 32%). Thus, only GαoB(C351I) transfection in the presence of PTX pretreatment could restore the response to levels observed after INSL3 stimulation alone. This effect did not change with time and was also observed after H2 relaxin stimulation of RXFP2 (data not shown), suggesting that RXFP2 can couple only to Gs and inhibitory GαoB.
There was no effect of transient transfection of any of the PTX-insensitive G proteins [Gαi1(C351I), Gαi2(C351I), Gαi3(C351I), GαoA(C351I), or GαoB(C351I)] on basal or forskolin-stimulated cAMP levels in either HEK-RXFP1 or HEK-RXFP2 cells, or in the parent HEK293T cell line (data not shown). None of the PTX-insensitive G protein isoforms had any effect on the pEC50 values when transiently expressed in HEK-RXFP1 or HEK-RXFP2 cells.
Discussion
The identification of the relaxin receptor (RXFP1) as a GPCR (Hsu et al., 2002) followed by expression in mammalian cells revealed a Gs-coupled receptor that when activated increased intracellular levels of cAMP (Hsu et al., 2002). Although this result is unequivocal, there is a lack of consensus regarding the importance of cAMP as a physiological signaling mechanism in target tissues (Bathgate et al., 2006). In terms of RXFP2-INSL3 signaling, most studies focus on cAMP, but in different cell types both stimulation and inhibition have been reported (Hsu et al., 2002; Kumagai et al., 2002; Kawamura et al., 2004). The present study examines cAMP signaling by these receptors in detail, with a specific focus on the G proteins involved.
Recent reports indicate that relaxin activates RXFP1 to cause a biphasic accumulation of cAMP with time (Nguyen et al., 2003; Halls et al., 2005a; Nguyen and Dessauer, 2005). Initial studies confirmed this finding in HEK-RXFP1 cells. cAMP accumulation was biphasic, and the PI3K inhibitors LY294002 and wortmannin significantly decreased cAMP at the longer time points. These studies enabled the selection of time points at which to construct concentration-response curves: 3 min to capture the immediate phase and 30 min to also capture the delayed phase. Concentration-response studies at these time points confirmed that the delayed pathway has characteristics similar to those identified in THP-1 cells (Nguyen et al., 2003; Nguyen and Dessauer, 2005), in that the cAMP response is blocked by inhibitors of PI3K and PKCζ. This suggests that PI3K and PKCζ are involved only in the delayed phase of cAMP accumulation.
PI3K is often activated by G-βγ after receptor stimulation (Stephens et al., 1994), suggesting that these subunits could be involved in the activation of the delayed pathway in this system. Cotransfection with the G-βγ scavenger βARK-ct inhibited the RXFP1 response only at 30 min, to the same degree observed with the PI3K and PKCζ inhibitors. Thus, the delayed pathway is mediated by G-βγ subunits. Time-course studies also suggested that G-βγ subunits were derived from Gi/Go proteins, because pretreatment of cells with PTX also blocked the delayed response.
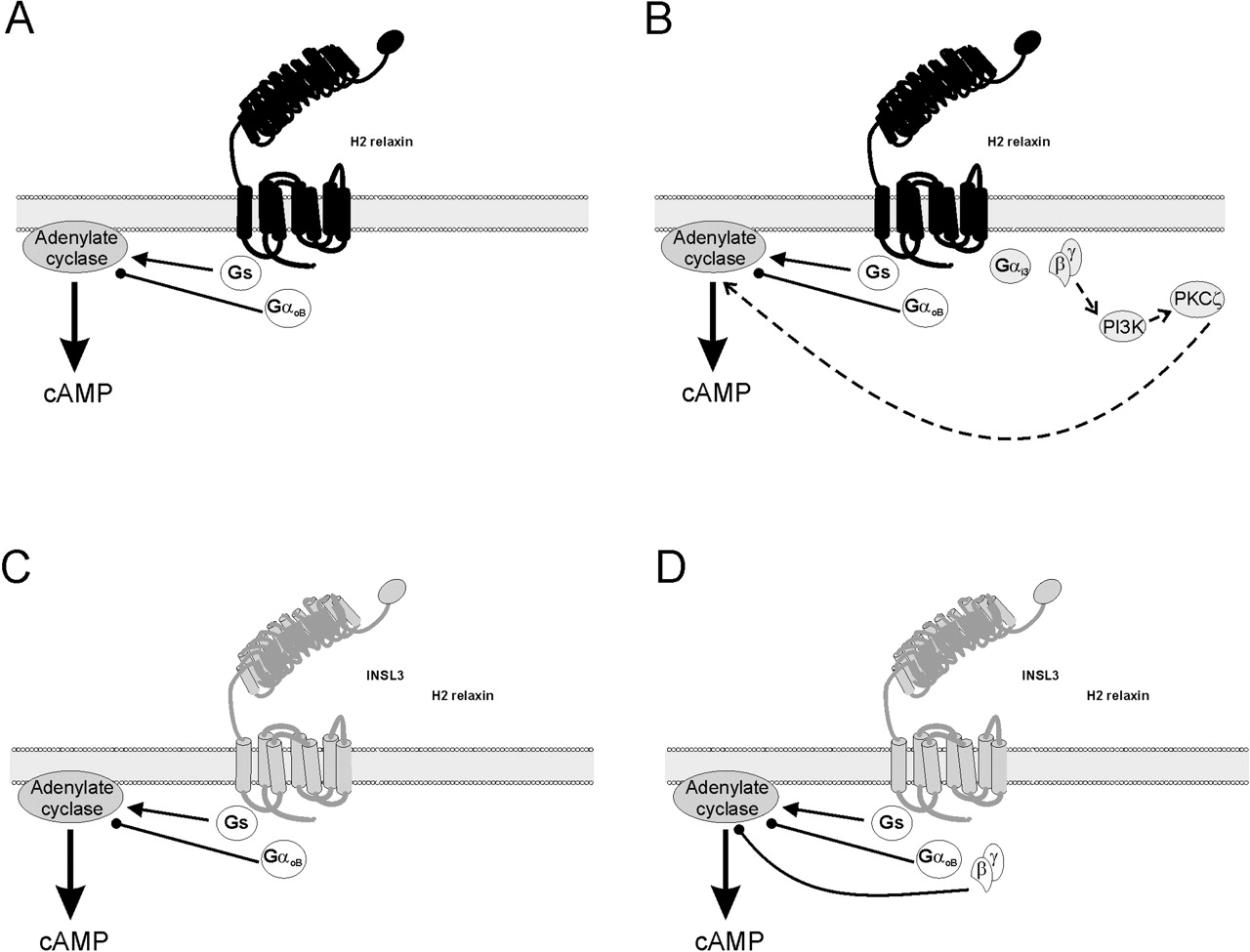
It is noteworthy that, at 3 min, PTX significantly increased the cAMP response compared with H2 relaxin treatment alone. This indicated that the receptor also couples to an inhibitory Gi/Go pathway. This pathway also contributes to the overall cAMP response at 30 min, suggesting the existence of three pathways: a Gs-mediated stimulation of cAMP (H2 relaxin in the presence of PTX), a Gi/Go-mediated inhibition (shown by the significant difference in response between PTX-treated and βARK-ct-treated cells), and a Gi/Go-derived G-βγ-mediated delayed cAMP increase via PI3K and PKCζ (as demonstrated by effect of βARK-ct, wortmannin, or chelerythrine chloride alone). Use of PTX-resistant Gi/Go mutants revealed differential Gi/Go coupling with time, providing further evidence for the activation of three pathways. The only isoform to restore signaling at 3 min to the characteristics of the wild-type was GαoB(C351I). This suggested that RXFP1 couples to GαoB initially to inhibit adenylate cyclase and decrease cAMP. However, at 30 min the only isoform that restored signaling in the presence of PTX was Gαi3(C351I). Thus, RXFP1 seems to initially couple to Gαs and GαoB (Fig. 8A) but then recruits Gαi3 to release G-βγ subunits that activate the delayed PI3K-PKCζ pathway (Fig. 8B).

cAMP signaling pathways activated by stimulation of RXFP1 and RXFP2. Binding of H2 relaxin to RXFP1 initially results in coupling to Gs to activate adenylate cyclase and increase cAMP and also to GαoB to inhibit adenylate cyclase and decrease cAMP accumulation (A). After a period of approximately 5 to 20 min, RXFP1 can recruit Gαi3 (B). The release of G-βγ from Gαi3 allows the activation of PI3K and activation and translocation of PKCζ to further stimulate adenylate cyclase and cAMP production. Activation of RXFP2 by either INSL3 or H2 relaxin involves only the stimulation of adenylate cyclase to increase cAMP via Gs, and the inhibition of cAMP production through adenylate cyclase by GαoB (C) and derived G-βγ subunits with time (D). Lines with arrows represent cAMP stimulatory pathways, those with circles, inhibitory pathways, and dotted lines, delayed pathways. Components of the immediate response at each receptor are unshaded, whereas components of the delayed response are shaded.
The involvement of Gi/Go proteins in the RXFP1 response to relaxin has been reported previously (Kompa et al., 2002). In functional assays, PTX pretreatment significantly decreased the maximum inotropic and chronotropic responses to relaxin in rat isolated atria, without alteration of potency. PTX pretreatment also significantly decreased cAMP in the same tissue. Thus, it seems that the model of cAMP accumulation presented here also exists at a physiological level. We suggest therefore that differences in relaxin-mediated signaling observed across a variety of cell types can be explained by the differential expression patterns of Gs, GαoB, and Gαi3, and thus by differential emphasis placed upon the three signaling pathways.
The mechanism of the recruitment of Gαi3 by RXFP1 is as yet unclear. The delay may potentially reflect the time required for translocation of PKCζ to the cell membrane. In rat-1 fibroblasts expressing α1A adrenoceptors, stimulation with phenylephrine caused significantly increased levels of PKCζ in particulate fractions after 15 min (Parmentier et al., 2004). Relaxin-mediated PKC translocation has been shown to occur as early as 2 min in PHMI-31 and THP-1 cells (Nguyen and Dessauer, 2005), and by 10 min in cells isolated from secretory endometrium (Kalbag et al., 1991). PKCζ translocation and activation of adenylate cyclase may also explain why responses to relaxin are persistent and continue for longer than 6 h with constant washing in physiological systems (Summers et al., 1995). In mouse pubic symphysis in vivo injection of relaxin for either 15, 30, or 60 min all resulted in significantly increased cAMP levels, and cAMP was still elevated at 120 min (Braddon, 1978). In studies with HEK-RXFP1 cells in the cytosensor microphysiometer (which measures overall cellular activity via extracellular changes in pH), the effects of H2 relaxin are not removed by washing for up to 1 h, and there is no further increase in response after restimulation (B. Nithianantharajah, unpublished observations). This suggests that the recruitment of Gαi3 and translocation of PKCζ may occur under physiological conditions.
It is noteworthy that our concentration-response studies have also revealed bell-shaped concentration-response relationships at both time points for RXFP1 and at 30 min for RXFP2. The decline in the response to H2 relaxin or INSL3 at higher concentrations does not seem to involve either desensitization or internalization (R. A. D. Bathgate et al., unpublished observations). However, it may reflect negative cooperativity as demonstrated for other glycoprotein hormone receptors (Urizar et al., 2005), whereby the binding of one ligand molecule decreases the affinity of another binding site, thus decreasing binding and therefore receptor activation with increasing concentrations of ligand.
In HEK-RXFP2 cells, time-course studies showed a different pattern of cAMP accumulation compared with RXFP1. Although there is 60% amino acid sequence homology between RXFP1 and RXFP2 (Hsu et al., 2002), stimulation of RXFP2 with INSL3 or H2 relaxin revealed no effect of the PI3K inhibitors wortmannin or LY294002, and this was confirmed in the concentration-response studies, suggesting that RXFP2 does not use the delayed signaling pathway identified for RXFP1. It is noteworthy that, and in contrast to the RXFP1 receptor, both PTX and βARK-ct increased the RXFP2 cAMP response. Pretreatment of HEK-RXFP2 cells with PTX increased basal levels of cAMP, but there was no effect of PTX on basal cAMP accumulation in HEK-RXFP1 or parent HEK293T cells, suggesting some coupling of the receptor to inhibitory Gi/Go proteins even at rest. The βARK-ct construct only effectively increased the cAMP response at 30 min, perhaps indicative of the time required for G-βγ subunits to effectively inhibit cAMP production. There is evidence for G-βγ inhibition of adenylate cyclase, specifically types I (Tang and Gilman, 1991), V and VI (Bayewitch et al., 1998b), and VIII (Steiner et al., 2006). Thus, this is likely the mechanism responsible for the observed cAMP inhibition by G-βγ subunits. When PTX pretreatment and βARK-ct transfection were assessed in combination at 30 min after INSL3 or H2 relaxin stimulation, there was no additive effect. This suggests that the inhibitory G-βγ subunits are derived from PTX-sensitive Gi/Go proteins.
The strong inhibitory component in the RXFP2 response is reflected by the lower levels of cAMP generated compared with RXFP1, despite equivalent levels of expression of both receptors in the stable cells (Sudo et al., 2003). Thus, RXFP2 seems to stimulate adenylate cyclase via Gs and to inhibit adenylate cyclase activity by both Gi/Go and G-βγ (Fig. 8B). This is in accord with previous reports of RXFP2 activating both a Gs stimulatory pathway (Hsu et al., 2002; Kumagai et al., 2002) and a Gi/Go inhibitory pathway (Kawamura et al., 2004), and it is similar to the initial RXFP1 response. This was confirmed by the effects of the PTX-insensitive Gi/Go isoforms transfected in HEK-RXFP2 cells. The only PTX-insensitive isoform to restore signaling in the presence of PTX, at both time points, was GαoB(C351I). Thus, it follows that the major difference between the cAMP signaling pathways used by these two receptors is the ability of RXFP1 to recruit Gαi3 over time. It would therefore be interesting to determine the exact domains of the receptors responsible for their G protein specificity, whether it is an inherent property of RXFP1 to recruit Gαi3 coupling, or whether RXFP2 inhibits its own coupling to the Gαi3 isoform.
Thus, RXFP1 and RXFP2 not only couple to different PTX-sensitive G proteins but also exhibit variation in their association with G-βγ subunits, because G-βγ from GαoB inhibits cAMP in HEK-RXFP2 cells, whereas G-βγ from Gαi3 activates PI3K-PKCζ to increase cAMP in HEK-RXFP1 cells. So far, there is no established basis for specificity between G-βγ and effectors, G-βγ and Gα subunits, or G-βγ and receptor, although there is emerging evidence to support this. In COS-7 cells, transfection of G-β1γ2 but not G-β5γ2 activated MAPK (Zhang et al., 1996). In addition, G-β1γ2-stimulated adenylate cyclase type II but inhibited adenylate cyclase type I, whereas G-β5γ2 inhibited both subtypes (Bayewitch et al., 1998a). Furthermore, there is some evidence for specificity of G-βγ for both Gα subunits and receptors: in GH3 cells, GαoA-β2γ2 couples preferentially to galanin receptors (Kalkbrenner et al., 1995), whereas GαoA-β3γ4 and GαoB-β1γ3 couple preferentially to muscarinic and somatostatin receptors, respectively (Kleuss et al., 1993).
RXFP1 and RXFP2 receptors clearly display different agonist-stimulated cAMP responses. Both receptors, upon activation, are initially able to couple to a classic Gs-adenylate cyclase pathway to increase cAMP production, but they also couple to an inhibitory GαoB-mediated pathway to modulate accumulation of cAMP. Differences between the receptors stem from the ability of RXFP1 to recruit Gαi3. Thus, only RXFP1 displays the biphasic pattern of cAMP accumulation, the second phase of which is mediated by the release of Gαi3 G-βγ subunits, which then activate the PI3K-PKCζ-adenylate cyclase pathway to further increase accumulation of cAMP.
Acknowledgments
We thank Dr. John Wade for the synthesis and supply of relaxin family peptides. We also thank Sharon Layfield for generation of the stable cell lines.
Footnotes
-
R.A.D.B. is an R. D. Wright Fellow of the National Health and Medical Research Council. This work was supported in part by National Health and Medical Research Council Block Grant Reg Key 983001 to the Howard Florey Institute, Project Grant 300012, and ARC Linkage Grant LP0560620. M.L.H. is an Australian Postgraduate scholar and recipient of a Monash University Faculty of Medicine, Nursing and Health Sciences Excellence Award.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.021691.
-
ABBREVIATIONS: H, relaxin human gene; INSL, insulin/relaxin-like peptide; GPCR G protein-coupled receptor; RXFP, relaxin family peptide receptor; MAPK, mitogen-activated protein kinase; HEK, human embryonic kidney; PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; PTX, pertussis toxin; LY294002, 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride; HRP, horseradish peroxidase; βARK-ct, β-adrenergic receptor kinase I-C terminus; FBS, fetal bovine serum.
- Received December 14, 2005.
- Accepted March 28, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}