


Abstract
In common with other members of the Cys-loop family of pentameric ligand-gated ion channels, 5-hydroxytryptamine type 3 receptors (5-HT3Rs) are activated by the binding of a neurotransmitter to an extracellular orthosteric site, located at the interface of two adjacent receptor subunits. In addition, a variety of compounds have been identified that modulate agonist-evoked responses of 5-HT3Rs, and other Cys-loop receptors, by binding to distinct allosteric sites. In this study, we examined the pharmacological effects of a group of monoterpene compounds on recombinant 5-HT3Rs expressed in Xenopus oocytes. Two phenolic monoterpenes (carvacrol and thymol) display allosteric agonist activity on human homomeric 5-HT3ARs (64 ± 7% and 80 ± 4% of the maximum response evoked by the endogenous orthosteric agonist 5-HT, respectively). In addition, at lower concentrations, where agonist effects are less apparent, carvacrol and thymol act as potentiators of responses evoked by submaximal concentrations of 5-HT. By contrast, carvacrol and thymol have no agonist or potentiating activity on the closely related mouse 5-HT3ARs. Using subunit chimeras containing regions of the human and mouse 5-HT3A subunits, and by use of site-directed mutagenesis, we have identified transmembrane amino acids that either abolish the agonist activity of carvacrol and thymol on human 5-HT3ARs or are able to confer this property on mouse 5-HT3ARs. By contrast, these mutations have no significant effect on orthosteric activation of 5-HT3ARs by 5-HT. We conclude that 5-HT3ARs can be activated by the binding of ligands to an allosteric transmembrane site, a conclusion that is supported by computer docking studies.
Introduction
5-hydroxytryptamine type 3 receptors (5-HT3Rs) are members of the Cys-loop family of ligand-gated ion channels (Lester et al., 2004; Nys et al., 2013), a neurotransmitter receptor family that also includes nicotinic acetylcholine receptors (nAChRs), GABAA receptors (GABAARs), glycine receptors, and invertebrate glutamate-gated chloride channels. Human 5-HT3Rs are thought to play a role in gastrointestinal function, in nociception, neurodevelopment, and in a variety of psychiatric disorders (Lummis, 2012; Engel et al., 2013; Thompson, 2013). As a consequence, they are important targets for therapeutic drug discovery.
In common with other Cys-loop receptors, 5-HT3Rs are pentameric complexes and are activated by neurotransmitter (5-HT) binding to an extracellular site, located at the interface of adjacent subunits (Thompson et al., 2010; Nys et al., 2013). The agonist binding site of 5-HT3Rs and other Cys-loop receptors, which is also the binding site for competitive antagonists, is referred to as the orthosteric binding site and has been characterized extensively (Arias, 2000). In addition, a number of compounds have been identified that modulate agonist-evoked responses by binding to distinct allosteric sites on Cys-loop receptors (Hogg et al., 2005; Davies, 2011; Yevenes and Zeilhofer, 2011).
In addition to competitive antagonists, a variety of noncompetitive antagonists (negative allosteric modulators) of 5-HT3Rs have been identified that, typically, are thought to bind in the transmembrane region (Thompson, 2013). Examples of such compounds include verapamil (Hargreaves et al., 1996), cannabinoids (Barann et al., 2002), cannabidiol (Yang et al., 2010), hydrocortisone (Corradi et al., 2011), menthol (Ashoor et al., 2013), and PU02 (6-[(1-naphthylmethyl)thio]-9H-purine) (Trattnig et al., 2012). A variety of positive allosteric modulators of 5-HT3Rs have also been identified, including alcohols (Zhou and Lovinger, 1996), 5-hydroxyindole (van Hooft et al., 1997) colchicine (de Oliveira-Pierce et al., 2009), and 5-chloroindole (Newman et al., 2013).
Studies conducted with other Cys-loop receptors have also identified a variety of allosteric ligands displaying diverse pharmacological properties. For example, a large number of positive and negative allosteric modulators of nAChRs have been identified and there is evidence that at least some of these interact with an intrasubunit transmembrane site (Young et al., 2008; Collins et al., 2011). More recently, a group of nAChR allosteric agonists have been identified that cause receptor activation in the absence of an orthosteric agonist and have been proposed to act via an intrasubunit transmembrane site (Gill et al., 2011, 2012, 2013). Similarly, there is evidence for allosteric activation of other Cys-loop receptors including GABAARs (Amin and Weiss, 1993) and glutamate-gated chloride channels (Cully et al., 1994, 1996). Here we examined the pharmacological properties of agonists acting on recombinant receptors containing the 5-HT3A subunit (homomeric 5-HT3ARs) expressed in Xenopus oocytes.
As is discussed below, there is increasing evidence that monocyclic terpenes such as carvacrol, menthol, propofol, and thymol act as allosteric modulators of several Cys-loop receptors. Typically, such compounds lack direct agonist activity but can modulate agonist-evoked responses, acting as either positive or negative allosteric modulators. In this study, we provide evidence that human 5-HT3ARs can be activated by monocyclic terpenes (carvacrol and thymol) via an allosteric transmembrane site. Carvacrol and thymol are structural isomers (see Fig. 1) and are components of naturally occurring essential oils from plants such as thyme and oregano. They have been reported to have antibacterial, anti-inflammatory, antioxidant, and vasorelaxant effects (Baser, 2008; Peixoto-Neves et al., 2010). In addition, both carvacrol and thymol are used as acaricides to control mite (Varroa destructor) infestation of honeybee hives (Damiani et al., 2009). There is evidence that carvacrol and thymol are positive allosteric modulators of both invertebrate and mammalian GABAARs (Priestley et al., 2003; García et al., 2006; Reiner et al., 2013). In addition, carvacrol has also been reported to inhibit the binding of nicotine to nAChRs via a site that is distinct from the conventional orthosteric binding site (Tong et al., 2013). Carvacrol and thymol have close chemical similarity to propofol, an intravenous anesthetic that acts as both a positive allosteric modulator and a direct activator of GABAARs (Hales and Lambert, 1991) and has been shown to interact with a transmembrane binding site in both GABAARs (Chiara et al., 2013; Yip et al., 2013) and nAChRs (Jayakar et al., 2013). In addition, propofol has been shown to inhibit 5-HT3Rs (Barann et al., 2000).

Chemical structures of the monoterpene compounds examined in this study.
In summary, we provide evidence that carvacrol and thymol are species-selective 5-HT3Rs agonists, activating human but not mouse receptors. By means of subunit chimeras and site-directed mutagenesis, we have obtained evidence indicating that carvacrol and thymol interact with a transmembrane site and act as allosteric agonists on human 5-HT3Rs.
Materials and Methods
Compounds.
Carvacrol (5-isopropyl-2-methylphenol), chlorothymol (4-chloro-2-isopropyl-5-methylphenol), linalool (3,7-dimethylocta-1,6-dien-3-ol), menthol (2-isopropyl-5-methylcyclohexanol), p-cymene [1-methyl-4(1-methylethyl)benzene], propofol (2,6-diisopropylphenol), thymol (2-isopropyl-5-methylphenol), and p-thymol (4-isopropyl-3-methylphenol) were obtained from Sigma-Aldrich (Gillingham, UK). Citral (3,7-dimethylocta-2,6-dienal) was obtained from Merck (Nottingham, UK), α-terpineol [2-(4-methyl-1-cyclohex-3-enyl)propan-2-ol] was obtained from Acros-Organics/Thermo Fisher Scientific (Loughborough, UK), and tropisetron was from LKT Laboratories (St. Paul, MN).
Plasmids.
Human 5-HT3A and 5-HT3B subunits in plasmid pcDM8 (Davies et al., 1999) were a gift from Ewen Kirkness (Institute for Genomic Research, Rockville, MD). Mouse 5-HT3A subunit cDNA (Maricq et al., 1991) was a gift from David Julius (University of California, San Francisco, CA) and was subcloned into plasmid pRK5, as previously described (Harkness and Millar, 2001). A construct containing the human α7 nAChR subunit in plasmid pSP64GL was previously described (Broadbent et al., 2006). Subunit chimeras containing the extracellular domain of either the rat or human nAChR α7 subunit, fused to the transmembrane and C-terminal domains of the mouse 5-HT3A subunit, have also been previously described (Cooper and Millar, 1998; Craig et al., 2004).
Construction of 5-HT3A Subunit Chimeras.
Human/mouse (h/m) and mouse/human (m/h) 5-HT3A subunit chimeras were constructed, which contained the extracellular N-terminal domain of the 5-HT3A subunit from one species fused to the transmembrane and intracellular domains of the 5-HT3A subunit from the other species (referred to as h/m 5-HT3A and m/h 5-HT3A subunit chimeras, respectively). The m/h 5-HT3A subunit chimera in expression vector pRK5 was constructed by using an existing BclI site in the mouse 5-HT3A subunit cDNA (immediately upstream of the first transmembrane domain) and a BamHI site that was introduced into the human 5-HT3A subunit cDNA by site-directed mutagenesis. The h/m 5-HT3A subunit chimera in expression vector pcDNA3.1 was constructed from an existing rat α7(V201)/mouse 5-HT3A subunit chimera (Cooper and Millar, 1998) and the human 5-HT3A subunit cDNA containing a BamHI site, introduced as described above. Construction of the h/m 5-HT3A chimera resulted in a codon for valine being changed to a codon for glycine at the junction of the human and mouse 5-HT3A cDNAs. Site-directed mutagenesis was performed on the h/m 5-HT3A subunit chimera to recreate the valine codon.
Site-Directed Mutagenesis and cRNA Synthesis.
Site-directed mutagenesis was performed on human and mouse 5-HT3A subunit cDNAs using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) and verified by nucleotide sequencing. The numbering of amino acids changed by site-directed mutagenesis is based on that described previously for the human 5-HT3A subunit (National Center for Biotechnology Information accession number BAA08387) and for the mouse 5-HT3A subunit (National Center for Biotechnology Information accession number NP_038589).
Xenopus Oocyte Electrophysiology.
Xenopus laevis oocytes were isolated and defolliculated, as previously described (Young et al., 2007). Expression of recombinant 5-HT3Rs was achieved by injection of either cRNA (6–12 ng) into the cytoplasm or by injection of plasmid cDNA constructs (10–30 ng) into the oocyte nuclei. In vitro synthesis of cRNA was performed using the mMessage mMachine SP6 or T7 kits (Ambion, Austin, TX). Oocytes were injected with a volume of 32.2 nl using a Drummond variable volume microinjector. Two-electrode voltage-clamp recordings were performed as previously described (Young et al., 2007). Agonists and allosteric modulators were applied to voltage-clamped oocytes using a computer-controlled gravity perfusion system (ALA Scientific Instruments, Farmingdale, NY). Typically, agonists were applied for 3–20 seconds and allosteric modulation of agonist-evoked responses was determined by preapplication of modulators for 2 minutes, followed by their coapplication with agonists.
Cell Culture and Transfection.
The mammalian cell line tsA201, derived from the human embryonic kidney 293 cell line, was obtained from Dr. William Green (University of Chicago, Chicago, IL). Cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Paisley, UK) containing 2 mM L-GlutaMAX (Invitrogen) plus 10% heat-inactivated fetal calf serum (Sigma, Poole, UK) with penicillin (100 U/ml) and streptomycin (100 μg/ml) and were maintained in a humidified incubator containing 5% CO2 at 37°C. Cells were transiently transfected with a plasmid expression vector encoding the human 5-HT3A subunit cDNA (pcDNA3) using Effectene transfection reagent (Qiagen, Crawley, UK) according to the manufacturer’s instructions. Cells were transfected overnight and assayed for expression approximately 40–48 hours after transfection.
Radioligand Binding.
Radioligand binding studies were performed with [3H]GR65630 (70.1 Ci/mmol; Perkin Elmer, Seer Green, UK), essentially as previously described (Baker et al., 2004). Membrane preparations were isolated from transfected tsA201 in 10 mM phosphate buffer (pH 7.2) containing the protease inhibitors leupeptin and aprotinin (2 μg/ml) and pepstatin (1 μg/ml). Binding experiments were performed in buffer containing 0.5% bovine serum albumin to reduce nonspecific binding. Samples were incubated with radioligand (10 nM [3H]GR65630) in a total volume of 300 μl for 2 hours at 4°C. Competition studies were carried out using [3H]GR65630 (10 nM) and varying concentrations (0.01 to 1 mM) of competing ligands (5-HT, carvacrol, or thymol). Samples were assayed by filtration onto Whatman GF/B filters presoaked in 0.5% polyethylenimine, followed by rapid washing with ice-cold 10 mM phosphate buffer using a Brandel cell harvester and levels of bound radioligand determined by scintillation counting as previously described (Baker et al., 2004).
Statistical Analysis.
Unpaired t tests were used throughout (unless otherwise stated). Equal variance between control and experimental groups was assumed.
Computer Docking Simulations.
Computer docking simulations with carvacrol and thymol were performed using AutoDock 4 (Scripps Research Institute, La Jolla, CA) (Morris et al., 1998) with a homology model of the transmembrane domain of two adjacent subunits of the human 5-HT3AR (Supplemental Material), based on the 3.5 Å resolution X-ray structure of the mouse 5-HT3AR (Hassaine et al., 2014). A grid was defined that encompassed the entire transmembrane domain of two adjacent subunits in the 5-HT3AR homology model, together with the corresponding intersubunit transmembrane region. A “blind docking” approach was employed (Hetényi and van der Spoel, 2006) in which no assumptions were made concerning where within this transmembrane region ligands might bind. Predicted Gibbs free energy of binding (ΔG) was calculated as described (Morris et al., 1998; Huey et al., 2007).
Results
Activation of 5-HT3ARs by Carvacrol and Thymol.
Two-electrode voltage-clamp recording was used to examine the pharmacological properties of a series of monocyclic terpenes (Fig. 1) on 5-HT3Rs expressed in Xenopus oocytes. Two phenolic monoterpenes (carvacrol and thymol) resulted in activation of homomeric 5-HT3Rs containing the human 5-HT3A subunit (Fig. 2). By contrast, no agonist activation was observed on human 5-HT3ARs with p-thymol (a structural isomer of carvacrol and thymol) (Fig. 2) or with any of the other monoterpenes examined (see Fig. 1 for details of all compounds tested). With mouse 5-HT3ARs, none of the compounds tested (Fig. 1), including carvacrol and thymol, had any agonist activity, indicating that the agonist effect of carvacrol and thymol on 5-HT3ARs is species selective. In contrast with the rapid activation of human 5-HT3ARs by the endogenous agonist 5-HT, activation by carvacrol and thymol occurred with a significantly slower onset (Fig. 2). Whereas responses to 5-HT reached a plateau within 0.6 ± 0.04 seconds (n = 18), activation by carvacrol or thymol was significantly slower (P < 0.001 for both compounds), reaching a plateau within 62 ± 7 seconds (n = 7) and 48 ± 3 seconds (n = 7), respectively (Fig. 2). Agonist responses evoked by 5-HT, carvacrol, and thymol were blocked completely by the orthosteric antagonist tropisetron. Long applications of 5-HT (>7 minutes) resulted in complete desensitization of human 5-HT3ARs (Fig. 3). Similarly, both carvacrol and thymol caused receptor desensitization over a similar timescale (Fig. 3). After complete desensitization of human 5-HT3ARs with 5-HT, a subsequent coapplication of either carvacrol or thymol failed to reactivate desensitized receptors (not shown).
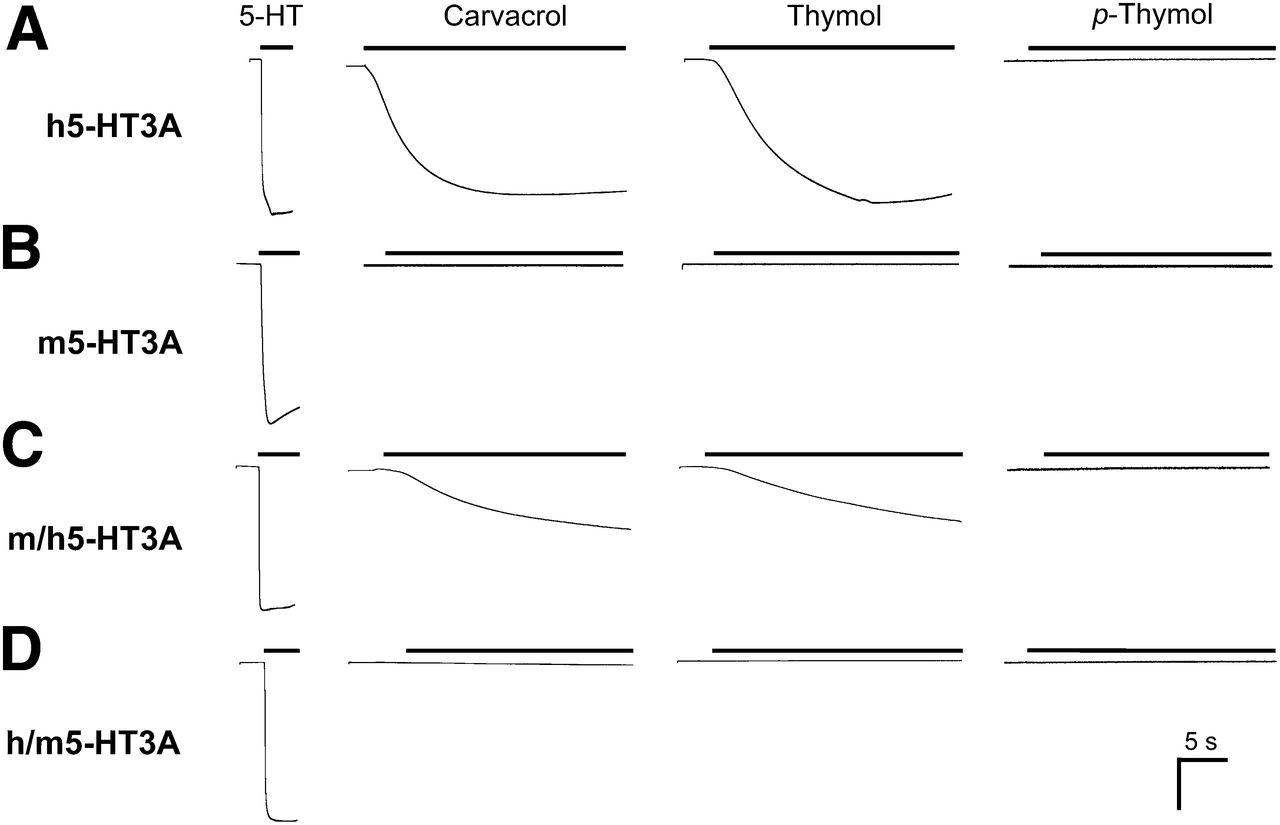
Agonist responses with human and mouse 5-HT3ARs expressed in Xenopus oocytes. Representative responses are shown with homomeric 5-HT3Rs containing the human 5-HT3A subunit (A), mouse 5-HT3A subunit (B), m/h 5-HT3A subunit chimera (C), and h/m 5-HT3A subunit chimera (D). In each case, responses are shown with 5-HT (100 μM; left), carvacrol (100 μM; left middle), thymol (100 μM; right middle), and p-thymol (100 μM; right). Bar, 1 μA in (A) and (B); 0.5 μA in (C) and (D).

Desensitization of human 5-HT3ARs. Representative traces are shown, illustrating complete desensitization of human 5-HT3ARs expressed in Xenopus oocytes in response to long applications (>7 minutes) of maximal concentrations of 5-HT (100 μM; black trace) and thymol (300 μM; red trace). Despite differences in the rates of activation, complete desensitization was observed over a broadly similar timescale (approximately 8 minutes) with 5-HT and thymol.
Potentiation of 5-HT3ARs by Carvacrol and Thymol.
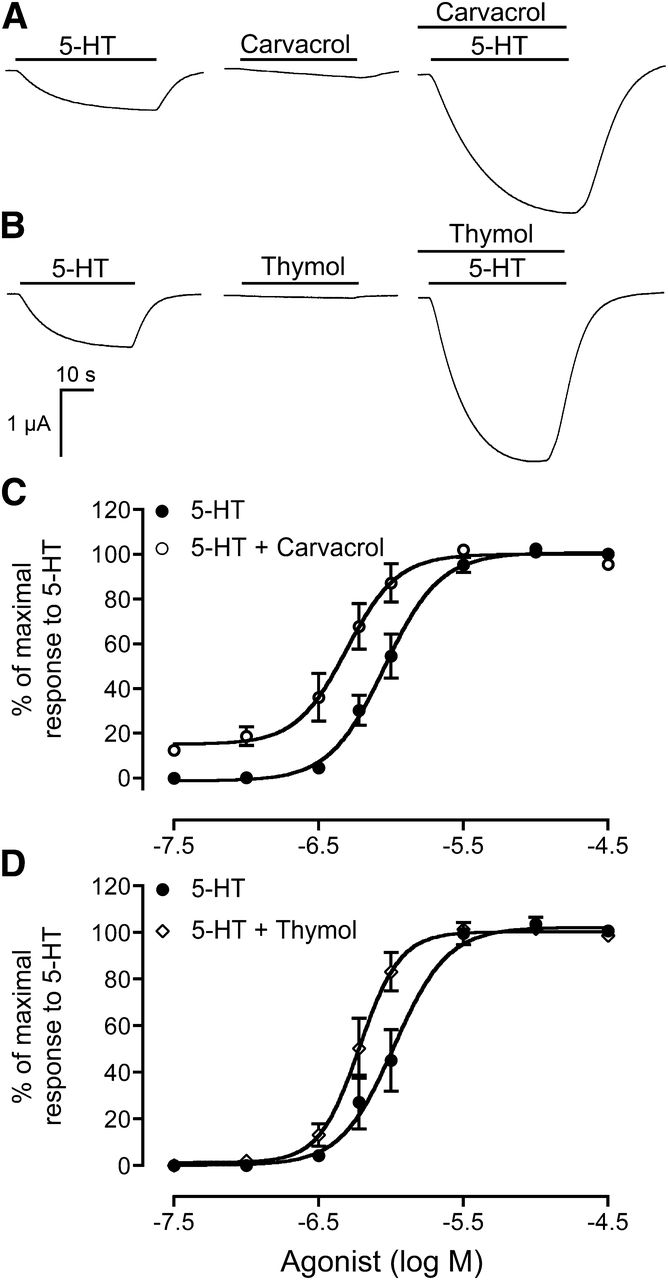
At lower concentrations (<10 μM), carvacrol and thymol did not display clear agonist activity on human 5-HT3ARs but both compounds potentiated responses to submaximal concentrations of 5-HT (Fig. 4, A and B), resulting in a leftward shift in the dose-response curve for 5-HT (Fig. 4, C and D). With an approximate EC25 concentration of 5-HT (0.6 μM), coapplication of either carvacrol (10 μM) or thymol (10 μM) to human 5-HT3ARs significantly potentiated responses evoked by 5-HT (2.6-fold ± 0.2-fold change in EC50 for 5-HT with both compounds; P < 0.001, n = 9). However, when carvacrol or thymol (at either a submaximal or maximal concentration) was coapplied with a saturating concentration of 5-HT to human 5-HT3ARs, no potentiation of the maximal current response was observed (Fig. 4). As was observed for the agonist activation, potentiation by carvacrol and thymol of responses evoked by submaximal concentrations of 5-HT was species selective. No evidence of potentiation of 5-HT–evoked responses was observed on the mouse 5-HT3AR with a range of concentrations of carvacrol or thymol (1–300 μM). Therefore, both the agonist and potentiating effects of carvacrol and thymol on 5-HT3ARs are species selective. In addition, none of the other compounds examined in this study (Fig. 1) had any activity in potentiating responses to 5-HT on either human or mouse 5-HT3ARs.

Potentiation by carvacrol and thymol of submaximal concentrations of 5-HT. (A and B) Representative traces illustrating potentiation by carvacrol (A) and thymol (B). In each case, responses are shown to an approximate EC25 concentration of 5-HT (0.6 μM; left), to either carvacrol or thymol (10 μM; middle), and to 5-HT (0.6 μM) coapplied with carvacrol or thymol (10 μM; right). (C and D) Dose-response curves for 5-HT in the absence (closed circles) or presence of 10 μM carvacrol (C; open circles) or 10 μM thymol (D; open diamonds) with human 5-HT3ARs expressed in Xenopus oocytes (means of four independent experiments). To note, the curve illustrating potentiation with carvacrol (C) has its origin at approximately 16%. This reflects the fact that carvacrol (at 10 μM) has weak agonist effects (see Fig. 5A).
Human/Mouse 5-HT3A Subunit Chimeras.
With the aim of investigating the mechanism of receptor modulation by carvacrol and thymol, artificial 5-HT3A subunit chimeras were constructed. We generated h/m and m/h subunit chimeras that contained the N-terminal extracellular domain of one subunit fused to the transmembrane and C-terminal domain of the other subunit (h/m 5-HT3A and m/h 5-HT3A subunit chimeras). Carvacrol and thymol (but not their structural isomer, p-thymol) displayed agonist activity on the m/h 5-HT3A subunit chimera (Fig. 2C) but had no agonist effect on the h/m 5-HT3A subunit chimera (Fig. 2D). This would imply that the agonist activity of carvacrol and thymol is mediated via the C-terminal (transmembrane) domain of the 5-HT3A subunit. Carvacrol displayed slightly higher potency (P < 0.001) on human 5-HT3A than on the m/h 5-HT3A chimera (Fig. 5; Table 1), whereas there was no significant difference between in the potency of thymol on these receptors. As had been observed with human 5-HT3ARs, activation of receptors containing the m/h 5-HT3A subunit chimera was significantly slower with carvacrol and thymol than when activated by 5-HT (Fig. 2, C and D). Agonist dose-response curves (with 5-HT, carvacrol, and thymol) were constructed for 5-HT3Rs containing wild-type and chimeric subunits (Fig. 5) and EC50 values are summarized in Table 1.

Agonist dose-response curves. Dose-response curves for 5-HT (filled circles), carvacrol (open circles), and thymol (open diamonds) are illustrated for homomeric 5-HT3Rs containing the human 5-HT3A subunit (A), mouse 5-HT3A subunit (B), m/h 5-HT3A subunit chimera (C), and h/m 5-HT3A subunit chimera (D). Data are means of four to five independent experiments.
Agonist effects of 5-HT, carvacrol, and thymol on wild-type, chimeric, and mutated 5-HT3Rs
Data are means ± S.E.M. Responses to carvacrol and thymol are expressed as a percentage of the maximal response to 5-HT. Significant differences between maximal responses (% of 5-HT) of mutated receptors and the corresponding wild-type receptor (mouse or human), determined by unpaired t test, are indicated
Mutagenesis of the Human 5-HT3A Subunit.
Examination of the amino acid sequences of the human and mouse 5-HT3A subunits revealed a high degree of conservation throughout the subunit, with only five amino acid differences in the four transmembrane domains (TM1–TM4; Fig. 6). Given the evidence that monocyclic terpenes bind within the transmembrane domain of other Cys-loop receptors (see Discussion for more details), together with our studies of 5-HT3A subunit chimeras, we examined the effect of mutating nonconserved amino acids within the transmembrane regions of human and mouse 5-HT3A subunits. The five nonconserved amino acids in the transmembrane regions of human 5-HT3A were mutated individually to their corresponding amino acid in the mouse subunit and their effect on agonist activation examined (Table 1). The most dramatic effect was seen with the mutation of a single methionine to a valine (M259V) in the first transmembrane domain (TM1), which resulted in the almost complete abolition of agonist activation by carvacrol and thymol (Fig. 7; Table 1). By contrast, the M259V mutation had no significant effect on activation by the orthosteric agonist 5-HT (Fig. 7; Table 1).

Amino acid sequence alignment of human and mouse 5-HT3A subunit transmembrane regions. Alignments are presented of the four transmembrane domain regions: TM1, TM2, TM3, and TM4. Amino acid differences in the transmembrane regions of the human and mouse 5-HT3A subunits are indicated by asterisks.

Characterization of human 5-HT3ARs containing a single point mutation (M259V) in TM1. (A) Representative responses are shown with 5-HT (100 μM; left), carvacrol (100 μM; middle), and thymol (100 μM; right). (B) Dose-response curves are shown for 5-HT (filled circles), carvacrol (open circles), and thymol (open diamonds). Data are means of 4–8 independent experiments.
Mutagenesis of the Mouse 5-HT3A Subunit.
Mutations were also constructed in the mouse 5-HT3A subunit with the aim of examining whether a minimal set of amino acid changes could be identified that conferred on mouse 5-HT3ARs the ability to be activated by carvacrol and thymol. A single mutation (V264M), analogous to the M259V mutation in human 5-HT3A subunit, had no significant effect on the pharmacological effects of carvacrol or thymol (Table 1). However, progressively larger agonist responses were observed with carvacrol and thymol in mouse 5-HT3ARs containing two, three, or four amino acid mutations located within the TM1 domain (Table 1). For example, a mouse 5-HT3A subunit containing just four transmembrane mutations in TM1 (A251V, V264M, C270Y, and D274N) generated a homomeric 5-HT3AR in which agonist responses to carvacrol and thymol were 32 ± 3% and 32 ± 1%, respectively, of the maximal response to 5-HT (Fig. 8).

Characterization of mouse 5-HT3ARs containing four point mutations (A251V, V264M, C270Y, and D274N) in TM1. (A) Representative responses are shown with 5-HT (100 μM; left), carvacrol (300 μM; middle), and thymol (300 μM; right). (B) Dose-response curves are shown for 5-HT (filled circles), carvacrol (open circles), and thymol (open diamonds). Data are means of 4–10 independent experiments.
Heteromeric 5-HT3A/3BRs.
Although 5-HT3A subunits form homomeric 5-HT3ARs efficiently in Xenopus oocytes, they are also able to coassemble with 5-HT3B subunits to generate heteromeric 5-HT3A/3BRs (Davies et al., 1999). Consequently, the effect of carvacrol and thymol was examined on human heteromeric 5-HT3A/3BRs, by coexpression of human 5-HT3A and 5-HT3B subunits in Xenopus oocytes. As had been found with human homomeric 5-HT3ARs, both carvacrol and thymol acted as agonists on heteromeric 5-HT3A/3BRs. However, compared with responses evoked by the endogenous agonist 5-HT, carvacrol and thymol both generated significantly smaller maximal agonist responses on the heteromeric receptor (P < 0.05 and P < 0.001, respectively; Table 1).
Radioligand Binding.
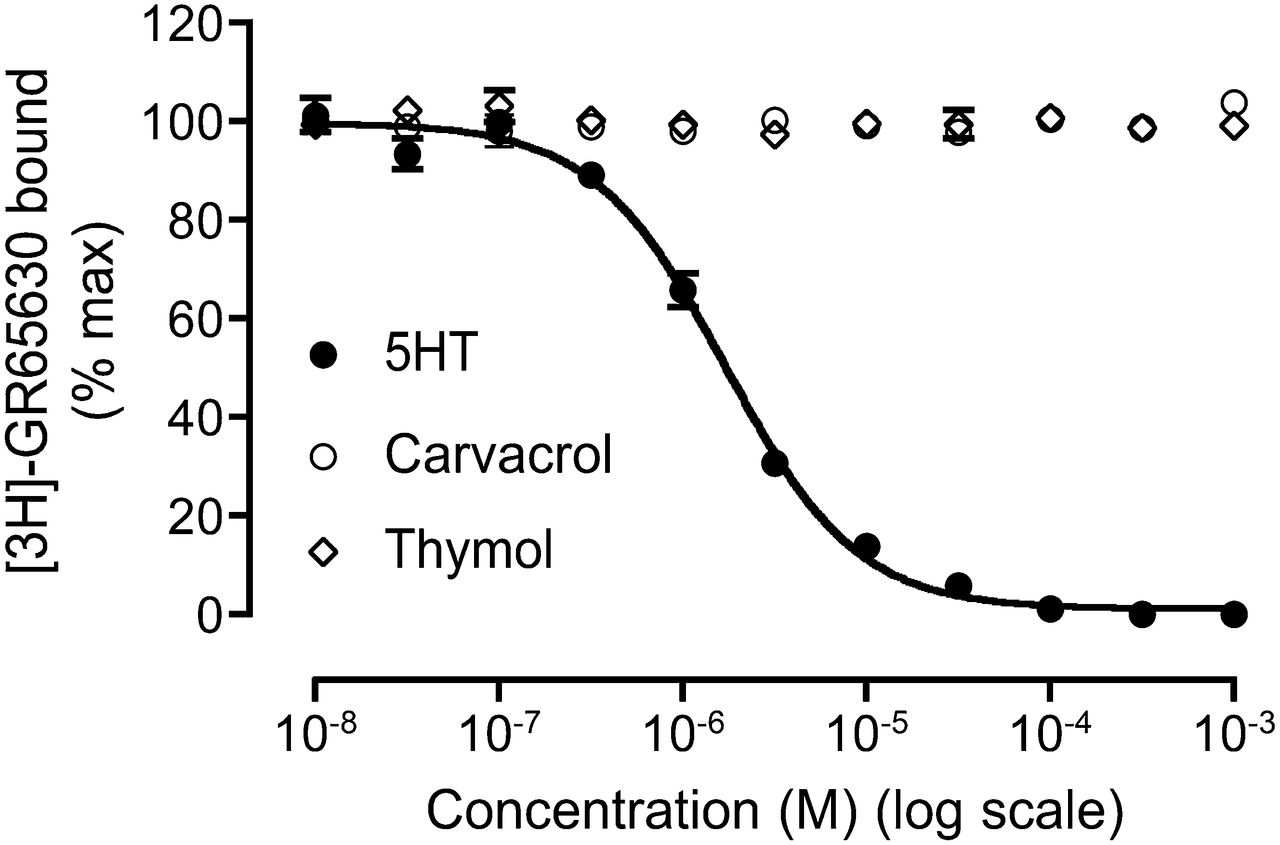
Competition radioligand binding studies were performed to examine the ability of agonists (5-HT, carvacrol, and thymol) to displace the binding of [3H]GR65630, a 5HT3R-selective ligand, from its extracellular binding site (Fig. 9). In agreement with previous binding studies (Barann et al., 2002), the orthosteric agonist 5-HT caused complete displacement of [3H]GR65630 in a concentration-dependent manner, with an IC50 of 1.70 ± 0.14 μM (n = 3). By contrast, neither carvacrol nor thymol displaced [3H]GR65630 binding, even at the highest concentrations tested (Fig. 9).
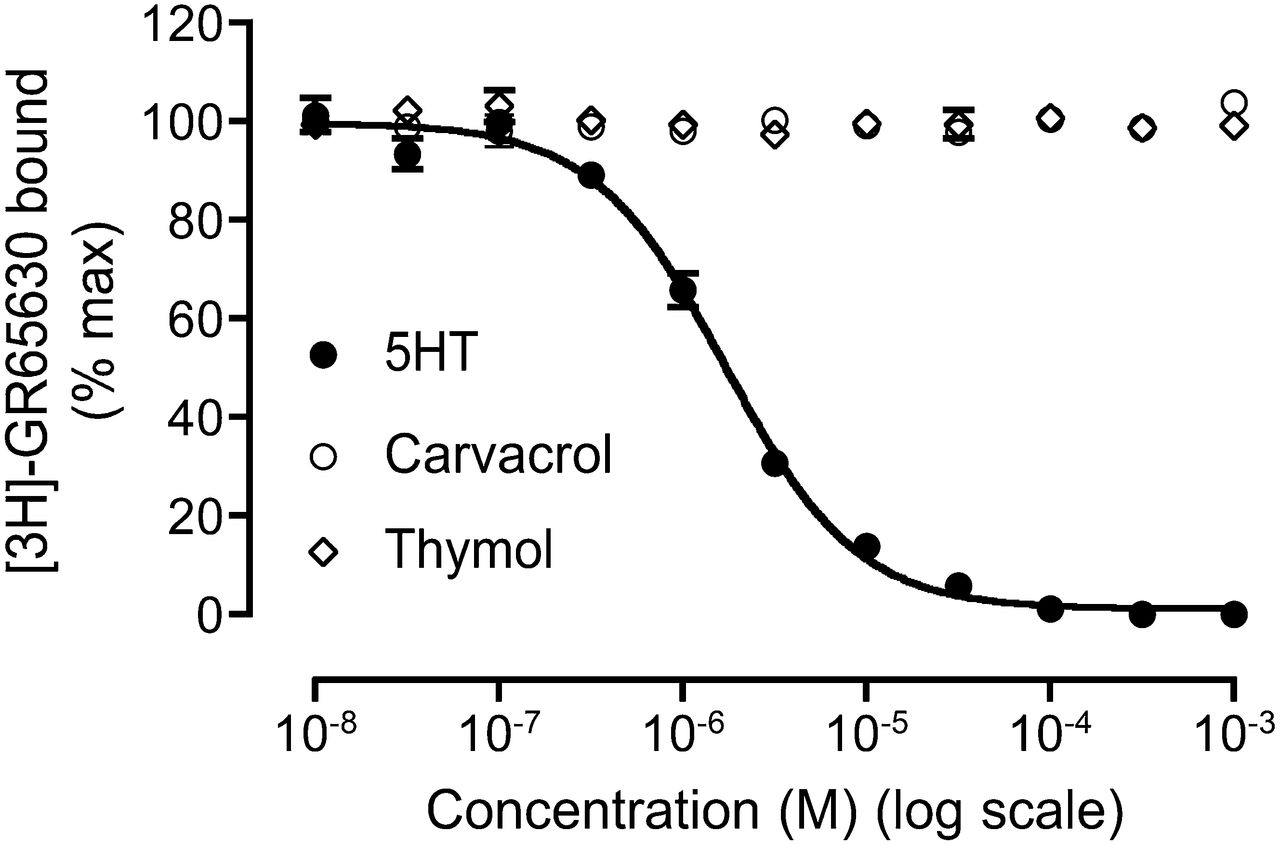
Competition radioligand binding. 5-HT caused complete displacement, in a concentration-dependent manner, of the orthosteric ligand [3H]GR65630 from its extracellular binding site. By contrast, no significant displacement of [3H]GR65630 was observed with either carvacrol or thymol. All points are means ± S.E.M. from three independent experiments.
Computer Docking.
A plausible explanation for the activation of human 5-HT3Rs by carvacrol and thymol is that this occurs via a transmembrane binding site, rather than via the conventional orthosteric binding site. Computer docking studies were performed to examine the interaction of carvacrol and thymol with a 5-HT3AR homology model. In both cases, the lowest energy docked conformations of the two ligands (−6.1 kcal/mol for carvacrol and −5.8 kcal/mol for thymol) were in very close proximity (within 6 Å) to the side chain of M259, the amino acid in TM1 of the human 5-HT3A subunit that, when mutated to valine, largely abolished agonist activation by carvacrol and thymol. The position of the lowest energy docked conformation of the two ligands was almost identical and was located in the intersubunit cavity between TM1 and TM2 helices of one subunit and the TM3 and TM4 helices of the adjacent subunit (Fig. 10).

Computer docking simulations of carvacrol and thymol into a homology model of the human 5-HT3AR. Docking studies were performed with a homology model containing the transmembrane domain of two adjacent subunits from the human homomeric 5-HT3AR. The top panel is an image showing all five subunits of the pentameric receptor, viewed from the extracellular side, in which the backbone α-helices of the two subunits that were used for computer docking studies are shaded in light blue and green. A close-up view of the predicted ligand-binding site is also shown viewed from above (middle panel) and from the side (bottom panel). The lowest energy docked conformation of two ligands (−6.1 kcal/mol for carvacrol and −5.8 kcal/mol for thymol) was in very close proximity (within 6 Å) to the side chain of M259 in TM1 of the human 5-HT3A subunit. The position of the lowest energy docked conformation of the two ligands was almost identical and was located in the intersubunit cavity between TM1 and TM2 helices of one subunit and the TM3 and TM4 helices of the adjacent subunit. The side chain of M259 is shown in yellow.
Discussion
It is well established that conventional agonists activate Cys-loop receptors, such as the 5-HT3R, by binding to an extracellular site located at the interface of two adjacent subunits (Thompson et al., 2010). This binding site for agonists and competitive antagonists is commonly referred to as the orthosteric site. However, there is increasing evidence indicating that Cys-loop receptors can be activated by allosteric ligands, interacting at sites that are distinct from the orthosteric site. For example, nAChRs can be activated by ligands binding to a transmembrane allosteric site in the absence of orthosteric agonists such as acetylcholine (Gill et al., 2011, 2012). In addition, ivermectin, a macrocyclic lactone, activates glutamate-gated chloride channels in the absence of the endogenous agonist glutamate (Cully et al., 1994, 1996) via a transmembrane binding site (Hibbs and Gouaux, 2011). Similarly, pentobarbital, a barbiturate anesthetic, causes allosteric activation of GABAARs (Amin and Weiss, 1993). In addition to being an allosteric modulator of GABAARs, propofol causes direct receptor activation at higher concentrations (Hales and Lambert, 1991). Activation by both orthosteric and allosteric agonists has also been reported for G protein–coupled receptors (Langmead and Christopoulos, 2006).
Previous studies with 5-HT3Rs have demonstrated allosteric modulation (both potentiation or inhibition) of responses evoked by orthosteric agonists, such as 5-HT (Thompson, 2013). For example, positive allosteric modulation of 5-HT3Rs has been observed with alcohols (Machu and Harris, 1994), volatile anesthetics (Machu and Harris, 1994), colchicine (de Oliveira-Pierce et al., 2009), and 5-chloroindole (Newman et al., 2013). In addition, menthol (a monoterpene) has been shown to act as an allosteric inhibitor of human 5-HT3Rs (Ashoor et al., 2013). Inhibitory effects have also been observed on human 5-HT3Rs with propofol, an intravenous anesthetic with close chemical similarity to thymol and carvacrol (Barann et al., 2008).
Thymol was previously reported to act as a positive allosteric modulator of GABAARs (Priestley et al., 2003; García et al., 2006). Similarly, carvacrol and chlorothymol have been reported to influence radioligand binding to GABAARs in a manner that is consistent with them acting as positive allosteric modulators (Reiner et al., 2013). Carvacrol has also been reported to inhibit the binding of nicotine to nAChRs via a site that is distinct from the orthosteric binding site (Tong et al., 2013). Therefore, these previous studies with Cys-loop receptors are consistent with our data from 5-HT3Rs, indicating that modulation by thymol and carvacrol occurs via an allosteric site.
Given the chemical similarity of carvacrol and thymol, it seems probable that the two compounds we have identified with allosteric agonist activity on human 5-HT3Rs and also related phenol monoterpenes, such as propofol, might interact at a broadly similar binding site on Cys-loop receptors. A binding site for propofol has been identified by photolabeling in the transmembrane domain of GABAARs (Chiara et al., 2013; Yip et al., 2013) and of nAChRs (Jayakar et al., 2013). Similarly, a transmembrane binding site for propofol has been identified in the bacterial pentameric Gloeobacter violaceus ligand-gated ion channel by means of X-ray crystallography (Nury et al., 2011). Electrophysiological studies with a variety of pentameric ligand-gated ion channels provide additional evidence that propofol interacts via a transmembrane site (Ghosh et al., 2013; Lynagh and Laube, 2014). This evidence, indicating that propofol interacts with a transmembrane binding site of pentameric receptors, is consistent with our experimental data indicating that carvacrol and thymol may interact with a transmembrane site in 5-HT3Rs. We have shown, for example, that carvacrol and thymol act as agonists of receptors containing m/h 5-HT3A subunit chimeras but not with human/mouse subunit chimeras. Carvacrol and thymol activate human 5-HT3Rs with a significantly slower onset than the endogenous orthosteric agonist 5-HT (P < 0.001). A similar difference in the onset of receptor activation has been observed with nAChRs activated by either the orthosteric agonist acetylcholine or by allosteric agonists acting via transmembrane binding site (Gill et al., 2011, 2012).
Our studies with subunit chimeras suggest that the transmembrane region may be important in mediating the agonist action of carvacrol and thymol on human 5-HT3ARs. In addition, we have found that a single point mutation in TM1 (M259V), which has no significant effect on agonist activation by 5-HT, results in the almost complete block of the agonist activity of carvacrol and thymol. It is not uncommon to find that a single mutation can abolish a pharmacological effect but a single mutation (or even a small number of mutations) may be insufficient to confer a pharmacological property. Nevertheless, we have found that a few as two mutations in TM1 can confer on mouse 5-HT3ARs the ability to be activated by carvacrol and thymol and that increasingly robust agonist activation is seen receptors containing three or four mutations in TM1.
In agreement with previous studies (Barann et al., 2002), competition radioligand binding experiments with the orthosteric ligand [3H]GR65630 demonstrated that the endogenous agonist 5-HT caused complete displacement of the radioligand in a concentration-dependent manner. By contrast, no significant displacement of [3H]GR65630 binding was observed with either carvacrol or thymol. This provides evidence that carvacrol and thymol do not bind the conventional orthosteric agonist binding site and is consistent with them acting at a distinct allosteric site.
Computer docking studies predicted that the lowest energy docked conformation of carvacrol and thymol was in very close proximity to M259, the amino acid that, when mutated, had a profound effect on agonist activation by carvacrol and thymol on human 5-HT3ARs (but had no significant effect on activation by 5-HT). We are mindful that computer docking studies using receptor homology models should be interpreted with a degree of caution. Nevertheless, the results of these docking studies are consistent with the hypothesis that carvacrol and thymol are interacting at a transmembrane site.
We found that human heteromeric 5-HT3A/3BRs were activated significantly less strongly by carvacrol and thymol than human homomeric 5-HT3ARs. Comparison of the amino acid sequence of the human 5-HT3B subunit, with that of the 5-HT3A subunit, revealed that at the position analogous to the M259V mutation in the human 5-HT3A subunit, the human 5-HT3B subunit contained a valine. In this respect, the human 5-HT3B subunit more closely resembles the mouse, rather than the human, 5-HT3A subunit and may provide an explanation for the lower efficacy of carvacrol and thymol on human homomeric 5-HT3ARs.
An interesting aspect of this study is that carvacrol and thymol were found to have species-selective effect on 5-HT3Rs (acting as agonists on human 5-HT3Rs but not on mouse 5-HT3Rs). In this respect, these findings are similar to a previous study that examined the effects of colchicine on 5-HT3Rs. Colchicine acts as a positive allosteric modulator of human 5-HT3Rs but as an inhibitor of mouse 5-HT3Rs (de Oliveira-Pierce et al., 2009). However, whereas we have obtained evidence that is consistent with thymol and carvacrol interacting with a transmembrane site, colchicine has been proposed to act via an extracellular allosteric binding site (de Oliveira-Pierce et al., 2009).
Previous studies with α7 nAChRs have identified positive allosteric modulators (PAMs) that can be classified as being either type I or type II PAMs, depending on their effects on receptor desensitization (Bertrand and Gopalakrishnan, 2007). Type I PAMs have little or no effect on receptor desensitization, whereas type II PAMs cause a marked reduction in the otherwise very rapid rate of agonist-evoked desensitization of α7 nAChRs. In addition, type II PAMs (but not type I) as well as nondesensitizing α7-selective allosteric agonists facilitate reactivation of desensitized α7 nAChRs (Young et al., 2008; Collins et al., 2011; Gill et al., 2011). The failure of carvacrol and thymol to reactivate desensitized 5-HT3Rs indicates that they have properties that are more similar those of type I PAMs of α7 nAChRs.
Our primary focus in this study was to investigate the phenomenon of agonist activation of human 5-HT3ARs by carvacrol and thymol. Our main conclusion is that these compounds are acting as allosteric agonists at a site that is distinct from the extracellular orthosteric agonist binding site for the endogenous agonist 5-HT. In addition, our data are consistent with the binding site for carvacrol and thymol being in the transmembrane domain. Although it is not the primary focus of this study, it is possible that the allosteric binding site for carvacrol and thymol could be exploited as a potential target for the development of novel receptor-selective ligands and therapeutic drugs.
In summary, a combination of factors suggests that the most plausible explanation for the agonist activation of human 5-HT3Rs by carvacrol and thymol is that this occurs via an interaction with the transmembrane domain, rather than at the conventional orthosteric binding site. The onset of activation by these compounds is significantly slower than with activation by the orthosteric agonist 5-HT, a finding that has been reported with orthosteric and allosteric (transmembrane) agonists of nAChRs (Gill et al., 2011, 2012). It is also consistent with studies obtained with chimeric and mutated 5-HT3R subunits. Computer docking simulations with a 5-HT3ARs homology model are also consistent with this interpretation and suggest that allosteric activation by carvacrol and thymol may occur via an intersubunit binding site. Interestingly, an intersubunit binding site has been proposed as the site of action of several negative allosteric modulators of 5-HT3Rs including hydrocortisone (Corradi et al., 2011) and PU02 (Trattnig et al., 2012), albeit at a site located somewhat higher in the transmembrane domain.
Authorship Contributions
Participated in research design: Lansdell, Millar.
Conducted experiments: Lansdell, Sathyaprakash, Doward.
Performed data analysis: Lansdell, Sathyaprakash, Millar.
Wrote or contributed to the writing of the manuscript: Lansdell, Millar.
Footnotes
- Received June 30, 2014.
- Accepted October 22, 2014.
This research was supported by the Research Councils UK Medical Research Council (MRC) via the Confidence in Concept Scheme [Grant MC_PC_12024]. A.D. was supported by an MRC Collaborative Award in Science and Engineering Ph.D. studentship that was cofunded by the MRC and Eli Lilly.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- GABAAR
- GABAA receptor
- 5-HT
- 5-hydroxytryptamine
- h/m
- human/mouse
- m/h
- mouse/human
- nAChR
- nicotinic acetylcholine receptor
- PAM
- positive allosteric modulators
- PU02
- 6-[(1-naphthylmethyl)thio]-9H-purine
- TM
- transmembrane domain
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}