


Abstract
An agonist at a specific G protein-coupled receptor may exhibit a range of efficacies for any given response in a cell-specific manner. We report that the relationship between different states of agonism is regulated by the type of G protein expressed in the cell. In NIH-3T3 α2-adrenergic receptor (AR) transfectants, the α2-AR agonists clonidine, oxymetazoline, UK-14304, and epinephrine increased [35S]guanosine-5′-O-(3-thio)triphosphate binding in a dose-dependent manner from a basal value of 101.2 ± 6.5 fmol/mg to a maximal response (100 μM) of 196.6 ± 9.8, 182.3 ± 2, 328.1 ± 11.2, and 340.6 ± 3 fmol/mg, respectively. Thus, clonidine and oxymetazoline behaved as partial agonists. Receptor-mediated activation of G proteins in membrane preparations was blocked by cell pretreatment with pertussis toxin, indicating receptor coupling to the subgroup of pertussis toxin-sensitive G protein (Giα2,3) expressed in NIH-3T3 cells. Ectopic expression of Goα1 but not Giα1 increased the relative efficacy of clonidine and oxymetazoline such that the two ligands now behaved as close to full agonists in this assay system. The relationship between full and partial agonists in the different genetic backgrounds was not altered by progressive reduction in the amount of G protein available for coupling to receptor. The increased efficacy observed for clonidine in the Goα1 transfectants was not due to changes in the relative affinities or amounts of high-affinity, Gpp(NH)p-sensitive binding of agonist. These data suggest that there is little difference in the ability of clonidine to interact with or stabilize α2-AR–Giα2/Giα3 versus α2-AR–Goα1 complexes, but that the subsequent step of signal transfer from receptor to G protein is more readily achieved for the clonidine/α2–AR/Goα1 complex. Such observations have important implications for receptor theory and drug development.
Mechanisms of partial agonism for receptors coupled to heterotrimeric G proteins remain unresolved. Explanations of such ligand behavior must incorporate concepts of inverse agonism and receptor G protein precoupling, the “energy landscape” generated by multiple conformations of the receptor and the cell-specific manifestation of this phenomenon. For example, a specific drug can behave as a partial agonist in one tissue, a full agonist in another, and an antagonist in a third system (Steer and Atlas, 1982; Kenakin, 1984; North and Surprenant, 1985; Surprenant et al., 1990; Gollasch et al., 1991; Hoyer and Boddeke, 1993; Eason et al., 1994). The cellular responses mediated by the great majority of G protein-coupled receptors are also cell specific. The realization that a single receptor molecule functions in a cell-specific manner is a simple point but has broad implications. First, these observations indicate that the signaling system is dynamic and likely developmentally regulated and responsive to physiological and nonphysiological challenges. Second, the action of an agonist/antagonist at a receptor in one cell may be different from that observed for the same receptor in another cell type. Third, the action of an agonist/antagonist in a normal cell can be quite different from its action in the same cell after initiation of a disease process.
The realization that there are multiple receptor subtypes at which drug behavior is system-dependent necessitated thought revision on several issues related to concepts of agonism and the basic tenants of receptor theory (Ariëns, 1954; Kenakin and Morgan, 1989; Black, 1989;Weiss et al., 1996; Kenakin, 1997). The classification of partial versus full agonists at G protein-coupled receptors often depends on whether the readout is proximal [i.e., [35S]guanosine-5′-O-(3-thio)triphosphate (GTPγ35S) binding] or distal (i.e., adenylyl cyclase activity and contraction/relaxation of smooth muscle) within the signal transduction cascade. The degree of receptor reserve for a specific signal transduction pathway also influences data interpretation, and several observations suggest selective activation of different signal transduction pathways by ligand-induced stabilization of specific conformations of the receptor protein (Kenakin, 1995; Perez et al., 1996; Berg et al., 1998; Bonhaus et al., 1998). For receptors capable of coupling to multiple G proteins, the relationships between partial and full agonists may be influenced by the type of G proteins expressed in the cell, stoichiometric considerations, and/or by accessory proteins that regulate the transfer of signal from receptor to G protein and further downstream to various effectors (Coupry et al., 1992; Kataoka et al., 1993; Sato et al., 1995, 1996; Watson et al., 1996; McLatchie et al., 1998;Cismowski et al., 1999; A. Takesono, M. Cismowski, M. Bernard, C. Ribas, P. Chung, E. Duzic, and S.M.L., unpublished observations). As an initial approach to this issue, we determined the relationship between full and partial agonists for a typical G protein-coupled receptor before and after altering the population of G proteins expressed in the cell.
Materials and Methods
Experimental Procedures.
Techniques used for cell culture, membrane preparations, and radioligand binding studies were conducted as described previously (Coupry et al., 1992; Kataoka et al., 1993;Sato et al., 1996). For [35S]GTPγS binding, cell membrane preparations were resuspended in assay buffer (5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, 100 mM NaCl, 1 μM guanosine diphosphate, 1 μM propranolol, 50 mM Tris-HCl, pH 7.4). The reaction was initiated by adding membranes (10 μg in 10 μl) to tubes containing 90 μl of assay buffer containing [35S]-GTPγS (0.2 nM; 1250 Ci/mmol) and agonist or vehicle. Samples were incubated at 24°C for various times and the reactions terminated by rapid filtration through nitrocellulose filters with 4 × 4 ml of wash buffer (100 mM NaCl, 50 mM Tris-HCl, 5 mM MgCl2, pH 7.4, 4°C). Radioactivity bound to the filters was determined by liquid scintillation counting. Nonspecific binding was defined by 100 μM GTPγS.
NIH-3T3 fibroblasts were transfected by calcium phosphate coprecipitation [16 μg of pMSV.α2A/D-adrenergic receptor (AR), 4 μg of pNEO resistance plasmid]. For cotransfection of cells with the receptor and Gα subunits, Goα1 or Giα1 cDNAs were inserted in the drug-resistant plasmid downstream of the mouse metallothionein promoter, which allows both basal and inducible expression of downstream cDNAs as previously described (Coupry et al., 1992; Kataoka et al., 1993). We specifically used the powerful promoter activity of the long terminal repeat of MSV to achieve maximum expression of the α2A/D-AR, whereas G protein transcription was driven by the weaker metallothionein promoter to achieve expression levels similar to those observed endogenously for the two Gα subunits. G418-resistant clones were screened for receptor expression by radioligand binding assays using the α2-AR selective radioligand [3H]RX821002. Receptor/G protein cotransfectants were selected by screening receptor-expressing transfectants for expression of Goα or Giα1 by immunoblotting with selective antisera. Antisera were kindly provided by Drs. Eva Neer (Harvard Medical School), John Hildebrandt (Medical University of South Carolina), and Tom Gettys (Medical University of South Carolina), respectively. For radioligand binding studies, 10 μg of membrane was incubated (30 min, 24°C) in a total volume of 100 μl containing increasing concentrations of the selective α2-AR antagonist [3H]RX821002 (0.025–20 nM) or the selective α2-AR agonists [3H]clonidine (0.025–12 nM) and [3H]UK14304 (0.025–12 nM). Binding reactions were carried out in a 96-well filtration plate (Millipore Corp., Bedford, MA) and terminated by vacuum filtration. Nonspecific binding was determined in the presence of 10 μM rauwolscine and saturation binding studies were analyzed by the RADLIG data analysis software (version 4 of KINETIC, EBDA, LIGAND, LOWRY; BIOSOFT-1994) in which EBDA incorporates nonlinear curve-fitting. At radioligand concentrations near the K d, specific binding represented 85 to 95% of total binding. Several independently isolated transfectants expressing a range of receptor densities were used in these studies as indicated in the text. Receptor densities were determined in each membrane preparation used for evaluation of agonist efficacy because there is a certain variability in receptor density from different cell isolates. Experiments involving Gα transfectants for evaluation of agonist efficacy were also similarly evaluated by immunoblotting of each membrane preparation.
Results and Discussion
Agonism was quantitated at the level of G protein itself. We used a cellular system engineered to express a typical G protein-coupled receptor (α2-AR) at varying receptor densities. We specifically focused on a transfected cell model so that we could control the relative amounts of receptor and provide the same microenvironment for analysis of receptor-mediated activation of G protein subunits. Such an approach overcomes the difficulties presented by cell-specific signaling events and the heterogeneity of signaling proteins when one attempts to address specific events at the receptor–G protein interface. NIH-3T3 fibroblasts were stably transfected with an AR subtype, rat α2A/D-AR, and G protein activation was determined by measuring agonist-induced increases in the binding of the nonhydrolyzable GTP analog GTPγ35S. In membranes prepared from α2-AR transfectants, the adrenergic agonist epinephrine and the selective α2-AR agonist clonidine activated G proteins in a time- and concentration-dependent manner (Fig. 1A and B). The action of both ligands required expression of the α2-AR. It is important to realize that under these incubation conditions in membrane preparations, a significant component of GTPγ35S binding is reversible and thus an equilibrium is achieved (Q.Y. and S.M.L., unpublished observations; also refer to discussion in Breivogel et al., 1998). In this system, the major rate-limiting step for GTPγ35S binding to membrane G proteins in response to agonist is an agonist-induced decrease in the affinity of Gα for GDP (Lorenzen et al., 1996; Selley et al., 1997; Breivogel et al., 1998). The ability of an agonist to promote the latter event is intimately related to its efficacy (Lorenzen et al., 1996; Selley et al., 1997).
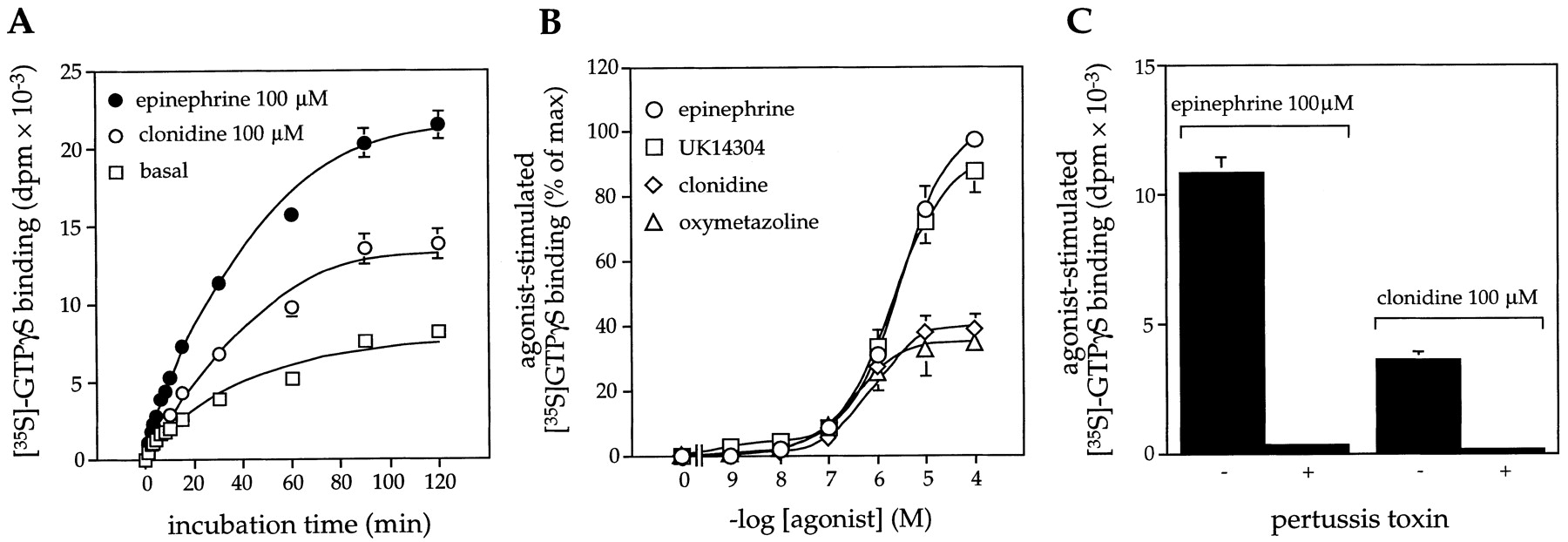
Receptor-mediated activation of G proteins in NIH-3T3 fibroblasts expressing α2 -AR. G protein activation was determined by measuring agonist-induced increases in the binding of GTPγ35S in membrane preparations. A, G protein activation was determined in the presence of vehicle (basal), epinephrine (100 μM), or clonidine (100 μM) for various incubation times at 24°C. B, G protein activation was determined in the presence and absence of increasing concentrations of epinephrine, UK14304, clonidine, and oxymetazoline (24°C for 30 min). Data are expressed as the percentage of maximal response elicited by epinephrine (100 μM). C, influence of pertussis toxin pretreatment (100 ng/ml, 18 h) of cells on G protein activation by agonist. Receptor density (α2-AR no. 1) = 5.3 ± 0.5 pmol/mg membrane protein. Data are expressed as means ± S.E. generated from three (A and B) or four separate experiments (C).
In NIH-3T3 α2-AR transfectants, the α2-AR agonists clonidine and oxymetazoline behaved as partial agonists, whereas the selective α2-AR agonist UK-14304 and the adrenergic agonist epinephrine behaved as full agonists. Receptor-mediated activation of G proteins in membrane preparations was blocked by cell pretreatment with pertussis toxin, indicating receptor coupling to a subgroup of pertussis toxin-sensitive G protein (Giα1–3 and Goα1,2; Fig. 1C). Immunoblotting indicated that within the subgroup of pertussis toxin-sensitive G proteins, NIH-3T3 cells expressed Giα2 and Giα3, but not Giα1 or Goα (Duzic et al., 1992), and thus the signal evaluated must be mediated through Giα2/Giα3.
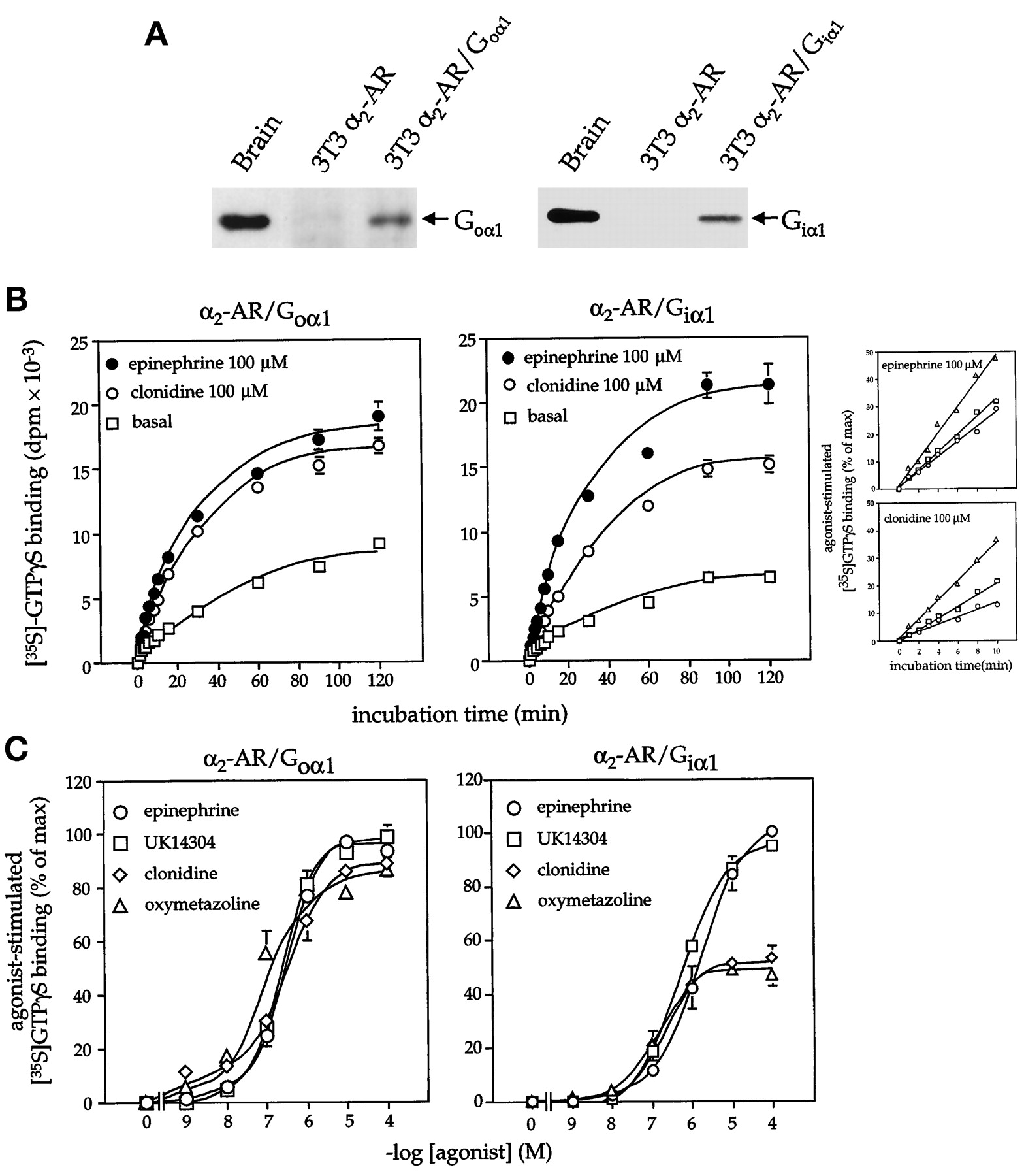
Although clonidine and oxymetazoline are generally classified as partial agonists at α2-AR, their relative efficacy is somewhat tissue and effector specific (Steer and Atlas, 1982; North and Surprenant, 1985; Surprenant et al., 1990;Gollasch et al., 1991; Eason et al., 1994). To determine the influence of G protein type on the relationship between partial and full agonists, we re-evaluated α2-AR-mediated activation of G proteins following cotransfection of NIH-3T3 fibroblasts with the receptor and two functionally distinct G protein α subunits (Giα1 or Goα1). In α2-AR/Goα1 cotransfectants, the amount of G protein activated by the ligands clonidine and oxymetazoline was identical with that elicited by the full agonists epinephrine and UK-14304 (Fig. 2). The enhanced agonist-induced activation of G protein in α2-AR/Goα1 cotransfectants was apparent after only short incubation times (Fig. 2B, inset). Goα1 expression also increased the potency of each α2-AR agonist [EC50: epinephrine, 2.0 ± 0.3 versus 0.3 ± 0.02 μM ( p < .01); UK14304, 1.7 ± 0.2 versus 0.38 ± 0.02 μM ( p < .01); clonidine, 1.1 ± 0.1 versus 0.19 ± 0.01 μM (p < .01); oxymetazoline, 0.4 ± 0.03 versus 0.05 ± 0.01 μM ( p < .01); pvalues refer to α2-AR/Goα1 (8.74 ± 0.48 pmol receptor/mg membrane protein) versus α2-AR transfectants (5.3 ± 0.5 pmol receptor/mg membrane protein]. In contrast to the results obtained in α2-AR/Goα1 cotransfectants, clonidine and oxymetazoline still behaved as partial agonists in cells stably transfected with the α2-AR and the G protein Giα1, even though receptor expression was 3 to 15 times greater than that achieved in the receptor/Goα1 transfectants (Fig. 2 and Table1). (Expression of Gα subunits in NIH-3T3 fibroblasts did not alter basal GTPγ35S binding and did not increase the maximal response elicited by the full agonist epinephrine under these assay conditions. This likely reflects several factors including the use of subsaturating concentrations of GTPγ35S, the inclusion of GDP in the assay system and the reversible binding of GTPγ35S in this system.) Previous studies indicated that the α2-AR is capable of coupling to Giα1, and that the expressed Giα1 protein in NIH-3T3 fibroblasts is functional based on its ability to alter cell responsiveness to transforming growth factor-β (Kataoka and Lanier, 1993; Bahia et al., 1998). The differences in the relationship of partial and full agonists in cells expressing α2-AR/Goα1 versus α2-AR alone were also not altered by a progressive reduction in the amount of G protein available for coupling to the receptor (Fig. 3). These data indicated that the increased efficacy of clonidine and oxymetazoline following the expression of Goα1 was not simply an issue of G protein or receptor levels, but was related to the type of G protein available for coupling to receptor. Indeed, the influence of Goα1 on the relative efficacy of clonidine and epinephrine in our system parallels the behavior of clonidine as a full agonist at α2-ARs in systems where the receptor likely couples to Goα heterotrimer (e.g., receptor-mediated inhibition of calcium channels; Surprenant et al., 1990; Gollasch et al., 1991; Trombley, 1992).

Receptor-mediated activation of G proteins in α2 -AR/Goα1 and α2-AR/Giα1 transfectants. A, Transfectants were evaluated for Goα1 or Giα1 expression by immunoblotting with selective antiserum (left panel: membrane protein-rat brain = 10 μg, NIH-3T3 = 50 μg; right panel: membrane protein-rat brain = 20 μg, NIH-3T3 = 100 μg). G protein activation by α2-AR agonists was determined by measuring agonist-induced increases in GTPγ35S binding to membranes prepared from α2-AR/Goα1 no. 2 (receptor density = 8.7 ± 0.48 pmol/mg membrane protein) or α2-AR/Giα1 (receptor density = 36 ± 0.2 pmol/mg membrane protein) transfectants. B, GTPγ35S binding was determined in the presence of epinephrine (100 μM) or clonidine (100 μM) for various incubation times at 24°C. C, GTPγ35S binding (24°C for 30 min) was determined in the presence of increasing concentrations of agonists. Data in B are presented as means ± S.E. derived from two to three experiments. Inset, comparison of the time course of G protein activation by clonidine and epinephrine in α2-AR (○), α2-AR/Goα1 (▵), and α2-AR/Giα1 (■) transfectants. Data from the first 10 min of the time course presented in Figs. 1A and 2B are compared for the three model systems. Data are expressed as the percentage of the maximal response achieved by each individual receptor agonist. Data in C are expressed as the percentage of the maximal response achieved with the selective α2-AR agonist UK14304 or the adrenergic agonist epinephrine and are presented as means ± S.E. derived from two experiments.
Influence of receptor density and G protein expression on relative efficacy of clonidine

Receptor-mediated activation of G proteins in α2 -AR and α2-AR/Goα1 transfectants after cell pretreatment with pertussis toxin. Progressive reduction in the amount of G protein available for receptor coupling was achieved by cell pretreatment (18 h) with increasing concentrations of pertussis toxin. GTPγ35S binding was determined in the absence and presence of epinephrine (100 μM) or clonidine (100 μM) at 30-min incubation time points (24°C). Receptor density: α2-AR no. 1 = 5.3 ± 0.5 pmol/mg membrane protein, α2-AR/Goα1 no. 2 = 8.7 ± 0.48 pmol/mg membrane protein. Data are presented as means ± S.E. of two experiments with duplicate determinations.
These observations have significance relative to concepts of conformational induction versus conformational selection in terms of agonist efficacy and the development of cell-specific drug efficacy. Relative to the data reported in this article, three mechanistic concepts of partial agonism should be considered. First, full and partial agonists may stabilize different receptor conformations varying in their affinity for G protein or in their ability to activate G protein (a type of conformational induction by agonists). In this situation, receptor conformations stabilized by the partial agonist clonidine or the full agonist epinephrine would differ in their ability to activate Giα, but would be functionally equivalent in terms of activating Goα. Second, full and partial agonists may stabilize the receptor in the same active conformation, but the two types of compounds generate quantitatively different amounts of this receptor conformation. In the Giα, but not Goα1 genetic background, the relative amount of such a complex would then be rate limiting in terms of the total amount of G protein that can be activated.
Third, for receptors that are precoupled, the receptor that is precoupled to G protein X may be stabilized in a conformation that is different from that of a receptor that is precoupled to G protein Y (a type of conformational induction by G proteins). Full and partial agonists may differ in their affinity for these two conformations of receptor. Alternatively, these two receptor conformations may differ in the efficiency in which they mediate signal transfer from receptor to G protein when the agonist site is occupied. In one scenario, full agonists would interact with all precoupled R–G complexes (e.g., both G protein X and G protein Y), whereas a partial agonist would only interact with a subgroup of precoupled R–G complexes (e.g., G protein X or G protein Y). This concept can be further extended if one assumes that the precoupled receptor G protein complex is totally responsible for the readout of agonist activation. In such a case, the differences between clonidine and epinephrine observed in the two genetic backgrounds may simply reflect a greater propensity of Goα1 to generate the precoupled complex as compared with Giα.
As one approach to these issues, we determined whether expression of Goα1 or Giα1 influenced the population of receptors interacting with agonist. The conformation of receptors for which agonists exhibit high affinity is stabilized by interaction with G proteins and this complex is disrupted in radioligand binding assays by the addition of a nonhydrolyzable analog of GTP. We simultaneously evaluated the radioligand binding properties of the antagonist ([3H]RX821002) and agonist ([3H]clonidine and [3H]UK14304) radioligands in the three genetic backgrounds (control and Giα1 and Goα1 transfectants). Both [3H]clonidine and [3H]UK14304 exhibited high affinity, Gpp(NH)p-sensitive binding in each cell line and the amount of high-affinity, Gpp(NH)p-sensitive binding was related to the receptor densities (as determined with the antagonist radioligand [3H]RX821002) of the individual cell lines (α2-AR no. 1 versus α2-AR no. 2; α2-AR/Goα1 no. 1 versus α2-AR/Goα1 no. 2; Fig. 4 and Table 2). In each genetic background, the number of Gpp(NH)p-sensitive binding sites for [3H]UK14304 was greater than that observed with [3H]clonidine. Thus, the conversion of clonidine from a partial to full agonist by expression of Goα1 was not due to an increase in the Gpp(NH)p-sensitive, high agonist-affinity receptor population. There was also no change in the affinities exhibited by UK14304 and clonidine at this Gpp(NH)p-sensitive binding site identified in the radioligand binding studies in the different genetic backgrounds (Table 2).

Gpp(NH)p-sensitive binding of [3H]clonidine and [3H]UK14304 in α2-AR, α2-AR/Goα1, and α2-AR/Giα1 transfectants. Saturation binding isotherms were performed on five cell lines using the α2-AR-selective antagonist [3H]RX821002 (0.025–20 nM) and the α2-AR-selective agonists [3H]UK14304 and [3H]clonidine (0.025–12 nM). For each radioligand, parallel binding studies were conducted in the absence and presence of the nonhydrolyzable GTP analog Gpp(NH)p (100 μM). [3H]RX821002 binding was not influenced by addition of Gpp(NH)p. In contrast, ∼80 to 85% of the specific binding of [3H]clonidine and [3H]UK14304 (at ligand concentrations of 2 nM) was Gpp(NH)p-sensitive. For both [3H]clonidine and [3H]UK14304, the specific binding obtained in the presence of Gpp(NH)p was subtracted from that obtained in the absence of Gpp(NH)p at each radioligand concentration to generate the presented data. The data are representative of two to three experiments using different preparations of cell membranes.K d and B maxvalues are presented in Table 2.
Ligand recognition properties of α2-AR in different genetic backgrounds
These data suggest that there is little difference in the ability of clonidine to interact with or stabilize α2-AR–Giα2/Giα3 versus α2-AR–Goα1 complexes, but that the subsequent step of signal transfer from receptor to G protein is more readily achieved for the clonidine/α2–AR/Goα1 complex. This interpretation may reflect structural aspects of the interaction between the α2-AR and Giα2/Giα3 versus Goα1 or perhaps the differences in the nucleotide binding properties of the G proteins themselves such that Goα1 is “easier” to activate. Relative to Giα2,3, Goα1 has a higher basal rate of nucleotide exchange (Ferguson et al., 1986; Carty et al., 1990), and in this sense one might conclude that Goα1 is actually “primed” for activation by receptors.
The influence of G protein type on the relationship between full and partial agonism indicates that agonism, or efficacy, is a dynamic phenomenon that is intimately related to the array of proteins expressed by a cell at any given stage of development or in specific disease states. Thus, the effects of a particular drug would be maximized in a tissue where it behaves as a full agonist and diminished in tissues where it behaved as a partial agonist. The same thoughts may apply to inverse agonists. Such information may lead to the development of drugs that display a spectrum of agonistic behavior in a cell type-specific manner such that these distinctions work to therapeutic advantage.
Acknowledgments
We thank Dr. T.P. Kenakin (Glaxo Wellcome Research, Research Triangle Park, NC) and Dr. John D. Hildebrandt (Department of Pharmacology, Medical University of South Carolina) for helpful discussions and encouragement. We appreciate the expert technical assistance of John Daw and Peter Chung.
Footnotes
- Received May 14, 1999.
- Accepted June 14, 1999.
-
Send reprint requests to: Stephen M. Lanier, Ph.D., Department of Pharmacology, Medical University of South Carolina, 171 Ashley Ave., Charleston, SC 29425. E-mail:laniersm{at}musc.edu
-
↵1 Visiting scientist from the Institute of Cardiovascular Basic Research, Beijing Medical University, Beijing, Peoples Republic of China.
-
This work was supported by the National Institutes of Health Grant NS24821 (to S.M.L.).
Abbreviations
- GTPγ35S
- [35S]guanosine-5′-O-(3-thio)triphosphate
- AR
- adrenergic receptor
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}