


Abstract
Small molecules provide powerful tools to interrogate biological pathways but many important pathway participants remain refractory to inhibitors. For example, Cdc25 dual-specificity phosphatases regulate mammalian cell cycle progression and are implicated in oncogenesis, but potent and selective inhibitors are lacking for this enzyme class. Thus, we evaluated 10,070 compounds in a publicly available chemical repository of the National Cancer Institute for in vitro inhibitory activity against oncogenic, full-length, recombinant human Cdc25B. Twenty-one compounds had mean inhibitory concentrations of <1 μM; >75% were quinones and >40% were of thepara-naphthoquinone structural type. Most notable was NSC 95397 (2,3-bis-[2-hydroxyethylsulfanyl]-[1,4]naphthoquinone), which displayed mixed inhibition kinetics with in vitroK i values for Cdc25A, -B, and -C of 32, 96, and 40 nM, respectively. NSC 95397 was more potent than any inhibitor of dual specificity phosphatases described previously and 125- to 180-fold more selective for Cdc25A than VH1-related dual-specificity phosphatase or protein tyrosine phosphatase 1b, respectively. Modification of the bis-thioethanol moiety markedly decreased enzyme inhibitory activity, indicating its importance for bioactivity. NSC 95397 showed significant growth inhibition against human and murine carcinoma cells and blocked G2/M phase transition. A potential Cdc25 site of interaction was postulated based on molecular modeling with these quinones. We propose that inhibitors based on this chemical structure could serve as useful tools to probe the biological function of Cdc25.
Mammalian cell communication and growth is regulated by protein phosphorylation, which is the product of a dynamic balance between the enzymatic activity of protein kinases and phosphatases. Small molecule inhibitors have provided valuable tools to decode the role of kinases and phosphatases participating in specific cellular signaling pathways, because they are generally reversible, nonquantal and cell permeative. Natural product inhibitors of serine/threonine protein phosphatases, such as okadaic acid and calyculin A, have been extremely valuable reagents to probe serine/threonine phosphatase function (Wera and Hemmings, 1995). In contrast, small anions, such as vanadate, have been the most commonly used inhibitor of the other major mammalian phosphatase class, the protein tyrosine phosphatases (PTPase), because of the dearth of readily available potent and selective inhibitors (Pestell et al., 2000). The PTPases are defined by the active site signature sequence motif HCX5R, where H is a highly conserved histidine residue, C is the catalytic cysteine, the five X residues form a loop in which all of the amide nitrogens hydrogen-bond to the phosphate of the substrate, and R is a highly conserved arginine that hydrogen-bonds to the phosphorylated amino acid of the substrate (Denu et al., 1996). The dual specificity protein phosphatase subfamily retains some of the structural attributes of PTPases but is unique in its ability to hydrolyze both phosphoserine/threonine as well as phosphotyrosine residues on the same protein substrate. Important members of the dual specificity phosphatase family are the Cdc25 phosphatases, which control cell cycle progression by activating cyclin-dependent kinases (Cdk) (Nilsson and Hoffman, 2000). Three Cdc25 homologs exist in humans: Cdc25A, Cdc25B, and Cdc25C (Sadhu et al., 1990; Millar et al., 1991; Nagata et al., 1991). Two splice variants of Cdc25A have been reported, whereas Cdc25B and C have at least seven and five each, respectively (Forrest et al., 1999; Wegener et al., 2000). The functional significance of these variants is currently unknown. Cdc25A and B have oncogenic properties (Galaktionov et al., 1995), are transcriptional targets of the c-myc oncogene (Galaktionov et al., 1996), and are overexpressed in many human tumors (Galaktionov et al., 1995; Cangi et al., 2000; Hernandez et al., 2001). Both Cdc25B and Cdc25C are thought to be regulators of G2/M transition through their ability to dephosphorylate and activate the Cdk1/cyclin B mitotic kinase complex, which is required for cell entry into mitosis (Nilsson and Hoffman, 2000). Cdc25A is likely to be important for G1/S phase transition and in preserving genomic integrity (Jinno et al., 1994), although Cdc25A may also have some role in the initiation of mitosis (Molinari et al., 2000). Cdc25A is rapidly degraded in response to DNA damage, which impairs the G1/S transition (Mailand et al., 2000).
Although two crystal structures have been published for the Cdc25A and B catalytic domains (Fauman et al., 1998; Reynolds et al., 1999), neither revealed the nature of interactions with small molecule inhibitors. Moreover, the protein substrate may initiate key conformational changes and provide an important catalytic acid (Stewart et al., 1999; Chen et al., 2000). Thus, rational design parameters for potential inhibitors are lacking. The current work was initiated based on the belief that selective Cdc25 inhibitors could be obtained by using a general and unbiased approach to evaluate a chemically diverse compound library. Thus, we probed a small subset of the National Cancer Institute's (NCI) 140,000 compound library for potential inhibitory lead structures and used this information to identify a pharmacophore yielding the most potent and selective inhibitor of Cdc25 reported to date. Molecular modeling studies using selected compounds suggest a binding motif consistent with our kinetic results.
Materials and Methods
Library Chemicals.
The NCI compounds evaluated were a generous gift from Jill Johnson (NCI, National Institutes of Health, Bethesda, MD) and include a 1,990 compound Diversity Set and 8,080 compounds from their 140,000 compound library. The NCI's NSC nomenclature was used throughout out this study for compound identification. The criteria for selecting the Diversity Set can be found at http://dtp.nci.nih.gov/and the compounds from both sets were pooled to enhance the power of the analysis.
Chemical Synthesis and Compound Analysis.
Both JUN 379 and JUN 390 were prepared by methods described previously (Draber, 1967;Nohara et al., 1974). JUN 255 and the corresponding isoquinone analog, JUN 260, were synthesized by treating a solution of 2-(tetrahydropyran-2-yloxy)ethanethiol (Li et al., 1999) and either 6,7-dichloroquinoline-5,8-dione or the corresponding isoquinoline (Ryu et al., 1999) in tetrahydrofuran at room temperature with triethylamine. The reaction mixture was stirred for 20 h, concentrated under reduced pressure, diluted with ethyl acetate, and washed with water. The organic layer was dried with Na2SO4 and concentrated under reduced pressure. The crude residue was purified by column chromatography on SiO2 (hexanes/ethyl acetate, 2:1). JUN 266, 276, 307, and 309 were synthesized using the appropriate corresponding diones and thiols in a similar manner. Chemical identity was confirmed for all compounds by melting point, infrared spectroscopy, 1H NMR, 13C NMR, and high-resolution mass spectrum analysis.
In Vitro Enzyme Assays.
The activities of the GST-fusion Cdc25A1, Cdc25B2, Cdc25C1, and VHR, as well as human recombinant PTP1B, were measured using O-methyl fluorescein phosphate (Sigma, St. Louis, MO) as substrate and a miniaturized, 96-well microtiter plate assay based on methods described previously (Rice et al., 1997). The final incubation mixtures (25 μl) were prepared with a Biomek 2000 laboratory automation workstation (Beckman Coulter, Inc., Fullerton, CA). Fluorescence emission from the product was measured after a 60-min incubation period at ambient temperature with a multiwell plate reader (Cytofluor II, excitation filter, 485/20; emission filter, 530/30; Applied Biosystems, Foster City, CA). Unbiased assignments for the best curve fit with the Lineweaver-Burk plots and single substrate/inhibitor kinetic model and for theK i values were determined by using the curve-fitting programs Prism 3.0 (GraphPad Software, Inc., San Diego, CA) and Sigma Plot 2000, Enzyme Kinetics Module 1.0 (SPSS, Inc., Chicago, IL). The best fit model determined by the Enzyme Kinetics Module was partial mixed inhibition described by the equation v =V max {(1 + βI / αK i) / [1 + I / (αK i)]} / {1 + (K m / S)(1 + I/K i) / [1 + I / (αK i)]}, although the correlation coefficients for a full mixed inhibition model were nearly equivalent. For the partial mixed inhibition model, I was the inhibitor compound concentration, the parameter α was defined asK m factor change when the inhibitor was bound to the enzyme/substrate complex and the parameter β was the Kp factor change when the inhibitor was bound to the enzyme/substrate complex. Two independent kinetic studies were performed with each Cdc25 isoform and similar results were obtained.
Reversibility assays were conducted by incubating 140 μg of (histidine)6-tagged Cdc25A catalytic domain (amino acid residues 336–523) with 200 μl of Ni-NTA Superflow resin (QIAGEN, Valencia, CA) on ice for 30 min in phosphate-buffered saline buffer in the absence of substrate. After centrifugation at 1000g for 5 min, the supernatant fraction was removed and the beads resuspended in 250 μl of 2× phosphatase assay buffer (Rice et al., 1997). We then added 75 μl of the resuspended beads to 10 μl of NSC 95397 (final concentration, 100 nM) or DMSO vehicle control and incubated at room temperature for 30 min. A sample was removed for a prewash Cdc25A enzyme activity determination as described previously (Rice et al., 1997; Tamura et al., 2000) and the remainder of the incubation mixture was centrifuged (1000g for 5 min), washed with phosphatase assay buffer, and assayed for Cdc25A activity as described previously (Tamura et al., 2000) after a further 60-min incubation at room temperature with shaking.
Antiproliferative Assays.
The proliferation of human MCF-7 breast and PC-3 prostate cells was measured by a colorimetric assay described previously (Vogt et al., 1998). Briefly, cells were treated with vehicle or compound continuously for 72 h and the medium was replaced with serum free medium containing 0.1% 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide. Plates were incubated for an additional 3 h in the dark and the total cell number was determined spectrophotometrically at 540 nm as described previously (Vogt et al., 1998). The proliferation of tsFT210 cells was measured 48 h after continuous compound exposure by microscopically counting Trypan blue negative cells with our method described previously (Tamura et al., 2000).
Flow Cytometric Analysis.
tsFT210 cells were plated at 2 × 105 cells/ml, maintained at 32.0°C and treated as described previously (Tamura et al., 2000). Briefly, cell proliferation was blocked at the G2 phase by incubation at 39.4°C for 17 h. To probe for G2/M arrest, we released synchronized cells by re-incubating at 32.0°C and immediately treated them for 6 h with various concentrations of NSC 95397, 1 μM nocodazole, or 0.5% DMSO vehicle. Cells were then harvested with phosphate-buffered saline and stained with a solution containing 50 μg/ml propidium iodide and 250 μg/ml RNase A. Flow cytometry analysis was conducted with an Epics XL flow cytometer (Beckman Coulter).
Molecular Modeling Studies.
The deposited Cdc25B catalytic domain crystal structure (Reynolds et al., 1999) (PDB identification code: 1QB0) served as the structural basis for forming the ligand/Cdc25B complexes in this study. First, the entire surface of the Cdc25B catalytic domain crystal structure was searched for possible binding site cavities. Site accessibility was probed for opening sizes of at least 4.0 Å or greater, using a grid resolution of 0.3 Å. Subsequently, the identified binding site cavities of Cdc25B were examined for optimal steric and chemical complementarity to thepara-quinones by order of their structure activity arrangement. This led to the determination that a pocket adjacent to the anion binding site located on the A chain of the protein (composed of residues T547, F543, Y428, L445, R479, M483, R482, E446, S549, W550, R544, P444, and L545) contained the best steric and chemical complementarity for the para-quinone ligands. Ligands were docked using a tethered minimization protocol, as described previously (Giannakakou et al., 2000; Gussio et al., 2000). Molecular mechanics potentials (CFF91 force field) (Maple et al., 1998) were used for all protein-ligand docking simulations. Further refinement of the Cdc25B/ligand was accomplished using various para-quinones complexes and molecular mechanics with a protocol of constraints and local geometry searches. Low-energy conformations of the ligands were collected from conformational searching algorithms using MSI Cerius2 and Catalyst programs (Accelrys, Inc., Princeton, NJ). Each conformer of each ligand was docked into the binding pocket, whereas the Cdc25B side chains were adjusted to the nearest geometry away from a ligand, until all van der Waals overlap greater than 0.25 Å between the amino acid residues of the binding site and a ligand was removed. This was followed by applying a tethered minimization protocol that consisted of the gradual removal of heavy atom constraints (5000 kcal mol−1Å−1 down to 0) from their initial positions over the course of an extended minimization procedure. Typically, the tethering force was reduced by a factor of 0.8 by minimization until the norm of the gradient was less than or equal to 1.0 kcal mol−1Å−1 on each cycle. These parameters resulted in ∼50 cycles of the relaxation procedure, at which point the tethering force approached 0 kcal mol−1Å−1. The resulting structure was checked against the original Cdc25B coordinates and subsequently rejected if the model deviated from the 1.91-Å quality of the original structure's coordinates. The best model of each ligand/Cdc25 complex was selected on the basis of chemical complementarily using the program HINT (Kellogg et al., 1991).
Results
Compound Library Analysis.
We evaluated 10,070 compounds obtained from the NCI's compound chemical repository for in vitro inhibitory activity against oncogenic, full-length, recombinant human Cdc25B, using a two-step approach. First, we performed a high throughput screen of the compound library at a single concentration of 10 μM and then retrospectively selected a criteria that would eliminate 99% of all compounds, namely >80% inhibition, which resulted in 114 candidate compounds. A secondary assay with 10 concentrations ranging from 5 nM to 10 μM was used to determine the in vitro IC50 values for Cdc25B. Only 21 compounds were found to have mean inhibitory concentrations of < 1 μM; >75% (16 of 21) were quinones and >40% (9 of 21) contained the para-naphthoquinone substructure (Fig.1, Table1). The four most potent compounds were NSC 95397, 139049, 135880, and 115447, which had IC50 values for Cdc25B of < 500 nM. Two of these compounds were para-quinones (NSC 95397 and NSC 115477). Surprisingly, NSC 95397 was a close congener of compound 5 (NSC 672121) (Fig. 2), which has been characterized extensively (Nishikawa et al., 1999; Tamura et al., 2000) but was not one of the compounds in this library subset. With an IC50 of 3.6 ± 0.6 μM for Cdc25B (Tamura et al., 2000), NSC 672121 is among the most potent inhibitors of the Cdc25 phosphatase family previously reported.
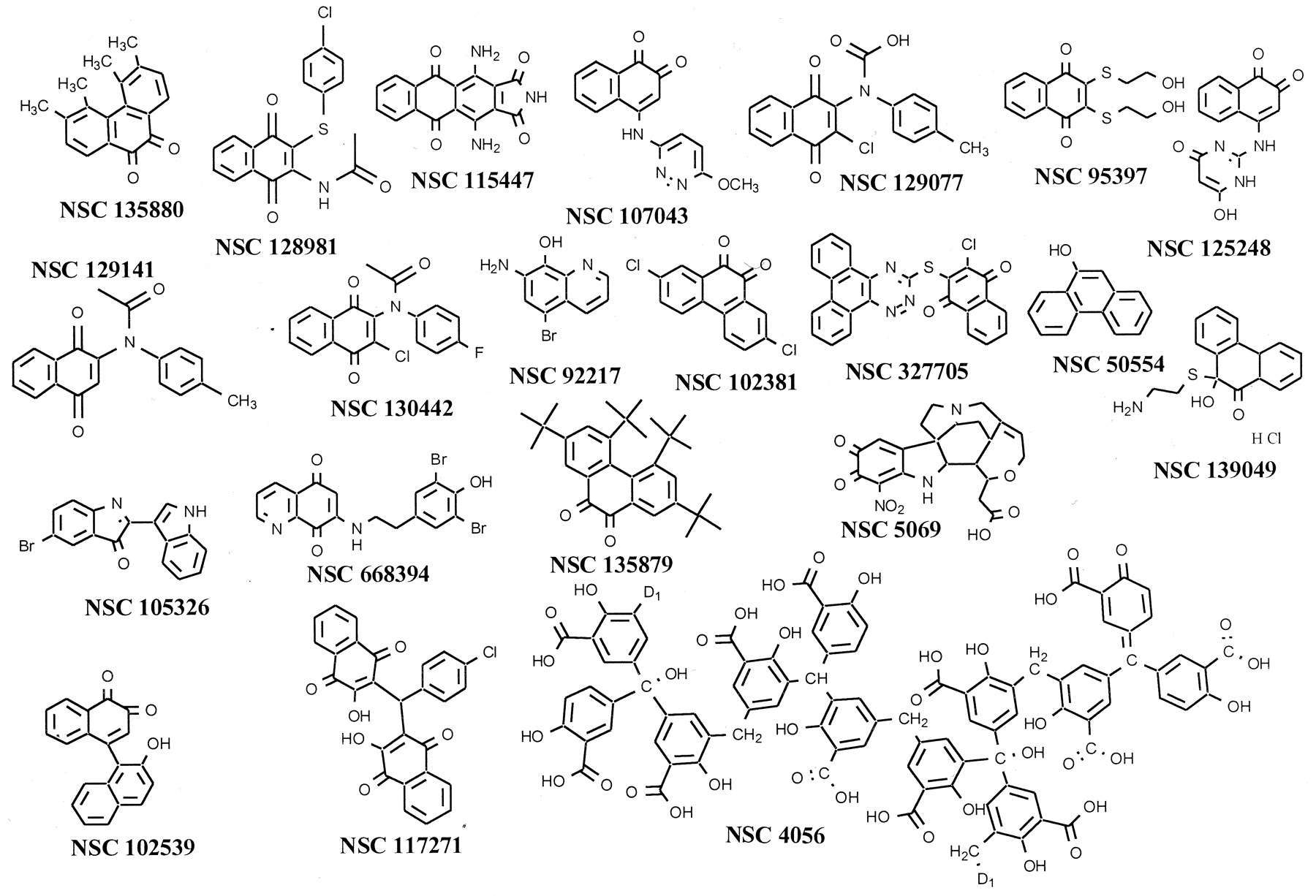
Chemical structures of NCI library compounds with IC50 values < 1 μM. NSC 4056, also known as aurintricarboxylic acid, is a polymer and the sites of polymer formation are indicated as D1.
In vitro inhibition of Cdc25B, VHR, and PTP1B by NCI compounds

Chemical structure of compound 5 or NSC 672121 with the chemical numbering for C2 and C3 indicated.
The importance of the para-quinone pharmacophore was emphasized by its overrepresentation among the 21 compounds with IC50 values < 1 μM compared with the inactive compound set of 10,049 compounds (p < 0.001) (Table 2). All four potent inhibitors showed >10-fold selectivity as inhibitors against Cdc25B compared with either the dual specificity phosphatase VHR or the protein tyrosine phosphatase PTP1B (Table 1). Indeed, all 21 Cdc25B inhibitors identified in the NCI compound library showed greater inhibitory activity toward Cdc25B compared with VHR or PTP1B. That thepara-quinone structure alone was insufficient to provide a potent Cdc25B inhibitor was revealed by the identification ofpara-quinones lacking marked Cdc25B inhibition at 10 μM (Fig. 3).
Frequency of the para-quinone substructures in inhibitors of Cdc25B

Chemical structures of para-quinone compounds found in the NCI library lacking marked inhibition against Cdc25B. The IC50 value (micromolar) or percent inhibition at 10 μM is indicated in the parentheses next to the NSC number, which is below the chemical structure.
We next examined the kinetics of inhibition against all three human Cdc25 isoforms. NSC 95397 had IC50 values against Cdc25A and Cdc25C of 22.3 ± 5.9 (SEM, n = 8) and 56.9 ± 17.7 (SEM, n = 7) nM, respectively. Using the small molecule substrate O-methyl fluorescein phosphate, all three isoforms showed best fit with a partial mixed inhibition kinetic model against NSC 95397 (R2 = 0.924–0.958) (Fig. 3). The correlation coefficients for both partial and full mixed inhibition models, however, were quite close. In two independent experiments, the mean K ivalues (± range) for NSC 95397 against Cdc25A, -B, and -C were 32 ± 15, 96 ± 12, and 40 ± 7 nM, respectively. In one representative experiment, the follow values (micromolar unless noted otherwise) were obtained: Cdc25A:V max, 5.60 × 10−2; K m, 42;K i, 1.65 × 10−2 (95% confidence intervals, 7.3–25.7 nM); α, 3.69; β, 1.22 × 10−9; Cdc25B:V max, 0.106;K m, 27.0;K i, 8.44 × 10−2 (95% confidence intervals,38.7–130 nM); α, 4.44; β, 5.028 × 10−9; Cdc25C:V max, 7.23 × 10−2; K m, 95;K i, 3.38 × 10−2 (95% confidence intervals,7.6–60.1 nM); α, 2.53; β, 4.90 × 10−9. Reversibility studies, however, suggested that NSC 95397, like NSC 672121 (compound 5), caused irreversible inhibition (Tamura et al., 2000), which may explain the mixed inhibition kinetics and could confound the interpretation of our kinetic model in the absence of rapid initial kinetic studies. Thus, preincubation with 100 nM NSC 95397 for 30 min in the absence of substrate resulted in 61% inhibition in enzyme washed free of buffer containing NSC 95397. Further incubation of the NSC 95397-pretreated, bead-bound, Cdc25A catalytic domain in the absence of NSC 95397 did not restore enzyme activity suggesting irreversible inhibition. Because the amino acids in the signature catalytic region, namely HC(X5)R, of all three isoforms are identical (HCEFSSER), we speculate that the modestK i isoform differences reflected slightly altered tertiary structures at or near the catalytic domain resulting from variable amino acids located outside of this sequence. These K i values for NSC 95397 compare quite favorably with our previously publishedK i values for NSC 672121 against Cdc25A, -B, and -C of 15, 1.7, and 1.3 μM, respectively (Tamura et al., 2000) and clearly place NSC 95397 as the most potent known in vitro inhibitor of the Cdc25 family.
Cytotoxicity Studies.
Because the Cdc25 phosphatase family has a central role in controlling cell cycle progression, we evaluated the effects of NSC 95397 on the growth of two breast and one prostate cancer cell lines. NSC 672121 inhibited the growth of human MCF-7 breast cancer cells with an IC50 of 7 μM, which was similar to the results seen by the NCI as part of their evaluation in the NCI 60 Tumor Cell Panel (http://dtp.nci.nih.gov/) (Fig. 5A); the overall average mean growth inhibitory concentration in all 60 tumor cells for NSC 672121 was 10 μM. An initial evaluation of NSC 95397 with the entire NCI 60 Tumor Cell Panel revealed a 10-fold lower overall mean growth inhibitory concentration of 1 μM. MOLT-4 leukemia, LOX IMVI melanoma, and SK-MEL-5 melanoma were the most sensitive (data not shown). We found a growth inhibitory IC50 of 2 μM when MCF-7 cells were exposed to NSC 95397, which was consistent with its enhanced potency against Cdc25 in vitro (Fig. 5A). We also compared the growth inhibitory effects of NSC 95397 and NSC 672121 with PC-3 human prostate and tsFT210 mouse mammary carcinoma cells to determine the generality of growth inhibition. As illustrated in Fig. 4B and C, the IC50 of NSC 95397 for growth inhibition was 3-fold lower than that of NSC 672121 with both tsFT210 cells and PC-3 cells.

Concentration-dependent growth inhibition by NSC 95397. Human MCF-7 (A) and PC-3 (B) cells were treated for 72 h with various concentrations of NSC 95397 or NSC 672121 and cell number determined with an MTT assay as described under Materials and Methods. Murine tsFT210 cells (C) were treated for 48 h with various concentrations of either NSC 95397 or NSC 672121 and viable cell number determined by Trypan blue exclusion as described under Materials and Methods.

Kinetic analysis of Cdc25 isoform inhibition by NSC 95397. Lineweaver-Burk plots for NSC 95397 inhibition of Cdc25A (A), Cdc25B (B), or Cdc25C (C) are shown. Concentrations (micromolar) are indicated in figure.
Cell Cycle.
The tsFT210 cells are a convenient model to study cell cycle effects because their growth can be synchronized without using chemicals. Because Cdc25B and C have a central role in regulating entry into mitosis, we used tsFT210 cells to assess cell cycle progression through the G2/M checkpoint. When tsFT210 cells were incubated at the permissive temperature of 32.0°C, they had a normal cell cycle distribution with 27.8% ± 1.0% (mean ± S.E.M., n = 7) of the total cells in G2/M phase (Fig. 6A). Incubation at the nonpermissive temperature of 39.4°C caused 56.8 ± 2.0% (n = 7) of the total cells to arrest in G2/M phase because of Cdk1 inactivation (Fig. 6B) (Tamura et al., 2000). When G2/M-arrested cells were cultured at the permissive temperature for 6 h with DMSO vehicle alone, 62.6 ± 0.7% (n = 3) of the cells entered G1 (Fig. 6C). In contrast, treatment with 1 μM nocodazole blocked cell passage through G2/M as expected (Fig. 6D). NSC 95397 had no effect at 0.1 μM (Fig. 6E), whereas at 1 μM, a small reduction in G2/M transition was noted (Fig. 6F). We observed only 20.5 ± 0.8% and 19.6 ± 0.5% (n = 3) of the total cells in G1 phase after treatment with 10 and 30 μM NSC 95397, respectively, indicating a profound inhibition (Fig. 6, G and H). These cell cycle data, which are similar to those seen with higher concentrations of NSC 672121 and other less potent inhibitors of Cdc25 (Tamura et al., 2000), support the hypothesis that NSC 95397 is acting to block Cdc25 phosphatase activity within cells.

Inhibition of tsFT210 cell cycle progression at G2/M phase by NSC 95397. tsFT210 cells were cultured at the permissive temperature of 32°C (A) and then incubated for 17 h at the nonpermissive temperature of 39.4°C (B). Cells were released from cycle arrest by shifting to the 32.0°C medium. The cells were incubated for 6 h in the presence of DMSO vehicle (C), 1 μM nocodazole (D), or various concentrations of NSC 95397 (E-H). The ordinate is relative cell number and the abscissa is DNA content in arbitrary units. The vertical bars on the abscissa indicate the relative fluorescence corresponding to 2N and 4N DNA content.
Molecular Modeling.
We next attempted to reconcile the structure-activity relationships found in the NCI compound library using the previously published crystal structure of the Cdc25B catalytic domain (Reynolds et al., 1999) and molecular modeling. Because there are no reported cocrystal structures with Cdc25 and a small molecule ligand, we exploited our results with the compound library and modeled interactions between our most potent inhibitors and the Cdc25B catalytic domain. After scanning the entire surface of the crystal structure of Cdc25B catalytic domain, the most accommodating for the active para-naphthoquinones was the secondary sulfate-binding site located adjacent to the catalytic site of Cdc25B phosphatase (Reynolds et al., 1999). Upon binding, the aromatic moiety of these compounds could desolvate through an opening approximately 11 Å2 flanked by the side chains of residues R544, R482, and Y428 (Fig.7). The interior of the binding site comprises the side chains T547, F543, Y428, L445, R479, M483, R482, E446, S549, W550, R544, P444, and L545. In the crystal structure, the free molecular volume of this site is occupied by a hydrogen-bonded network of water molecules that are by assumption displaced during small molecule occupation. We believe thepara-quinones exploit the large anionic electrostatic potentials on their carbonyl groups to form interactions with the guanidinium side chains of R482 and R544, which are <2.9 Å from the ligand carbonyls (Fig. 8). The catalytic C473 is estimated to be 8.5 Å from the ligand. R482 may play a particularly substantial role in the binding mode of these structures, because in the X-ray structures, the thermal factors for the R482 side chain are very high. This indicates that the side chain of R482 may not be involved in a consistently strong salt bridge with the side chain of nearby E446. Therefore, the R482 guanidinium may become quite stabilized when all of these ligands bind. This complex is further reinforced by the aromatic portion of the double ring system lodging further into the interior of the pocket in a hydrophobic area formed by the lipophilic portions of the side chains of residues R479, T547, Y428, and F543. This pocket accommodated all of the active structures we have studied. Other compounds that exhibit a lack of activity in vitro were also rationalized by electrostatic and steric restrictions that this pocket possesses.

Stereo view of a molecular model of NSC 95397 interactions with Cdc25B catalytic domain. The chemical bonds of NSC 95397 are indicated in green with carbon indicated in brown, sulfur in yellow and oxygen in red. The ligand size was enhanced to emphasize its location. The figure was made with the program RIBBONS (Carson, 1991).

Molecular model of NSC 95397 interactions with a binding site adjacent to the Cdc25B catalytic domain. NSC 95397 docked into the proposed binding site of the crystal structure of Cdc25B phosphatase. Proposed interactions between the diones of the inhibitor and the guanidinium side chains of R544, R482 and the phenolic OH of Y428 are shown. In the figure, nitrogen atoms are blue, oxygen atoms are red, and the chlorine atom of NSC 95397 and the catalytic cysteine 473 are yellow-green. The figure was made with the program RIBBONS (Carson, 1991).
To begin to test this molecular model, we synthesized several additional compounds (Table 3) based on the para-quinone structures identified herein as well as those from our more limited recent study demonstrating the isoquinoline 5,8-dione substructure (Lazo et al., 2001). The new compounds revealed that both steric and electrostatic considerations are substantial. Thus, the newly synthesized minimal 1,2-diazine dione, JUN 376, lacked both the requisite conjugated aromatic structure and substitutions to ensure good inhibition. Similarly the benzopyran JUN 390 also was inactive. The bis-thiol diones JUN 255, 260, 266, 276, and 379 revealed that additional bulk as well as replacement of the naphthoquinone moiety with an isoquinoline or quinoline did not improve potency.
Structures of synthesized compounds
Discussion
Small molecules have been invaluable as reagents to dissect functionality of enzymes. Most biologically interesting macromolecules, however, lack cell penetrating small molecule ligands that can disrupt their cellular functionality. Structural information required for the design of inhibitors is also absent for many attractive target macromolecules. Thus, alternative and generalizable approaches are needed. Our study illustrates the power of using preexisting discovery libraries as reagents to identify new small molecule inhibitors of a pharmacologically relevant class of enzymes. Cdc25 is an excellent prototype target for such an investigation because no potent or selective inhibitors are widely available at present, no enzyme-inhibitor crystal structure is available to permit rational inhibitor design, Cdc25 phosphatases are postulated to have critical roles in controlling cell cycle phase transition, and Cdc25 phosphatases have been implicated in cancer and Alzheimer's disease (Galaktionov et al., 1995; Cangi et al., 2000; Ding et al., 2000).
With a relatively small, publicly available compound library, we have identified the para-naphthoquinone scaffold as a promising lead structure for the design of inhibitors of Cdc25 dual specificity phosphatases. It seems that both electrostatic and steric issues are important for potent and selective inhibition of Cdc25. Although a complete structure-activity relationship has not been established, the compounds described here are the most potent and selective reported to date. Moreover, they clearly had antiproliferative actions and blocked G2/M transition, as would be expected of bona fide Cdc25 inhibitors that penetrate cells. Nonetheless, we have not yet established that these compounds inhibit Cdc25 within cells; this will require additional investigation. We also recognize that kinetic studies using a small molecule substrate, such as O-methyl fluorescein phosphate, should be viewed with some caution, because the results may not emulate activities with the natural cyclin dependent kinase substrates. For example, it has been suggested that the protein substrate may provide the catalytic acid needed for the dephosphorylation of the phosphoprotein substrate (Chen et al., 2000). Nonetheless, almost all previous attempts to identify inhibitors have used small molecule substrates. Thus, using small molecule substrates, such as O-methyl fluorescein phosphate, allows us to compare our experimental results and our computation model with previously reported inhibitors. Moreover, we can examine the selective effects of our inhibitor against several phosphatases using the same artificial substrate.
Our model for ligand binding provides a plausible explanation for the lack of activity for most of the NCI library para-quinones. For example, Fig. 3 illustrates some relatively inactivepara-quinones in the NCI library we have studied. All of the fused multicyclic structures and the para-alkyl–containing compounds cannot be accommodated by the small cleft. Because the proposed binding mode involved interactions between the ligand carbonyl oxygens and both R482 and R544 (Fig. 7 and 8), only long chainsortho to each other will fit conveniently and avoid steric restrictions within the site. Specific hydrogen bonding schemes for these interactions are currently not proposed as the protein model was held fixed during placement of the ligand, leading to less than optimal hydrogen bonding geometries. Nevertheless, the electrostatic interactions are favorable and, given the flexibility of the arginine side chains, minor rearrangements required to form strong hydrogen bonds are easily envisioned. Minimal substitutions at positions 2 and 3 of the para-quinones, such as NSC 43334, 72284, and JUN 379 (Fig. 3), revealed the importance of these positions. Thus, a simple halide, amine, or amide did not permit the favorable interactions produced by the bis-thiohydroxylethyl moiety. Similarly, bulky substituents at the naphthoquinone 2- and 3-positions, such as in NSC 5, 607, or 74702, markedly reduced inhibitory potential. The poorer inhibitory actions of NSC 101612 compared with NSC 95397 also illustrated the importance of the naphthoquinone pharmacophore.
Our proposed model of interaction at the secondary neighboring site in the Cdc25 catalytic domain not only provides information that might be useful in the design of new Cdc25 inhibitors but also suggests experimental approaches to validate the model. For example, mutations at R482, R544, and possibly Y428 in Cdc25B should diminish the inhibitory activity of the para-naphthoquinones. In the absence of a cocrystal with the inhibitor and Cdc25, such studies seem warranted to test the model.
In summary, our studies illustrate the power of using general-purpose discovery libraries to identify new pharmacologically interesting small molecules. We provide a molecular model for ligand interaction and speculate that NSC 95397 or analogs derived from its naphthoquinone structure could prove to be valuable reagents to examine the biological role of the Cdc25 phosphatase family.
Acknowledgments
We are grateful for the thoughtful comments of all of the members of the Lazo and Wipf Laboratories concerning these studies. We also thank Andrew P. J. Brunskill for his assistance with the structural graphics and Deepshikha Passey for her technical assistance supplying the histidine-tagged Cdc25A catalytic domain.
Footnotes
- Received September 28, 2001.
- Accepted January 8, 2002.
-
This work was supported by National Institutes of Health grants CA78039, CA52995, and CA82723 and the Fiske Drug Discovery Fund.
Abbreviations
- PTPase
- protein tyrosine phosphatase
- Cdk
- cyclin dependent kinase
- NCI
- National Cancer Institute
- JUN 379
- 4,5-dichloro-1,2-dimethyl-1,2-dihydropyridazine-3,6-dione
- JUN 390
- 2-amino-3-chloro-chromen-4-one
- JUN 255
- 6,7-bis-(2-(tetrahydropyran-2-yloxy)ethylsulfanyl)quinoline-5,8-dione
- JUN 260 6,7-bis-(2-(tetrahydropyran-2-yloxy)ethylsulfanyl)isoquinoline-5,8-dione
- VHR, VH1-related dual-specificity phosphatase
- DMSO
- dimethyl sulfoxide
- NSC 95397
- 2,3-bis-[2-hydroxyethylsulfanyl]-[1,4]naphthoquinone
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}