


Abstract
The peroxisome proliferator activated receptor α (PPAR) is a member of the steroid/hormone receptor superfamily that mediates the peroxisome proliferator-dependent transcriptional activation of genes encoding several peroxisomal and microsomal enzymes as well as peroxisome proliferation. Human liver is refractory to the pathological effects of peroxisome proliferators that are seen in mice. With the use of RNase protection assays, the ratio of hepatic PPARα mRNA to β-actin mRNA was found to be 1 order of magnitude lower in humans than that observed in mice. In addition, the isolation of human cDNA for PPARα that does not encode a functional PPAR because it lacks exon 6 as a result of alternate RNA splicing suggested that this process might also diminish the expression of PPARα. RNase protection analysis of total RNA revealed the presence of splice variants lacking exon 6 at significant levels in all 10 human liver samples examined. Supershift analysis using the CYP4A6-Z peroxisome proliferator response element and antisera specific for PPARα revealed easily detectable amounts of PPARα DNA binding activity in mouse liver lysates, whereas human liver lysates contained >10-fold lower amounts of PPARα DNA binding activity. In contrast to mouse lysates, the amount of PPARα binding in human lysates was generally less than that of other unidentified proteins. These results suggest that although humans retain the coding potential for a functional receptor, the low levels of PPARα expression in liver may be insufficient to compete effectively with other proteins that bind to peroxisome proliferator response elements.
A wide range of chemicals that cause an increase in the size and number of peroxisomes and ultimately lead to hepatocarcinogenesis in rodents are collectively known as peroxisome proliferators (Moody et al., 1991; Rao and Reddy, 1987). The increase in peroxisome size and number is accompanied by increases in peroxisomal fatty acid β-oxidation and microsomal ω-hydroxylation (Sharma et al., 1988). It has been proposed that the increased levels of H2O2 produced by increased peroxisomal β-oxidation leads to DNA damage and tumor formation (Reddy and Rao, 1989). Moreover, peroxisome proliferators elicit hepatomegaly and hyperplasia, which could contribute to tumorigenesis (Moody et al., 1991; Rao and Reddy, 1987). These pathological processes are evident in mouse and rat liver but have not been observed to any significant extent in primates (Lock et al., 1989). Furthermore, unlike in rodent hepatocytes, exposure to peroxisome proliferators did not have any significant effect on the peroxisomal β-oxidation in primary cultures of human hepatocytes (Bichet et al., 1990; Blaauboer et al., 1990;Elcombe and Mitchell, 1986).
The induction of the genes encoding the rat acyl CoA oxidase and the rabbit fatty acid ω-hydroxylase (CYP4A6) by peroxisome proliferators has been shown to be mediated by a member of the steroid hormone/nuclear receptor superfamily of transcription factors known as the PPARα [individual isoforms of PPAR, THR, and RXR are designated as α, β, γ, or δ. In addition, these designations are preceded by a single letter indicating the species of origin as mouse (m) or human (h)] (Kliewer et al., 1992; Muerhoff et al., 1992; Tugwood et al., 1992). This receptor is highly conserved between such distant organisms as mouse and frog (Dreyer et al., 1992; Issemann and Green, 1990), and it has been shown to activate transcription of responsive genes by binding to PPREs as a heterodimer with RXRs (Bardot et al., 1993;Gearing et al., 1993; Keller et al., 1993;Kliewer et al., 1992). Additional members of the same subfamily as PPARα have been described for several species (PPARβ, PPARγ, and PPARδ) (Dreyer et al., 1992; Kliewer et al., 1994; Schmidt et al., 1992); however, these receptors are relatively insensitive to peroxisome proliferators and compared with PPARα are expressed at low or undetectable levels in rodent liver (Braissant et al., 1996; Kliewer et al., 1994). In addition, the targeted disruption of the mouse PPARα gene has shown that the expression of PPARα is essential for peroxisome proliferator-mediated induction of hepatomegaly, peroxisome proliferation, peroxisomal β-oxidation, and microsomal ω-hydroxylation in mouse liver (Lee et al., 1995).
Human PPARα cDNAs have been isolated (Mukherjee et al., 1994; Sher et al., 1993) that encode a functional PPARα when tested in heterologous expression studies. The human PPARα exhibits a similar, but not identical, profile of activation by peroxisome proliferators as the murine PPARα (Mukherjee et al., 1994). These findings demonstrate that humans retain the coding potential for an intact receptor. The high amino acid sequence conservation of the human receptor and its functional similarity with PPARα from other species suggest other causes for the refractory nature of human liver to the pathological effects of peroxisome proliferators. In this report, we characterize the relatively low hepatic expression levels of PPARα in humans relative to levels found in mice, a responsive species exhibiting peroxisome proliferation, and document alternative splicing of a significant fraction of human PPARα RNA resulting in the deletion of exon 6 from the PPARα mRNA and premature termination of the translation product.
Experimental Procedures
Preparation of RNA and RNase protection analysis.
Total RNA was prepared from frozen human and CD-1 mouse livers according to the acid guanidinium thiocyanate-phenol-chloroform method (Chomczynski and Sacchi, 1987). RNase protection analysis was performed as described previously (Shephard et al., 1992). The riboprobe for mPPARα corresponded to nucleotides 164–504 of the mPPARα cDNA (Issemann and Green, 1990). The riboprobe for hPPARα corresponded to nucleotides 1239–1455 of the hPPARα cDNA (Sher et al., 1993). The riboprobe for mouse β-actin was derived from a template supplied by Ambion (Austin, TX). The riboprobe for human β-actin corresponded to nucleotides 29–183 of the human β-actin cDNA. The exon 6+ riboprobe corresponded to nucleotides 881-1108 of the hPPARα cDNA (Sher et al., 1993), whereas a second probe lacking exon 6 contained nucleotides 642–724 (exon 5) and nucleotides 928-1087 (exon 7). Protected fragments were separated by electrophoresis and analyzed using a Molecular Dynamics (Sunnyvale, CA) PhosphorImager SF. The measured intensities for each fragment were corrected for their respective contents of the labeled nucleotide.
Transient transfection experiments.
The HepG2 and Huh7 cell lines were obtained from American Type Culture Collection (Rockville, MD) and maintained in DMEM (HepG2) or RPMI-1640 (Huh7) (BioWhittaker, Walkersville, MD) supplemented with 10% fetal calf serum (Gemini, Calabasas, CA). The luciferase reporter plasmid, pLuc-TK-AB (Hsuet al., 1995), as well as the expression constructs pCMV-PPARα, pCMV-PPARα-G (Muerhoff et al., 1992), pRSV-hRXRα (Mangelsdorf et al., 1990), pRSV-hPPARα (Mukherjee et al., 1994), and pSVβGal (Promega, Madison, WI) have been described previously. The reporter and expression constructs were introduced into cells cultured in DMEM through a modification of the calcium phosphate coprecipitation procedure (Sambrook et al., 1989). After 16 hr, the DNA-containing culture medium was removed, and the cells were washed twice with DMEM and then exposed to culture medium containing Wy-14,643 (pirinixic acid; 50 μm) or the equivalent volume of solvent (DMSO, 0.25% v/v final concentration), which was replaced with identical medium after 24 hr. After an additional 24 hr, the cells were harvested, washed with phosphate-buffered saline (0.01 msodium phosphate, pH 7.4, 0.15 m NaCl), and then lysed by suspension in 0.1 m potassium phosphate buffer, pH 7.8, containing 1 mm dithiothreitol and 0.05% Triton X-100 followed by three cycles of freezing and thawing. Insoluble material was removed by centrifugation, and luciferase activity was determined using a Monolight 2010 luminometer (Analytical Luminescence Laboratory, San Diego, CA). β-Galactosidase activities were determined as described previously (Muerhoff et al., 1992). The luciferase activity obtained for individual cultures was expressed relative to the β-galactosidase activity obtained for the same lysate preparation.
Preparation of liver lysates and supershift analysis.
Frozen liver sections from 3 CD-1 or 3 Balb/C mice as well as 20 human liver samples were thawed and homogenized in phosphate-buffered saline solution containing 5 mm EDTA, 1 mmdithiothreitol, 0.2 mm4-(2-aminoethyl)benzenesulfonylfluoride (Calbiochem, San Diego, CA), 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 10% glycerol. The homogenates were then sonicated for 10 sec and centrifuged at 75,000 rpm in a Beckman Instruments (Palo Alto, CA) model TL-100 ultracentrifuge for 30 min at 4°. The protein content of the supernatant was determined using the BioRad Protein Assay (Bradford assay; Hercules, CA). For supershift analysis, 30 μl of lysate was combined with 1 μg (1 μl) of poly(dI/dC) and 1 μg (1 μl) of sheared salmon sperm DNA and incubated on ice for 10 min. After the addition of 25 fmol of radiolabeled, gel-purified, double-stranded oligonucleotide, the incubation was continued on ice for 10 min, after which 1 μl of either rabbit preimmune, rabbit anti-PPARα (Hsuet al., 1995), anti-RXR serum (a gift from R. Evans, Salk Institute for Biological Studies, La Jolla, CA), rabbit anti-ARP-1 serum (a gift from S. Malik, American Cyanamid, Pearl River, NY), or mouse monoclonal anti-THRβ1 antibody (clone J52; Affinity Bioreagents, Neshanic Station, NJ) was added, and the incubation was continued for an additional 30 min. After the addition of 1 μl of loading buffer (30% glycerol, 5 mg/ml bovine serum albumin, 0.005% bromphenol blue), the reaction mixture was loaded onto a 4% acrylamide/0.05% bisacrylamide gel containing 45 mm Tris borate buffer, pH 8.0, 1 mm EDTA, and 1.25% glycerol. Electrophoresis was performed at 160 V for 90 min at 4°. The gel was then dried and analyzed using a Molecular Dynamics PhosphorImager SF. For competition experiments, a 10–200-fold excess of competing oligonucleotide was added to the reaction.
EMSAs using in vitro transcribed/translated PPARs and RXRα.
Supercoiled plasmids containing the cDNAs corresponding to mouse PPARα (Muerhoff et al., 1992), PPARγ1 (Kliewer et al., 1994), human PPARα (Mukherjeeet al., 1994), Nuc1 (PPARδ) (Schmidt et al., 1992), and RXRα (Mangelsdorf et al., 1990) were used forin vitro transcription/translation in a TNT-coupled rabbit reticulocyte lysate system (Promega) at 30° for 90 min. Lysates containing each of the PPARs (1 μl) and/or human RXRα (1 μl) were incubated for 30 min at room temperature with 10 fmol of radiolabeled, gel-purified, double-stranded probe in 10 mm Tris, pH 8.0, 150 mm KCl, 6% glycerol, 0.05% Nonidet P-40, 1 mm dithiothreitol, and 125 ng/μl poly(dI/dC) and then electrophoresed through a 4% polyacrylamide (37.5:1) gel containing 45 mm Tris borate buffer, pH 8.0, 1 mm EDTA, and 1.25% glycerol at 130 V for 100 min at room temperature. When supershift assays were performed, 1 μl of rabbit anti-PPARα serum (Hsu et al., 1995) was added to the reaction.
Human liver specimens.
Human liver samples were obtained from the Liver Tissue Procurement and Distribution System (University of Minnesota, Minneapolis, MN), the International Institute for the Advancement of Medicine (Exton, PA), and the National Diabetes Research Interchange (Philadelphia, PA). All livers were frozen in liquid nitrogen within 10 hr of death, shipped overnight on dry ice, and stored at −70° until lysates were prepared. All tissue samples seemed to be in good condition. In some cases, low or moderate degrees of fatty liver were noted as indicated (see Table 2).
Relative expression of PPARα protein in mouse and human liver lysates
Results
RNA was prepared from human and mouse liver, as well as from two human hepatoma-derived cell lines, HepG2 and Huh7, and the human breast carcinoma cell line T47D. These RNA samples were analyzed by RNase protection to determine PPARα mRNA levels. As shown in Fig.1, top, the different exposure times and relative band intensities clearly indicate that human liver contains very low PPARα mRNA levels that are approximately 1 order of magnitude lower than those observed in mouse liver. This difference is unlikely to reflect a poor recovery of mRNA from the human liver samples because the levels of β-actin mRNA are similar in the mouse and human liver samples. PhosphorImager quantification of gels revealed that relative to β-actin, Huh7 cells contain levels of PPARα mRNA that approximate those seen in human liver, whereas HepG2 cells contain 4-fold lower levels, and T47D cells had no detectable PPARα mRNA (Fig. 1, bottom). In contrast, mouse liver lysates displayed significantly greater amounts of PPARα mRNA relative to β-actin.

RNase protection analysis of mouse and human PPARα mRNA. Top, total RNA was prepared from mouse and human liver samples, and 15 μg was hybridized with32P-labeled antisense riboprobes specific for human or mouse PPARα. The samples were then subjected to RNase digestion and electrophoresed through a 6% acrylamide/8 m urea gel. The protected fragments were visualized by autoradiography.35S-Labeled 1-kb ladder was included as a molecular weight marker (STD). A protection assay of yeast total RNA (YEAST) shows complete digestion of the probe.PROBE, undigested riboprobe. EXP, autoradiographic exposure time. Bottom, the level of PPARα mRNA in each sample is expressed as a percentage of fully protected β-actin mRNA. A Molecular Dynamics PhosphorImager was used to analyze RNase protection assays of total RNA prepared from either human and mouse liver (CD-1 strain) or from two human liver-derived cell lines, HepG2 and Huh7, and from the human breast cancer cell line T47D. The radioactivity of protected probe in each assay was determined by comparison with a standard curve of undigested riboprobe. The signal obtained with the PPARα riboprobe was normalized with the signal obtained for the same sample with a β-actin riboprobe.
When Huh7 or HepG2 cells are transfected with a peroxisome proliferator responsive reporter plasmid using the acyl-CoA oxidase PPRE upstream of the thymidine kinase promoter (pLuc-TK-AB), neither cell line displayed significant peroxisome proliferator-dependent luciferase expression (Fig. 2, pRSV and pCMV). However, cotransfection with expression plasmids containing cDNA for either human PPARα or murine PPARα led to a significant increase in the expression of the reporter gene in both cell lines without the addition of peroxisome proliferator to the medium (intrinsic activation). As reported previously, both receptors exhibit peroxisome proliferator-dependent transactivation in HepG2 cells (Hsu et al., 1995; Mukherjee et al., 1994). Although mPPARα exhibited a peroxisome proliferator-dependent transactivation in Huh7 cells, the intrinsic activation of hPPARα in this cell line is too great to see a significant, additional increase of transcription in the presence of Wy-14,643. A mutant of mPPARα bearing a glycine substitution for Glu284, PPARα-E284G, exhibits a much lower intrinsic activation, which may be related to a reduced affinity for agonists (Hsu et al., 1995; Forman et al., 1997). As shown in Fig. 2, expression of this PPAR in both cell lines reduced the degree of intrinsic activation and led to a greater effect of peroxisome proliferator on reporter gene expression. These results demonstrate that these human liver derived cell lines contain insufficient amounts of PPARα to fully activate transcription of the reporter and are unable to respond measurably when exposed to added peroxisome proliferators. However, supplementation of PPARα levels with either the murine or human receptor by heterologous expression dramatically increases transactivation of the reporter.

Effect of PPARα expression on reporter gene transcription in human liver cell lines. Huh7 and HepG2 cell lines were cotransfected with pSVβGal, pLuc-TK-AB, and either pCMV (CMV), pCMV-mPPAR-E284G (mPPAR-G), pCMV-mPPARα (mPPAR), pRSV, or pRSV-hPPARα (hPPAR). After incubation with DNA for 16 hr, the cells were washed and then solvent (DMSO; □) or Wy-14,643 (▪) was added in fresh medium for an additional 48-hr incubation. Individual luciferase activities were normalized to the β-galactosidase activity determined for the same sample and were expressed relative to the value obtained in the presence of DMSO for the pCMV expression vector without a cDNA insert. Values are mean plus standard deviation obtained from three independent transfections.
The potential that alternate processing of the human PPARα RNA might further diminish PPARα expression was suggested by the isolation from a human kidney cDNA library of a cDNA (hPPARsv)2that spanned the entire coding sequence of hPPARα but lacked sequences encoding exon 2 and exon 6 (Fig.3A) based on the organization of the mouse PPARα gene (Gearing et al., 1994). Exon 2 is part of the 5′ noncoding region, whereas exon 6 encodes a segment of the receptor polypeptide between the putative DNA and ligand-binding domains. Sequence analysis indicates that a frame shift terminates translation of the peptide immediately after the zinc finger domain of the receptor and before the carboxyl-terminal extension that participates in the binding of mPPARα to PPREs (Hsu MH, Palmer CNA, Song W, Griffin KJ, and Johnson EF, manuscript in preparation).

hPPARsv cDNA and the RNase protection probe used to characterize alternative RNA processing. A, The entire coding sequence of mPPARα was used to screen a human kidney cDNA library. Several partial clones encoding hPPARα were identified. The diagram shows the structure of one cDNA (hPPARsv) that spanned the entire coding sequence of the hPPARα but lacked sequences corresponding to exons 2 and 6 when compared with the genomic structure of mouse PPARα (Gearing et al., 1994). Exon 2 is part of the 5′ noncoding region, and exon 6 encodes a segment of the receptor polypeptide between the putative DNA and ligand binding domains (DBD and LBD, respectively). The deletion of exon 6 is predicted to result in a frame shift that terminates translation of the peptide immediately after the zinc finger region of the DNA binding domain. B, Location of the exon6+ RNase protection probe relative to the exon 6 deletion.
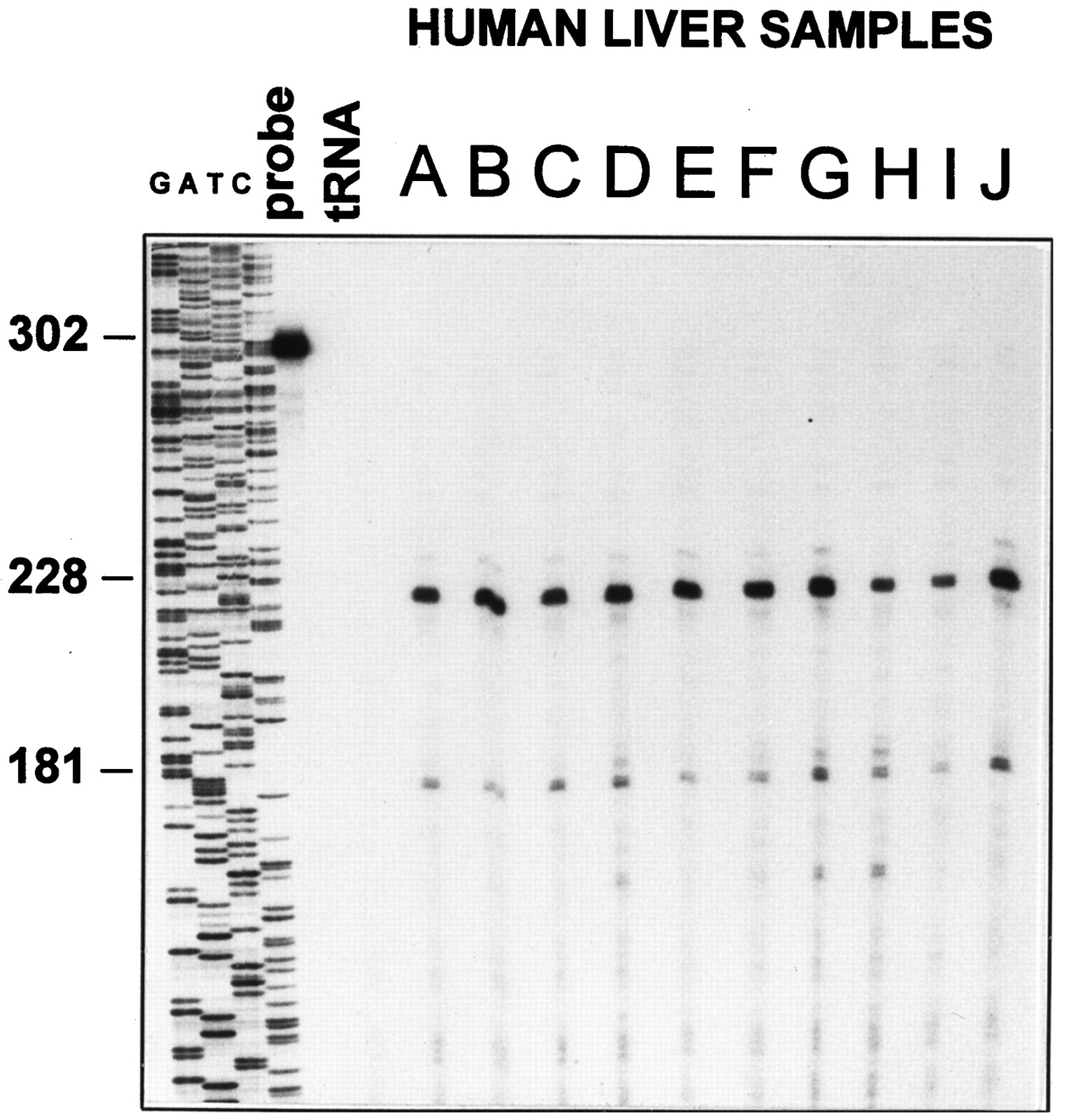
RNase protection assays were used to determine the existence and prevalence of this splicing variation for PPARα mRNA in human liver samples (Fig. 4). Total RNA was prepared from 10 human liver samples, and 30 μg of each was assayed for PPARα mRNA levels by RNase protection with a riboprobe derived from wild-type human PPARα that spans the junction of exons 6 and 7 (Fig.3B). A 228-nucleotide protected fragment is expected from RNase digestion of hybrids formed between the probe and properly processed mRNA (exon6+), and a 181-nucleotide protected fragment is expected for hybrids with splice variants that lack the portion of the probe corresponding to exon 6 (exon6−). Fragments corresponding to hybrids formed with transcripts contain exon 6 and those that do not are evident for RNA preparations from all 10 individuals (Fig. 4). Table1 summarizes the relative expression levels of both the intact and altered mRNAs in each of the human liver samples as quantified using a PhosphorImager and corrected for the respective contents of radionucleotide in each fragment. These results indicate the consistent presence of both mRNA species with a 3-fold interindividual variation in the expression level of the functional mRNA and less variation observed for the misspliced mRNA. The latter accounts for 28–42% of the protected fragments in each sample. Additional experiments (not shown) demonstrated that a probe corresponding to the sequence of the hPPARsv cDNA across the exon5/exon7 junction was protected, confirming the presence of the splice variant identified by the cDNA.

Analysis of the expression of the PPARα mRNA exon 6− splice variant. Total RNA (30 μg) from 10 human liver samples was assayed for PPARα mRNA levels by RNase protection with the exon6+ probe. The products of a sequencing reaction across the exon6+ probe using a primer corresponding to the terminus of the probe served as a reference. Left, predicted cleavage sites indicated by the expected size of each protected fragment. A 228-nucleotide band is expected for hybrids formed with the wild-type mRNA containing exon 6, and a 181-nucleotide band is predicted for hybrids formed with splice variants lacking exon 6. probe, undigested probe of 302 nucleotides. tRNA, complete digestion of the probe in the presence of yeast tRNA.
Relative expression of human liver PPARα mRNA
The low levels of PPARα in human liver lysates precluded the use of Western blots to examine the expression of PPARα protein. To increase sensitivity, human and mouse liver lysates were analyzed for binding to a 32P-labeled double-stranded oligonucleotide corresponding to the CYP4A6 Z PPRE (Palmer et al., 1995) using an EMSA. Assays were done in the absence of serum or in the presence of either preimmune, anti-mPPARα serum or antibody to hRXRs that are binding partners for PPARs. The anti-PPARα serum was raised against mouse PPARα (Hsu et al., 1995), and it also recognizes human PPARα. As shown in Fig.5, top, inclusion of PPARα antiserum greatly diminishes the amount of in vitrotranslated human or mouse PPARα/RXR complexes without appreciably affecting complexes containing NucI (PPARδ) or PPARγ, reflecting antibody specificity for the PPARα isoform. As expected, the antibody to RXR inhibits complex formation of all three PPARs (data not shown). The preimmune serum collected before PPARα antigen presentation does not affect PPARα/RXR heterodimer complex mobility.

Supershift analysis of PPARα and RXR in mouse and human liver lysates. Top, the anti-PPARα serum alters the mobility of both mouse and human PPARα/RXR (P/R) heterodimers but not those of mouse PPARγ1 or human NucI using in vitro translated proteins in an EMSA. probe, 32P-labeled CYP4A6-Z (Palmer et al., 1995). Bottom, human (K; Table 2) (225 μg of protein) and CD-1 mouse (150 μg of protein) liver lysates were analyzed for binding to32P-labeled double-stranded oligonucleotides corresponding to the CYP4A6-Z PPRE using an EMSA. Shown are assays done in the absence of serum or presence of preimmune, anti-PPARα, anti-RXR, anti-ARP-1, or anti-THRβ serum. I, Mobility of the band containing PPARα/RXR complexes. II, Unidentified complex of slower mobility formed with the CYP4A6-Z oligonucleotide.
Several electrophoretically distinct CYP4A6-Z PPRE protein complexes are evident for mouse and human liver lysates. Formation of the principal complex (Fig. 5, bottom, I) is extensively inhibited by the anti-mPPARα sera. Supershift experiments using anti-RXR antibody yielded results for complex I similar to those obtained with anti-PPARα. However, when human liver lysates were used, the RXR antibody exhibited a slightly greater inhibition in complex I formation, ≈125%, than that seen for anti-PPARα, suggesting that other RXR heterodimers, possibly including PPARγ or PPARδ, could be contributing to complex I in human liver lysates. The residual complex I that is not affected by either of the antibodies to PPARα or RXR suggests that other binding proteins form complexes with similar mobility to that of PPARα/RXR heterodimers. A second complex, II, was also detected that was not affected by anti-RXR or anti-PPARα sera. Complex II is more prominent in some human liver samples than complex I (Fig. 6, human liver samples H and R). Antibodies to nuclear receptors that recognize binding sites similar to PPREs, such as ARP-1 or THRβ (Fig. 5, bottom) as well as hepatocyte nuclear factor 4 or chicken ovalbumin upstream promotor transcription factor (data not shown), did not affect the intensity of either complex I or II.

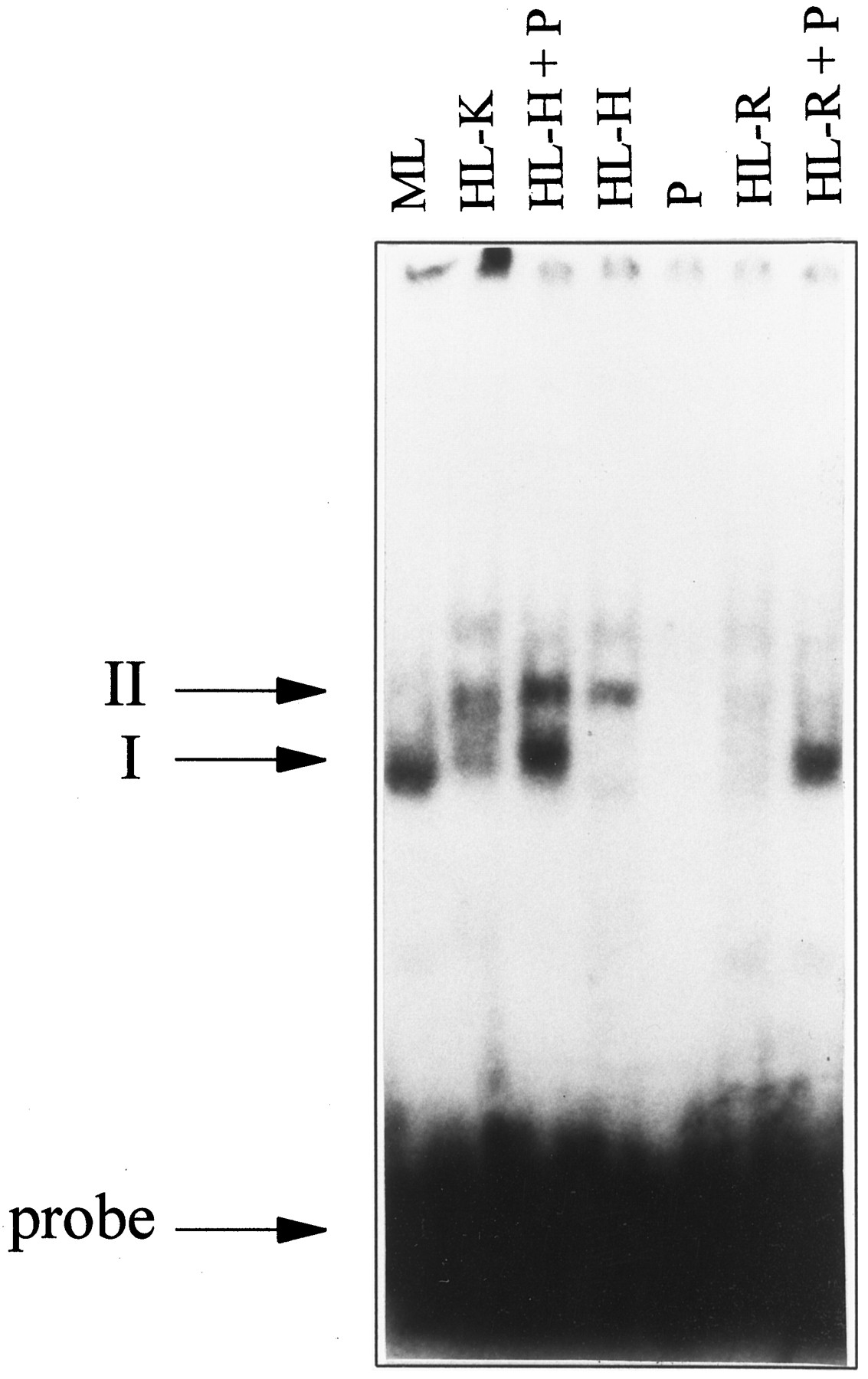
Addition of in vitro translated PPARα to human liver lysates in EMSA. In the absence of in vitro translated PPARα, CD-1 mouse liver (ML) lysate (100 μg of protein) and human liver lysate K (HL-K, 150 μg of protein) form PPARα/RXR heterodimer complexes (I) with the CYP4A6-Z PPRE. In contrast, human liver lysate H (HL-H, 100 μg of protein) and human liver lysate R (HL-R, 35 μg of protein) do not. When 1 μl of in vitro translated PPARα (P) is added to human liver lysates H and R, the intensity of the PPARα/RXR complexes is greatly increased.
The human liver lysate used in the experiment depicted in Fig. 5,bottom, exhibited the highest amount of PPARα/RXR complex of the 20 samples tested. This was determined by subtracting the intensity of complex I determined in the presence of antibody to PPARα from the intensity of the band determined in the presence of preimmune serum in the same experiment. In all experiments, intensities were measured using a PhosphoImager and were expressed relative to the intensity of complex I for human liver K determined in the presence of preimmune serum in the same experiment. As shown in Table2, ≈80% of complex I was shifted for each of six samples from mice, and the intensities of the mouse complex I band determined in the presence of preimmune serum was ≈5-fold that of the human reference, sample K (Fig. 5, bottom). The human samples showed a much greater variation. In 3 of 20 samples, significant complex formation could not be measured. In 10 of 20 samples, a significant effect of the antiserum to PPARα could not be detected (<20%), although complex I was evident. The remaining 7 samples exhibited intensities for complex I similar to that of human sample K (Fig. 5, bottom). In these samples, the antibody to PPARα diminished complex I formation by an average of 41%. Thus, in the 7 lysates in which PPARα could be detected by the assay, the amounts were ≈10-fold lower than those detected in mouse liver, and in the remainder of the samples the amount was below the level of detection (>20-fold less than mouse liver).
Competition experiments using increasing amounts of unlabeled CYP4A6-Z PPRE indicate the presence of both high and low affinity binding components in complex I (not shown). The estimated amount of the high affinity binding component is similar to the amount shifted by anti-PPARα and is not seen in samples in which the anti-PPARα does not diminish complex I, suggesting that the residual proteins in complex I are likely to bind with a lower affinity than PPARα/RXR.
To examine the competence of human liver lysates to support significant DNA binding in the presence of adequate amounts of PPARα, in vitro transcribed/translated human PPARα was added into human liver lysates H and R that displayed undetectable or low amounts of complex I for the conditions used in Fig. 6. Supplementation of these samples with hPPARα protein produced by in vitrotranscription/translation greatly increased the amount of CYP4A6-Z PPRE binding activity (Fig. 6, +P). This effect of hPPARα supplementation on binding is consistent with the transcriptional increase observed in hepatoma cells transfected with an expression vector for hPPARα to augment cellular expression of the receptor (Fig. 2). This observation further supports the hypothesis that the low levels of PPARα expression, rather than the absence or imperfection of other required accessory factors, may be insufficient to evoke the pathological effects of peroxisome proliferator exposure in human liver.
Discussion
The murine PPARα has been shown to mediate the peroxisome proliferator-dependent transcriptional activation of the genes encoding several hepatic enzymes, including the acyl CoA oxidase and fatty acid ω-hydroxylases (Kliewer et al., 1992; Muerhoff et al., 1992; Tugwood et al., 1992). Previous studies indicate that primary cultures of human hepatocytes do not display measurable peroxisome proliferator-dependent induction of these enzyme activities or peroxisome proliferation (Bichet et al., 1990;Blaauboer et al., 1990; Elcombe and Mitchell, 1986). Other investigators have demonstrated that humans retain the capacity to encode functional PPARα (Mukherjee et al., 1994; Sheret al., 1993). In this report, we have shown that human liver contains 10-fold lower levels of PPARα mRNA compared with the highly responsive mouse liver and that a fraction of this mRNA lacks exon 6 and does not encode a functional receptor. A report from a recent meeting (Tugwood et al., 1996) describes the isolation through polymerase chain reaction of several variant PPARα cDNAs from human biopsy samples, including one that lacks exon 6. Although the data were not shown, RNase protection assays were reported to detect the presence of this misspliced RNA in all 10 human liver samples that were tested, which is concordant with our results. However, the abundance of the splice variant RNA was estimated to be ≥1 order of magnitude below the level of the full-length transcript. In contrast, our results suggest higher levels for variant RNAs lacking exon 6 (Table 1). Detection of the splice variant in all of the individuals examined suggests that exon skipping is associated with the processing of the human PPARα pre-mRNA and that it does not reflect a rare allele. Exon skipping has been observed for transcripts of other genes and often leads to circular RNAs formed by the excised exons (Zaphiropoulos, 1997). However, neither the mechanisms leading to the excision of the exon and the formation of the circular RNA nor the factors that contribute to this process have been clearly defined.
Using supershift analysis of human and mouse liver lysates, we demonstrated that the CYP4A6 PPRE mainly forms complexes that contain PPARα with mouse liver lysates but not with human liver lysates. Direct comparisons of the amount of complex shifted by PPARα antibodies indicates that mouse liver lysates contain ≥1 order of magnitude more PPARα protein than human lysates. Reduced expression levels of functional human PPARα could allow PPREs to be occupiedin vivo by other nuclear receptors that bind to similar sequences, such as homodimers of RXR, ARP-1, or hepatocyte nuclear factor 4, and thus affect responsiveness to peroxisome proliferators. The lack of significant increases in smooth endoplasmic reticulum, number of peroxisomes, or enzyme induction as a result of peroxisome proliferator exposure observed in human hepatocytes is not due to an absence of functional PPARα but may reflect insufficient levels of PPARα to impose a response over other signaling pathways. This is corroborated by the finding that proteins other than PPARα contribute to the formation of complex I detected by EMSA as well as to a second electrophoretically distinct complex, II. These proteins predominate over the amount of PPARα/RXR in most human samples, and although they may bind with lower affinity, these proteins may compete more effectively with PPARα/RXR for binding to PPREs in human liver due to the relatively lower expression of PPARα compared with the levels found in mice. Supplementation of the level of PPARα in human liver lysates does confer significant PPRE binding activity, suggesting that sufficient levels of ancillary factors are present to support increased PPARα binding. Thus, under normal conditions, the low level of PPARα expression could make a primary contribution to human liver being refractory to the pathological effects of peroxisome proliferator exposure. It is not known whether PPARα expression can be induced by as-yet-unidentified factors in human liver as it is in rat liver by glucocorticoids (Lemberger et al., 1994). If so, elevated expression of hPPARα may increase the sensitivity of human liver to peroxisome proliferators.
Although the concentrations of PPARα seem to be low in human liver relative to mouse liver, the receptor is present and could modulate the expression of some genes. Fibrate drugs that are PPARα agonists are used therapeutically to lower serum triglycerides, and the induction of lipoprotein lipases and apolipoproteins are thought to contribute to these effects. PPREs have been characterized in the 5′ flanking sequences of the human lipoprotein lipase (Schoonjans et al., 1996) and apolipoprotein AII genes (Vu-Dac et al., 1995) as well as in the human peroxisomal acyl CoA oxidase gene (Varanasi et al., 1996). Although other tissues can contribute to the production of the lipoprotein lipase (Schoonjanset al., 1996), the expression of the apolipoprotein AII is largely restricted to liver and intestine (Vu-Dac et al., 1995). Fibrates have been reported to induce the expression of apolipoprotein AII in HepG2 cells as well as in primary cultures of human hepatocytes, whereas significant effects were not evident for the acyl CoA oxidase (Vu-Dac et al., 1995). As shown here, the levels of PPARα mRNAs are very low in HepG2 cells, and they are not adequate to regulate reporter gene transcription driven by the PPREs from the rat acyl CoA oxidase or P450 4A6 genes (Hsu et al., 1995). Whether these differences reflect characteristics of the PPREs or other pathways are involved is unclear. The lower levels of expression in human liver may permit PPARα to mediate some therapeutic responses to fibrates but limit the large, persistent, and extensive pathological changes that are observed in mice that involve the increased expression of a wider range genes.
Acknowledgments
We thank Dr. Dan Noonan (University of Kentucky, Lexington, KY) for providing the intact hPPARα cDNA, Dr. R. Vogel (Merck, Sharpe & Dohme Research Laboratories, West Point, PA) for providing the hNuc1 cDNA, Dr. S. Malik (American Cyanamid) for providing antiserum against ARP-1, and Dr. Ron Evans (Salk Institute for Biological Studies, La Jolla, CA) for providing the hRXRα and mPPARγ1 cDNAs and antiserum against hRXR.
Footnotes
- Received June 20, 1997.
- Accepted September 17, 1997.
-
Send reprint requests to: Eric F. Johnson, Ph.D., Division of Biochemistry, Dept. of Molecular and Experimental Medicine, The Scripps Research Institute, 10550 N. Torrey Pines Road, NX-4, La Jolla, CA 92037-9701. E-mail: johnson{at}scripps.edu
-
↵1 Current affiliation: Molecular Pharmacology Unit, Biomedical Research Center, Ninewells Hospital and Medical School, Dundee, Scotland.
-
↵2 A preliminary report of the characterization of this cDNA was presented at the 10th International Symposium on Microsomes and Drug Oxidations, Toronto, Canada, 1994.
-
This work was supported by United States Public Health Service Grants HD04445 (E.F.J.) and AA08990 (J.R.) and by the American Heart Association, California Affiliate, Postdoctoral Fellowship 93–96 (C.N.A.P.). Facilities for computer-assisted analysis and the synthesis of oligonucleotides are supported in part by General Clinical Research Center Grant M01-RR00833 and by the Sam and Rose Stein Charitable Foundation, respectively.
-
C.N.A.P. and M.-H.H. contributed equally to this work.
Abbreviations
- CoA
- coenzyme A
- ARP-1
- apolipoprotein regulatory protein 1
- PPRE
- peroxisome proliferator response element
- PPAR
- peroxisome proliferator activated receptor
- RXR
- retinoid X receptor
- THR
- thyroid hormone receptor
- DMEM
- Dulbecco’s modified Eagle’s medium
- DMSO
- dimethylsulfoxide
- EMSA
- electrophoretic mobility shift assay
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}