


Abstract
The effects of N-methyl-d-aspartate (NMDA) on opioid receptor-mediated G protein activation were explored in neuroblastoma X glioma hybrid (NG108–15) cells. Treatment of the cells with NMDA resulted in a remarkable attenuation of [35S]guanosine-5′-O-(3-thio)triphosphate binding stimulated by [d-Pen2,d-Pen5]-enkephalin (DPDPE), a δ-opioid receptor agonist. The effects of NMDA were dose and time dependent with an IC50 value of 5 nmand could be blocked by NMDA receptor antagonists. After NMDA treatment, the DPDPE dose-response curve shifted to the right (EC50 value increased ∼7-fold, from 6 to 40 nm), and the maximal response induced by DPDPE was reduced by ∼60%. The effects of NMDA were reversible, and the DPDPE response could recover within 60 min. The functional responses of δ-, μ-, and κ-opioid receptors in primarily cultured neurons also were attenuated significantly by NMDA treatment. The inhibitory effects of NMDA on opioid receptor-mediated G protein activation could be blocked by coadministration of the protein kinase C (PKC) inhibitors or by elimination of the extracellular Ca2+. Correspondingly, NMDA treatment of NG108 cells significantly elevated cellular PKC activity and stimulated Giα2 phosphorylation. Transient transfection into NG108–15 cells of the wild-type Giα2and a mutated Giα2 (Ser144Ala) resulted in a 2-fold increase in DPDPE-stimulated G protein activation. The DPDPE responses were greatly inhibited by NMDA treatment in the wild-type Giα2-transfected cells but much less affected in the mutant Giα2-transfected cells. In summary, NMDA attenuates opioid receptor/G protein coupling, and this process requires activation of PKC.
Opiates are the therapeutic mainstay of clinical pain management, and they have many other important physiological effects. Pharmacological studies have defined three major types of opioid receptors (designated δ, μ, and κ), all of which are coupled to PTX-sensitive G proteins (Gi/Go) and regulate adenylyl cyclase, Ca2+ channels, and potassium channels (Hescheler et al., 1987; McKenzie and Milligan, 1990). A prominent characteristic of opiates is their ability to induce drug tolerance and dependence, and the side effects greatly limit their therapeutic efficacy. Although it is suggested that receptor desensitization, internalization, and down-regulation may be involved, the mechanisms underlying opioid tolerance and dependence are not well understood (Louie et al., 1986; Nestler, 1996).
There is accumulating evidence that the NMDA receptor plays a role in opioid analgesia, tolerance, and dependence. In animal and clinical studies, various NMDA receptor antagonists profoundly attenuate opioid tolerance and dependence (Trujillo and Akil, 1991, 1994; Gutstein and Trujillo, 1993), and the NMDA receptor antagonist ketamine has been found to potentiate the analgesic effects of morphine in patients with cancer (Mercadante et al., 1995). These suggest that NMDA antagonists may be a promising weapon in combating the addictive and abuse characteristics of opiates, and it is worthwhile to investigate the cellular and molecular mechanisms of cross-talking between the NMDA receptor system and the opioid receptor system.
Neuroblastoma × glioma hybrid (NG108–15) cells have been used as a good model to study the interaction between the NMDA receptor and opioid receptors (Louie et al., 1986, Ohkuma et al., 1994). We demonstrated that activation of the NMDA receptor attenuated DOR-mediated inhibition of adenylyl cyclase (Cai et al., 1997), indicating the existence of cross-talk between NMDA and opioid receptor signaling pathways at cellular level. However, it is still not known, at the molecular level, how NMDA affects the signaling of opioid receptors. It has been suggested that NMDA may act on the upstream of adenylyl cyclase but not directly on opioid receptors (Cai et al., 1997). Therefore, in the current study, we focused on the opioid receptor/G protein coupling, and the results showed that NMDA attenuates opioid receptor-mediated G protein activation, possibly through PKC-mediated phosphorylation of the opioid receptor-coupled G proteins.
Materials and Methods
Cell culture.
NG108–15 cells were cultured with Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (GIBCO BRL, Gaithersburg, MD), 10% calf serum (GIBCO BRL), 0.1 mmhypoxanthine, 1 μm aminopterin, and 16 μmthymidine as described previously (Cai et al., 1996). Dissociated brain neurons obtained from neonatal mice were cultured in basal Eagle’s medium (GIBCO BRL) supplemented with 10% fetal calf serum (GIBCO BRL) and 10% calf serum (GIBCO BRL) as described previously (Eva et al., 1992).
Exogenous expression of Giα2 in NG108–15 cells.
Human Giα2 was cloned from human brain cDNA (Beals et al., 1987), and the influenza hemagglutinin epitope (YPYDVPDYA) recognized by monoclonal antibody 12CA5 was added to the amino terminus of Giα2through PCR. The 5′ primer sequence was GCG AAG CTT ATG TAC CCA GAC GTC CCA GAC TAC GCC GCG TGC ACC GTG AGC GCC, containing aHindIII restriction site before the start codon; and the 3′ primer sequence was CGC TCG AGC CTC AGA AGA GGC AGC AG, containing anXhoI restriction site after the stop codon. Giα2 antisense cDNA was generated by subcloning the wild-type Giα2 cDNA reversibly into pcDNA3. The mutant Giα2 constructs Giα2 (Ser144Ala) and Giα2 (Ser16Ala) were generated from the wild-type Giα2 by site-directed mutagenesis using PCR (Vallette et al., 1989) to introduce the mutation Ser144 or Ser16 to alanine. A nonsense cDNA was produced by shifting the reading frame of Giα2 cDNA. Wild-type Giα2, tagged-Giα2, and the mutated Giα2 cDNAs, which were verified with dideoxy DNA sequencing, were subcloned into pcDNA3 (InVitrogen, San Diego, CA) and transfected into NG108–15 cells individually using the Ca2+ phosphate precipitation method. The exogenous expression of Giα2 was determined by Western blot analysis using a specific anti-Giα2 antibody (NeoMarkers, Fremont, CA).
[35S]GTPγS binding assay.
The experiments were performed as described previously (Traynor and Nahouaki, 1995;Cheng et al., 1997). Briefly, cells were lysed in buffer containing 5 mm Tris·HCl, pH 7.5, 5 mm EDTA, and 5 mm EGTA, and the cell lysate was centrifuged at 30,000 × g for 10 min. Membranes (containing 50 μg of protein) were incubated in 50 mm Tris·HCl, pH 7.5, 5 mm MgCl2, 1 mm EGTA, 100 mm NaCl, 50 μm GDP, and 8 nm[35S]GTPγS (1200Ci/mmol; DuPont-New England Nuclear, Boston, MA) in a total volume of 0.1 ml at 30° for 1 hr. The reaction was terminated by dilution in phosphate-buffered saline and filtration through GF/C filters under a vacuum. Bound radioactivity was determined with a liquid scintillation counter. Basal binding was assessed in the absence of the agonists, and nonspecific binding was determined in the presence of 10 μm GTPγS. The percentage of agonist-stimulated [35S]GTPγS binding was calculated as [cpmagonist − cpmnonspecific]/[cpmbasal− cpmnonspecific].
cAMP assay.
The cells (∼1 × 105 cells) were challenged with control medium or medium containing NMDA (Sigma) at 37° for 5 min. Then, cells were treated further with agonists in the presence of 10 μmforskolin (Sigma) and 500 μm 1-methyl-3-isobutylxanthine (Sigma) at 37° for 10 min. The reaction was terminated with 1n perchloric acid and neutralized with 2 nK2CO3; the cAMP levels of each sample were measured using radioimmunoassay as described previously (Cai et al., 1997). Values were calculated as 100 × [cAMP(For+agonist) − cAMP(basal)]/[cAMP(For)− cAMP(basal)]. Where cAMP(For+agonist) is cAMP accumulation in the presence of forskolin and agonist, cAMP(basal) is accumulation in the absence of forskolin and agonist, and cAMP(For) is accumulation in the presence of forskolin alone. Protein content of each sample was determined using the modified Bradford-Pierce assay (Pierce Chemicals, Rockford, IL).
Measurement of PKC activity.
NG108–15 cells were challenged with 0.1 μm PMA, 1 μm NMDA, 1 μm NMDA plus 1 mm APV, or 1 μmNMDA plus 2 mm EGTA without Ca2+, all at 37° for 10 min. The cells were homogenized in the lysis buffer of 25 mm Tris·HCl, pH 7.5, 0.5 mm EDTA, 0.5 mm EGTA, 10 mm β-mercaptoethanol, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 0.5 mmphenylmethylsulfonyl fluoride. The cell membranes, which were obtained after centrifugation at 100, 000 × g, were solubilized in the lysis buffer containing 0.5% Triton X-100 for 1 hr. An aliquot of supernatant was measured after centrifugation for PKC activity. The activity of PKC was measured using the SignaTECT PKC Assay System (Promega, Madison, WI) according to manufacturer’s protocols.
Phosphorylation of Giα2.
Phosphorylation was carried out as described previously (Migeon et al., 1994;Strassheim and Malbon, 1994). At 48 hr after transient transfection with the tagged Giα2, NG108–15 cells were labeled at 37° for 60 min with 100 μCi/ml [32P]orthophosphate (5000 Ci/mmol; DuPont-New England Nuclear) in phosphate-free Dulbecco’s modified Eagle’s medium (GIBCO BRL). The labeled cells were stimulated without or with 1 μm NMDA, 1 μm NMDA, plus the PKC inhibitor or with 1 μm NMDA plus the NMDA receptor antagonist for 15 min at 37°. After stimulation, the cells were harvested by centrifugation (100 × g, 2 min). A pellet of cells (106/ml) was lysed by the addition of 1 ml of RIPA+ buffer containing 150 mm NaCl, 50 mm Tris, pH 8.0, 5 mm EDTA, pH 8.0, 1% (v/v) Nonidet P-40, 0.5% (w/v) deoxycholate, 10 mm NaF, 10 mm disodium pyrophosphate, 2 mmphenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 0.1% (w/v) SDS. Then, 800 μl of supernatant from a 15-min, 360,000 × g centrifugation was adsorbed through incubation with 100 μl of presoaked Protein A/Sepharose beads (Pharmacia Biotech, Uppsala, Sweden) and 1 μg of 12CA5 antibody. Beads were washed three times, and immunoprecipitated proteins were dissociated from beads with 30 μl of 1× SDS-polyacrylamide gel loading buffer (2% SDS, 25 mm Tris, pH 6.8, 5% glycerol, 0.5% 2-mecaptoethanol, and 0.005% bromphenol blue). Then, 20 μl of immunoprecipitated proteins was subjected to 10% SDS-polyacrylamide gel electrophoresis. The gels were dried and subjected to analysis with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA) to determine Giα2 phosphorylation. The amount of G proteins loaded onto each lane was examined by means of Western blot analysis in a parallel experiment using anti-Giα2 antibody (NeoMarkers, Fremont, CA).
Statistical analysis.
Each experimental point was measured in at least triplicate, and at least three experiments were carried out. Data are expressed as mean ± standard error of all determinations. Statistical significance of the experimental results was obtained with Student’s t test (StatView).p < 0.05 was accepted as denoting statistical significance.
Results
Attenuation of DPDPE-stimulated G protein activation by NMDA.
[35S]GTPγS binding assay has been proved to be an effective method to probe agonist-dependent activation of membrane-associated G proteins (Hilf et al., 1989; Lazarenoet al., 1993; Tian et al., 1994; Traynor and Nahouaki, 1995). As shown in Fig. 1, binding of [35S]GTPγS to the membranes in response to DPDPE increased ∼100% over basal. After the cells were challenged with various concentrations (10−11 to 10−5 m) of NMDA, a specific agonist of NMDA receptor, at 37° for 15 min, basal [35S]GTPγS binding did not change (data not shown), but the ability of DPDPE to stimulate [35S]GTPγS binding was attenuated remarkably. The effects of NMDA was dose and time dependent with an EC50 value of ∼5 nm (Fig.1), and the maximal effects were achieved in 15 min (Fig. 1B). The DPDPE dose-response curve shifted to the right, and the EC50 value of DPDPE increased ∼7-fold (from 6 to 40 nm) in response to NMDA treatment (Fig. 2A). After NMDA treatment, the DPDPE response recovered within 60 min (Fig. 2B), indicating that the inhibitory effects of NMDA were reversible.

A, Concentration-dependent effects of NMDA pretreatment of NG108–15 cells on DPDPE stimulation of [35S]GTPγS binding. Cells were incubated with different concentrations of NMDA (10−11 to 10−5 m) at 37° for 15 min; then, the membranes were prepared by lysis. Assays were performed in the presence of 1 μmDPDPE at 30° for 60 min, and the samples were filtered through glass-fiber filters. Data were expressed as percentages of basal [35S]GTPγS binding in the absence of agonists as described in Materials and Methods. B, Time-dependent effects of NMDA pretreatment of NG108–15 cells on DPDPE stimulation of [35S]GTPγS binding. Cells were incubated with NMDA for different times indicated, and assays were performed as described in A. Data are mean ± standard error of three separate experiments. Basal [35S]GTPγS binding was 29.88 ± 4.37 fmol/mg of protein in the absence of NMDA and 32.15 ± 3.19 fmol/mg of protein in the presence of NMDA. ∗, p < 0.05, compared with nontreated cells.

A, Effects of NMDA treatment of NG108–15 cells on the dose-response curve of [35S]GTPγS binding stimulated by DPDPE. Cells were incubated without (•) or with 1 μm NMDA (▪) at 37° for 15 min, and assays were performed in the presence of different concentrations (10−11 to 10−5 m) of DPDPE for 60 min at 30°. B, Recovery of DPDPE responses after NMDA treatment in NG108–15 cells. After treatment with NMDA (1 μm) for 15 min, the cells were washed three times with phosphate-buffered saline. DPDPE-stimulated [35S]GTPγS binding was carried out at different time indicated after NMDA treatment. Values are mean ± standard error of three separate experiments.
To examine whether the NMDA effects were mediated through NMDA receptor, the NMDA receptor antagonists APV and ketamine were used 5 min before NMDA application. In the presence of APV, which did not affect DPDPE response as shown in Fig.3A, the attenuation by NMDA of DPDPE-stimulated [35S]GTPγS binding was blocked (Fig. 3B). Another NMDA receptor antagonist, ketamine (100 μm), which did not alter basal DOR signaling, also blocked the inhibitory effects of NMDA (data not shown). These data indicate that the effects of NMDA were mediated through NMDA receptor.
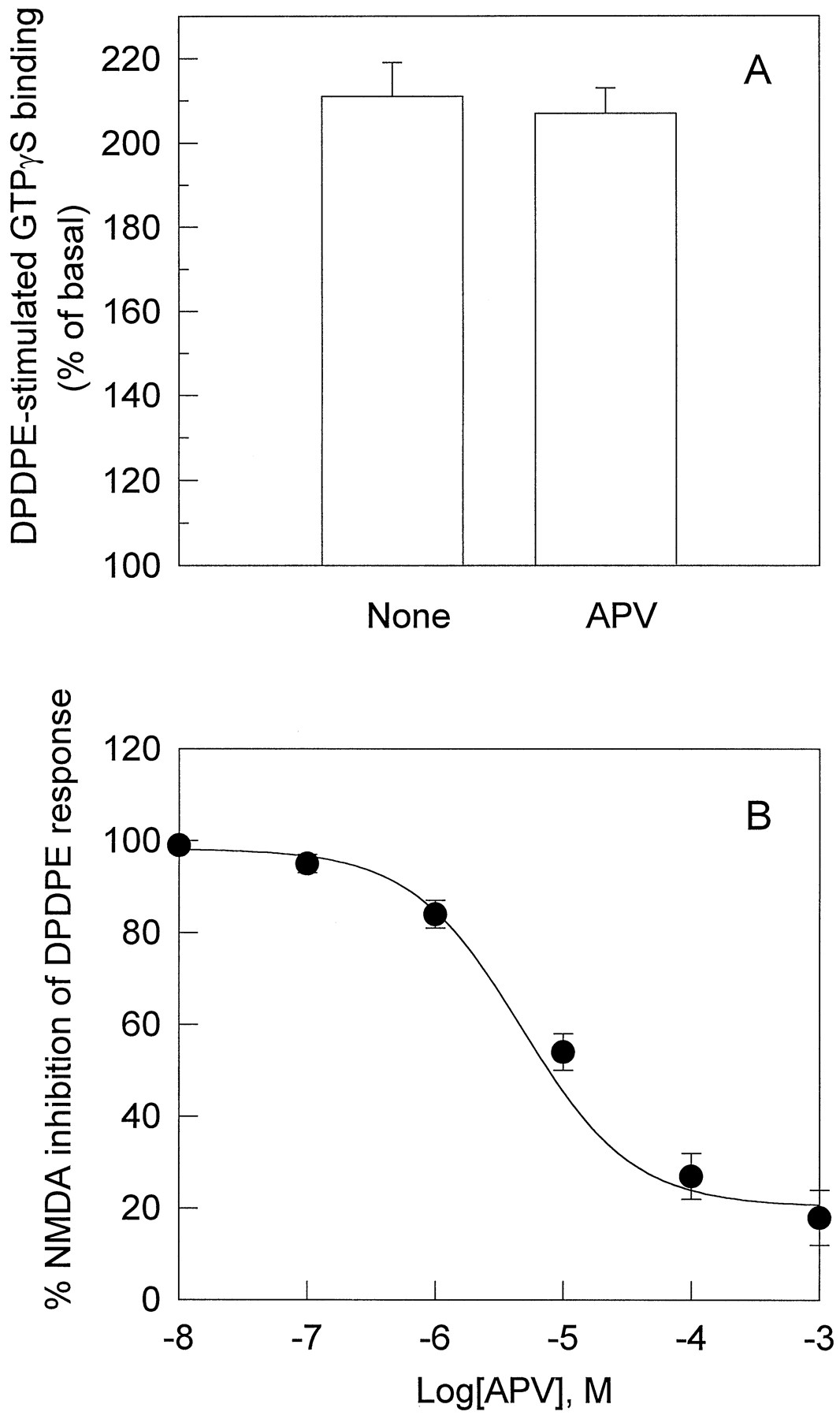
A, Lack of effects of APV on DPDPE-stimulated [35S]GTPγS binding in NG108–15 cells. Cells were treated with APV (100 μm) for 20 min; then, the membranes were prepared, and [35S]GTPγS binding was stimulated by DPDPE (0.1 μm) was carried out as described above. B, Blockade by APV of the inhibitory effects of NMDA on DPDPE response. NG108–15 cells were incubated without or with APV (10−7to 10−3 m) at 37° for 5 min and then at 37° for 15 min after the addition of 1 μm NMDA. Subsequent preparation of cell membranes and DPDPE-stimulated [35S]GTPγS binding assays were performed as described in the legend to Fig. 1. Values are mean ± standard error of three independent experiments.
Involvement of PKC in the inhibitory effects of NMDA on DPDPE-stimulated G protein activation in NG108–15 cells.
Next, we sought to determine whether two major protein kinases, PKC and PKA, were involved in the inhibitory effects of NMDA on opioid receptor-mediated G protein activation. In the parallel experiment, pretreatment of NG108–15 cells with 0.1 μm PMA, a PKC activator, displayed significant attenuation of DPDPE-stimulated [35S]GTPγS binding, which was comparable to that of NMDA (Fig. 4A). Furthermore, treatment with the specific PKC inhibitors chelerythrine chloride (15 μm) (Ki = 0.66 μm; Calbiochem, San Diego, CA) or Gö 6976 (0.2 μm) (Ki = 0.008 μm; Calbiochem) could totally block the inhibitory effects of not only PMA but also NMDA (Fig. 4A). However, PKA inhibitor H-89 (1.2 μm; Ki = 0.048 μm; Calbiochem) failed to block the effects of NMDA (Fig. 4A), suggesting an essential role of PKC in the inhibitory effects of NMDA on opioid-stimulated G protein activation.
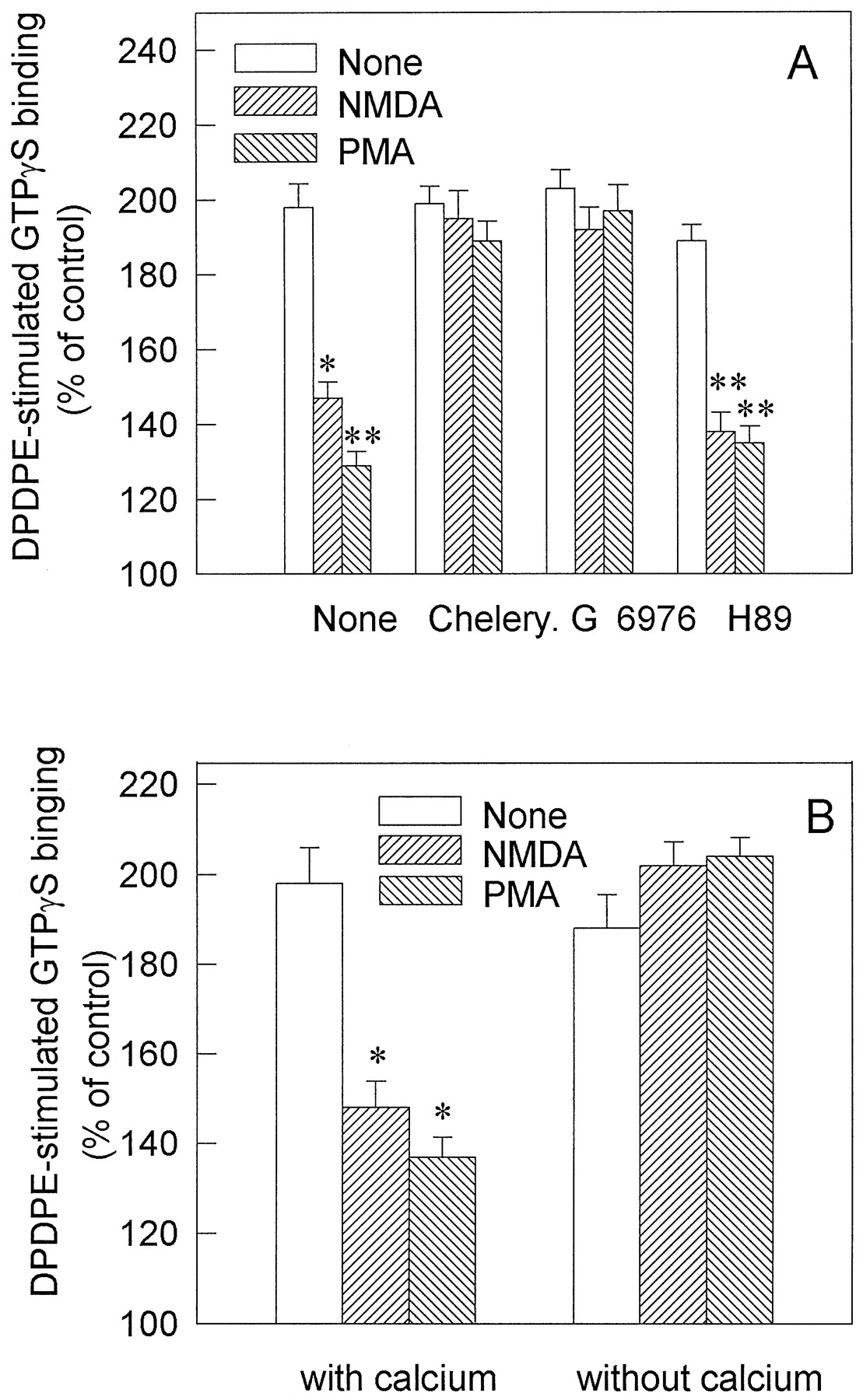
A, Blockade by PKC inhibitor of the inhibitory effects of NMDA on DPDPE-stimulated [35S]GTPγS binding. Cells were incubated at 37° with specific PKC inhibitors chelerythrine chloride (15 μm; Chelery) and Gö 6976 (0.2 μm) or specific PKA inhibitor (1.2 μm H-89) for 5 min; then, the cells were treated with 1 μm NMDA or 100 nm PMA for 15 min. The subsequent DPDPE stimulation of [35S]GTPγS binding was measured under the same conditions as that in the legend to Fig. 1. B, Removal of extracellular Ca2+ abolished NMDA attenuation of DPDPE-stimulated [35S]GTPγS binding. NG108–15 cells were treated without or with 1 μm NMDA or 100 nm PMA in the normal medium with Ca2+ or the medium without Ca2+ plus 2 mm EGTA at 37° for 15 min. Then, the membranes were prepared, and the subsequent DPDPE stimulation of [35S]GTPγS binding assays was performed as described in the legend to Fig. 1. Data are mean ± standard error of three independent experiments. There was no apparent interference on the basal [35S]GTPγS binding from the kinase inhibitors or EGTA. ∗, p < 0.05; ∗∗,p < 0.01, compared with nontreated cells.
NMDA receptor is a ligand-dependent ion channel of high Ca2+ permeability (MacDermott et al., 1986). It raises the possibility that NMDA exerts its effects by mobilizing Ca2+ and consequently activating Ca2+-dependent PKC. As shown in Fig. 4B, both NMDA- and PMA-mediated inhibition on DPDPE stimulated [35S]GTPγS binding to the cell membranes was completely abolished by removal of the extracellular Ca2+ with 2 mm EGTA. These results suggest that Ca2+ influx is required for the effects of NMDA, and Ca2+-dependent PKC may be involved.
Attenuation by NMDA of functional responses of μ-, δ-, and κ-opioid receptors in primarily cultured neurons.
Primarily cultured neurons from neonatal mice, which express NMDA receptor and all three opioid receptor subtypes, were used to determine whether NMDA also affects functional responses of δ-, μ-, and κ-opioid receptors. After the cells were treated with 1 μm NMDA at 37° for 15 min, the inhibition of cAMP accumulation induced by DPDPE, DAGO, or U69593, the specific agonist of δ-, μ-, and κ-opioid receptor, respectively, was significantly attenuated, which was blocked by Gö 6976 (0.2 μm) (Fig.5A). Stimulation of [35S]GTPγS binding by these three agonists also was reduced in the neuronal cells treated with NMDA for 15 min, and the inhibitory effects of NMDA also were blocked by coadministration of 0.2 μm Gö 6976 (Fig. 5B).
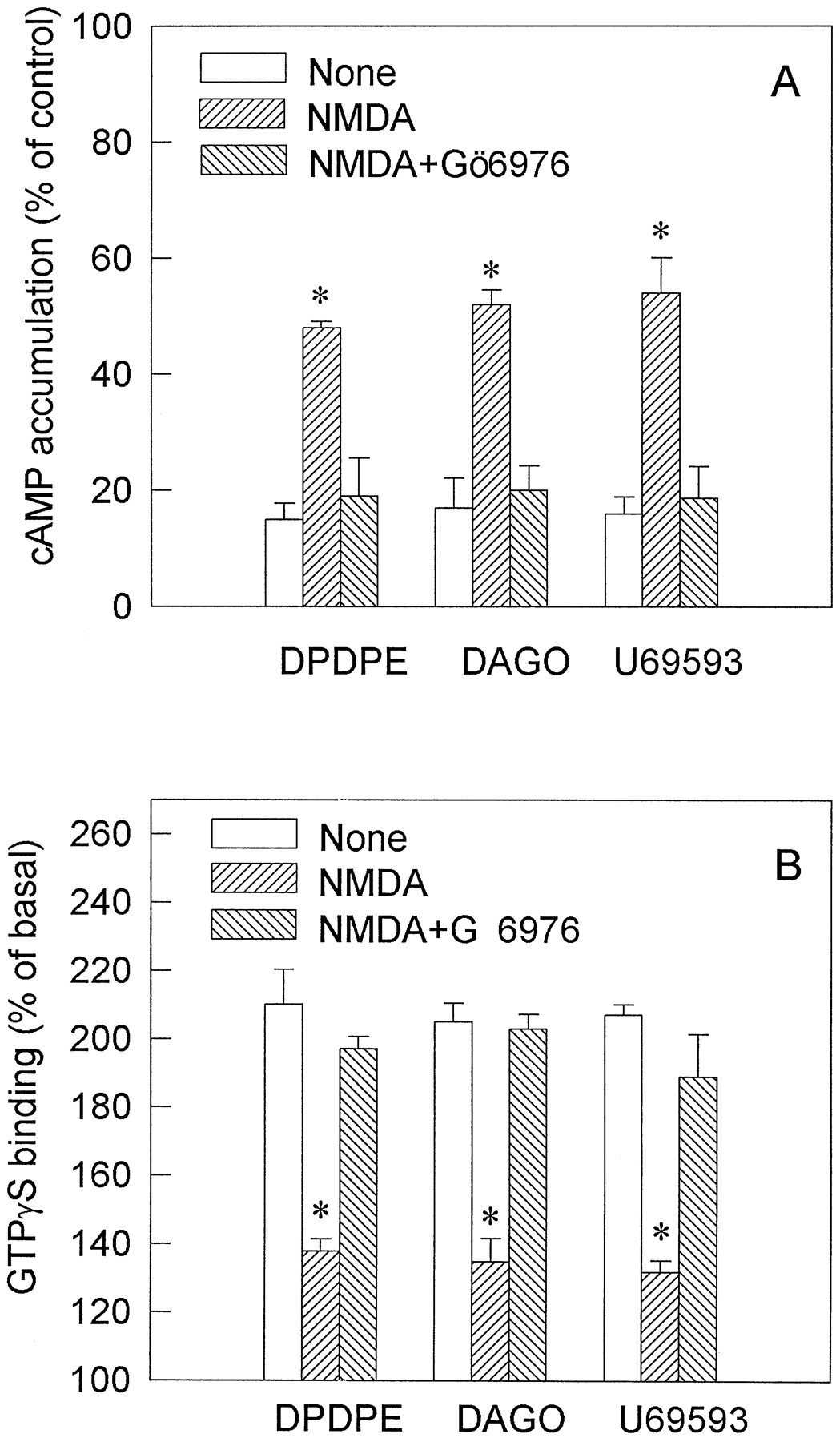
A, Attenuation by NMDA of opioid-mediated inhibition of cAMP accumulation in primary neuronal cultures. Primarily cultured mouse cortical neurons were pretreated without or with 1 μm NMDA or 1 μm NMDA plus 0.2 μm Gö 6976 at 37° for 5 min. Percentages of forskolin-stimulated cAMP production in the presence of 1 μm DPDPE, 1 μm DAGO, or 1 μmU69593 were measured as described in Materials and Methods. B, Attenuation by NMDA treatment of opioid-stimulated [35S]GTPγS binding in primary neuronal cultures. Cells were incubated without or with 1 μm NMDA or 1 μm NMDA plus 0.2 μm Gö 6976 at 37° for 15 min. The membranes were prepared, and the [35S]GTPγS binding was performed in the presence of 1 μm DPDPE, 1 μm DAGO, or 1 μmU69593 for 60 min at 30°. Values are mean ± standard error of three separate experiments. ∗, p < 0.05, compared with nontreated cells.
Activation of PKC by NMDA or PMA in NG108–15 cells.
The PKC activity after NMDA or PMA pretreatment of NG108–15 cells was measured directly. As shown in Fig. 6, challenge of the cells with NMDA resulted in a remarkable elevation of membrane-associated PKC activity (∼40% over basal), which was comparable to the elevation by PMA treatment (60% over basal). The stimulation of PKC activity by NMDA was significantly blocked by NMDA receptor antagonist APV (1 mm) or by the removal of extracellular Ca2+ with 2 mm EGTA (Fig. 6), indicating mediation through NMDA receptor and involvement of Ca2+ influx.

Activation of membrane-associated PKC by NMDA in NG108–15 cells. Cells were incubated at 37° for 5 min with 1 mm APV or 2 mm EGTA without Ca2+before 15-min treatment with 1 μm NMDA. PKC activity was measured as described in Materials and Methods. The results are mean ± standard error of four experiments and expressed as percentages of basal PKC activity in nontreated cells. ∗,p < 0.05, compared with nontreated cells.
Phosphorylation of Giα2 in response to NMDA treatment.
The above data demonstrate an essential involvement of PKC in the inhibitory effects of NMDA on opioid receptor/G protein coupling. Agonist binding to δ-opioid receptor was not changed and an increase in phosphorylation of δ-opioid receptor was not observed after NMDA treatment (Cai et al., 1997). G proteins that couple to opioid receptors are therefore a likely target of the NMDA effects. It has been shown that the functional response of DOR is mediated largely by Giα2 (McKenzie and Milligan, 1990). In NG108–15 cells pretreated with 100 ng/ml PTX for 24 hr, DPDPE-stimulated GTPγS binding was blocked completely (Fig.7A). Transfection with Giα2 antisense cDNAs resulted in a remarkable attenuation of the DOR response (Fig. 7B), whereas transfection with a nonsense cDNA did not affect the function and expression of Giα2 (Fig. 7, B and C), indicating that Giα2 was one of the major transducer of DOR. To explore the possible role of G protein phosphorylation in this cross-regulation, we metabolically labeled NG108–15 cells transiently transfected with the epitope tagged Giα2 with [32P]orthophosphate followed by immunoprecipitation with antibody 12CA5 and observed the effects of NMDA on phosphorylation of Giα2. NMDA treatment led to significant phosphorylation of Giα2(Fig. 8A, lane 2) in comparison with the control (Fig. 8A, lane 1). Fig. 8B showed that equal amounts of proteins were loaded onto each lane. As shown in Fig. 8C, [32P] incorporation into Giα2 increased >5-fold after NMDA treatment. The Giα2 phosphorylation was blocked by the PKC inhibitor chelerythrine chloride (Fig. 8A, lane 3) or by the NMDA receptor antagonist APV (Fig. 8A, lane 4).
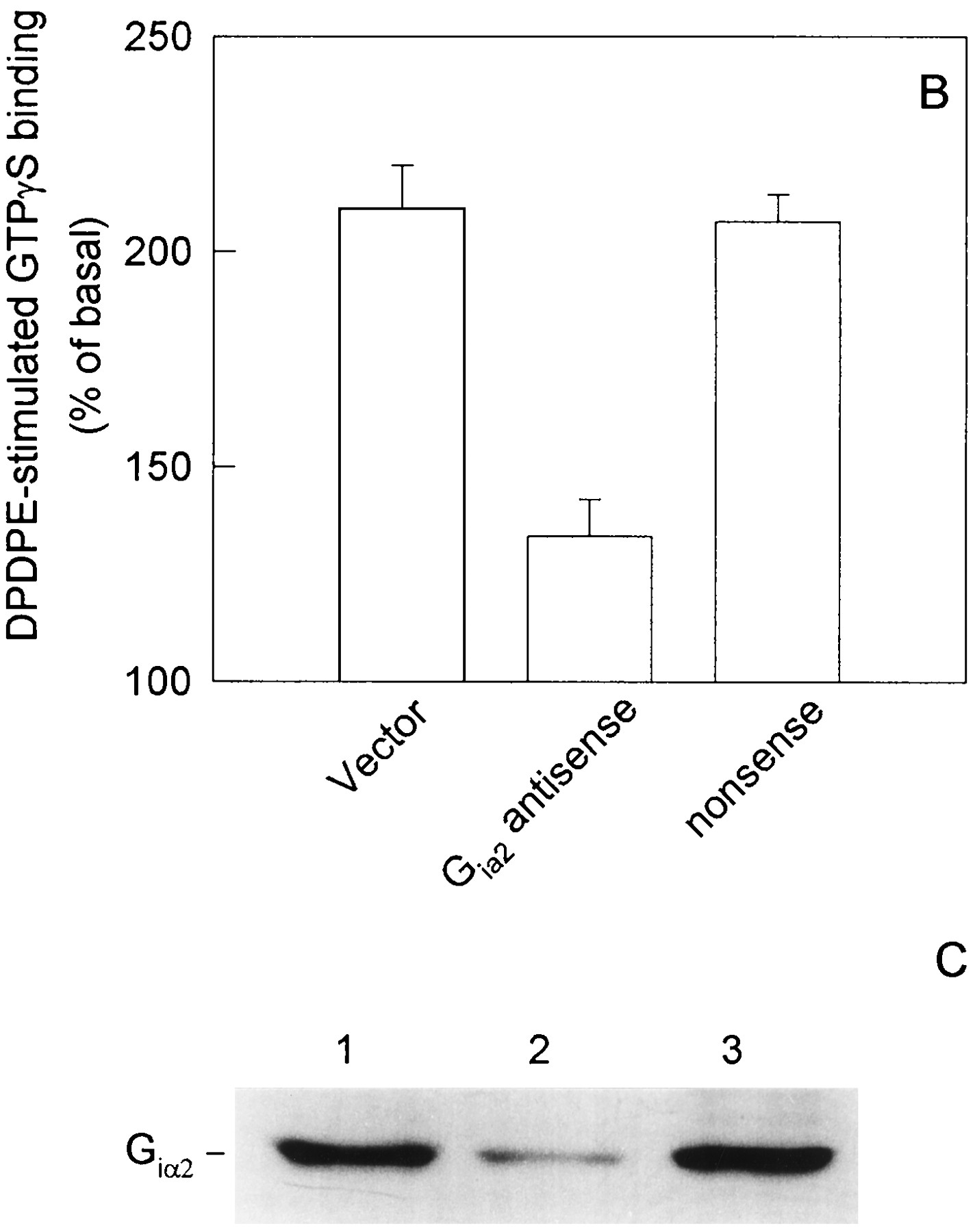
A, Abolishment by PTX treatment of DPDPE stimulation of [35S]GTPγS binding in NG108–15 cells. Cells were incubated with PTX (100 ng/ml) for 24 hr; then, the membranes were prepared by lysis. [35S]GTPγS binding assays were performed in the presence of 1 μm DPDPE at 30° for 60 min, and the samples were filtered through glass-fiber filters. B, Attenuation of DPDPE-stimulated [35S]GTPγS binding in NG108–15 cells transfected with Giα2antisense. NG108–15 cells were transfected with vector, Giα2 antisense cDNA, nonsense cDNA. At 48 hr after transfection, the cell membranes were prepared, and DPDPE-stimulated35S]GTPγS binding were performed as described in A. Data are expressed as percentages of the basal values and plotted as mean ± standard error of three separate experiments. C, Expression of Giα2 in NG108–15 cells transfected with Giα2 antisense cDNA. Cells were transfected with vector (lane 1), Giα2 antisense cDNA (lane 2), or nonsense cDNA (lane 3); then, Western blotting was performed as described in Materials and Methods.

A, Autoradiogram of phosphorylation of Giα2. NG108–15 cells express the tagged Giα2 were metabolically labeled with [32P]orthophosphate. The labeled cells were treated without (lane 1) or with (lane 2) 1 μm NMDA or with 1 μm NMDA plus 15 μm chelerythrine chloride (lane 3) or 1 μm NMDA plus 1 mm APV (lane 4). Cell extracts containing equal amount of proteins were subjected to immunoprecipitation with 12CA5 antibodies. Aliquots of the precipitates were resolved on SDS-polyacrylamide gels and subjected to phosphorimaging. B, Immunoblotting of the Giα2 content. In a parallel experiment, equal amount of the precipitates were subjected to SDS-polyacrylamide gels, followed by electrotransfer to nitrocellulose membranes and immunoblotting using antibody against Giα2. C, Quantification of [32P]phosphate incorporation in Giα2 for each lane as measured by phosphorimaging. Data are mean ± standard error of four independent experiments and expressed as percentages of the control values. ∗, p < 0.01, compared with NMDA-treated cells.
Attenuation of the NMDA effects in NG108–15 cells transfected with mutated Giα2 (Ser144Ala).
To assess a potential correlation between NMDA-induced G protein phosphorylation and the inhibitory effects of NMDA on opioid receptor function, two mutated Giα2 constructs (Ser144Ala or Ser16Ala) were generated using PCR. Transfection of both wild-type and the two mutated Giα2 constructs resulted in ∼10-fold overexpression of the proteins (data not shown), and the DPDPE responses were increased ≥60% in the transfected cells. Interestingly, the increased DPDPE responses were suppressed to the same levels by NMDA treatment in wild-type and Giα2 (Ser16Ala)-, but not Giα2 (Ser144Ala)-, transfected cells as that in control cells (Fig. 9). These results strongly support the hypothesis that phosphorylation of G proteins that couple to opioid receptors is responsible for the inhibitory effects of NMDA on the functional responses of opioid receptors.

Effect of NMDA on DPDPE-stimulated [35S]GTPγS binding in NG108–15 cells transiently transfected with wild-type and two mutated Giα2constructs [(Giα2 (S144A) and Giα2(S16A)]. Cells transfected without or with wild-type or the mutant Giα2 constructs were challenged without or with 1 μm NMDA at 37° for 15 min. Then, cell membranes were prepared, and subsequent DPDPE-stimulated [35S]GTPγS binding was performed as described in the legend to Fig. 1. Data are mean ± standard error of three independent experiments. Giα2 (Ser144Ala) represents the mutated Giα2, and Giα2 (Ser16Ala) represents the mutated Giα2. ∗, p < 0.01, compared with control.
Discussion
Based on our previous results that activation of NMDA receptor attenuated opioid receptor-mediated inhibition of adenylyl cyclase activity (Cai et al., 1997), we explored possible mechanisms for the inhibitory effects of NMDA on the functional response of opioid receptors. The fact that neither opioid binding to the receptors nor adenylyl cyclase activity was affected by NMDA treatment (Cai et al., 1997) suggests that NMDA may act on the upstream region of adenylyl cyclase in the signaling pathway of opioid receptors. In the current study, we clearly demonstrated that NMDA dose- and time-dependently attenuated DPDPE-stimulated activation of G proteins in NG108–15 cells and that NMDA also attenuated the responses mediated by μ-, δ-, and κ-opioid receptors in primarily cultured neurons. These data indicate that NMDA does attenuate opioid receptor/G protein coupling. At the concentration of NMDA used, the effects of NMDA were reversible and were blocked by the NMDA receptor antagonists, which excludes the possibility of toxic effects of NMDA on the functional responses of opioid receptors. The EC50 value of NMDA obtained in our experiment is apparently lower than that reported by other authors (Moran and Rivera, 1992), possibly due to different methods and different experiment conditions.
NMDA receptor is one type of ion channel that is highly permeable to Ca2+, and activation of NMDA receptor by its specific agonist induces Ca2+ influx in NG108–15 cells (MacDermott et al., 1986). Elevation of intracellular Ca2+ is required for activation of a family of PKC (Nishizuka et al., 1992). It is logical to hypothesize that activation of PKC via Ca2+ influx would be a prerequisite for the NMDA effects. This has been strongly supported by our current results that PKC activity was increased significantly in response to NMDA treatment, the inhibitory effects of NMDA on DPDPE-mediated activation of G proteins were blocked by PKC inhibitor but not by PKA inhibitor, and removal of extracellular Ca2+ abolished not only the inhibitory effects of NMDA but also the stimulation of PKC activity by NMDA. It can be suggested that PKC is essentially involved in the inhibitory effects of NMDA on opioid receptor-mediated G protein activation.
NMDA-mediated activation of PKC could attenuate opioid receptor/G protein coupling at the level of the receptor, G proteins, or both. Because agonist binding to DOR was not changed and phosphorylation of DOR was not observed after NMDA treatment (Cai et al., 1997), G proteins that couple to opioid receptors are a likely target of the effects of NMDA. Previous studies have shown that DOR couples to three types of G protein α subunits (Giα2, Goα2, and one isoform of Giα3) in NG108–15 cells (Roerig et al., 1992), whereas the functional response is mediated largely by Giα2 (McKenzie and Milligan, 1990). The current results further demonstrated that in NG108–15 cells transfected with an antisense Giα2 cDNA, DPDPE-stimulated G protein activation was attenuated remarkably, supporting that Giα2 is one of the major transducer of DOR, although because transfection of the Giα2 cDNA resulted in a ∼10-fold overexpression of the protein and DPDPE-stimulated G protein activation was enhanced ∼1.6-fold, it is suggested that Giα2 may not the limiting factor in promoting GTPγS binding after DPDPE stimulation.
Phosphorylation and inactivation of Giα2 by direct stimulation of PKC have been documented (Bushfield et al., 1990a, 1990b, 1990c; Strassheim and Malbon, 1994;). Metabolic labeling and immunoprecipitation of Giα2 from cell extracts provide an useful tool for us to address the role of protein phosphorylation on Giα2. Using this method, we observed phosphorylation of Giα2 in the cells after NMDA treatment and the blockade of NMDA-induced Giα2 phosphorylation by APV or chelerythrine chloride. These data indicated that NMDA-induced phosphorylation of Giα2 was mediated by NMDA receptor and through activation of PKC. It has been reported that on Giα2, Ser144, which is highly conserved in the α subunits of both Gi and Go proteins, is the major phosphorylation site of PKC (Morris et al., 1994). Transfection of the mutated Giα2 in which Ser144 was replaced by an alanine resulted in a remarkable attenuation of the inhibitory effects of NMDA on the DOR function. In contrast, in the cells transfected with another Giα2 mutant (Ser16Ala), in which Ser16 is not reported to be a PKC phosphorylation site, the inhibitory effects of NMDA were not changed significantly. Our data thus suggest a possible correlation between NMDA-induced PKC activation and attenuation of opioid-receptor-mediated activation of G proteins.
The current data demonstrated only the disruption by NMDA of DOR/G protein coupling in general. It will be of interest to determine whether NMDA treatment prevents the physical association of the G protein α with βγ subunits, the association of the heterotrimeric G protein with the DOR, or the binding of GTPγS to the receptor-bound G proteins. Although phosphorylation of Giα2may be an important step in mediating the effects of NMDA on DOR coupling, the incomplete abrogation of the effects of NMDA suggests alternate sites of regulation by NMDA beyond Ser144 on Giα2. Moreover, on the basis of the disparity between protein levels and DOR coupling when Giα2 was overexpressed as mentioned, it is postulated that Giα2 probably is not the limiting factor in promoting GTPγS binding to the cell membranes in the presence of DPDPE. It is necessary to explore further the possible involvement of other components, perhaps Gβγsubunits or Giα3 or Goα, in the effects of NMDA.
Acknowledgments
We thank Li-Zhen Jiang, Yan-Ping Wang, and Jian Qiu for technical assistance.
Footnotes
- Received July 18, 1997.
- Accepted January 12, 1998.
-
Send reprint requests to: Dr. Gang Pei, Shanghai Institute of Cell Biology, Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, People’s Republic of China. E-mail:gangpei{at}sunm.shcnc.ac.cn
-
This work was supported by research grants from National Natural Science foundation of China (39630130 and 39625015), Chinese Academy of Sciences, Shanghai Research Center of Life Sciences, Shanghai Educational Development Foundation and German Max-Planck Society.
Abbreviations
- Gi
- inhibitory G protein
- NMDA
- N-methyl-d-aspartate
- DOR
- δ-opioid receptor
- DPDPE
- [d-Pen2,d-Pen5]-enkephalin
- DAGO
- [d-Ala2,N-MePhe4,Gly-ol5]-enkephalin
- PKC
- protein kinase C
- PMA
- phorbol-12-myristate-13-acetate
- APV
- dl-amino-5-phosphonovaleric acid
- PKA
- cAMP-dependent protein kinase
- SDS
- sodium dodecyl sulfate
- PTX
- pertussis toxin
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}