


Abstract
G protein activation by different μ-selective opioid agonists was examined in rat thalamus, SK-N-SH cells, and μ-opioid receptor-transfected mMOR-CHO cells using agonist-stimulated guanosine-5′-O-(γ-thio)-triphosphate ([35S]GTPγS) binding to membranes in the presence of excess GDP. [d-Ala2,N-MePhe4,Gly5-ol]Enkephalin (DAMGO) was the most efficacious agonist in rat thalamus and SK-N-SH cells, followed by (in rank order) fentanyl = morphine ≫ buprenorphine. In mMOR-CHO cells expressing a high density of μ receptors, no differences were observed among DAMGO, morphine or fentanyl, but these agonists were more efficacious than buprenorphine, which was more efficacious than levallorphan. In all three systems, efficacy differences were magnified by increasing GDP concentrations, indicating that the activity state of G proteins can affect agonist efficacy. Scatchard analysis of net agonist-stimulated [35S]GTPγS binding revealed two major components responsible for agonist efficacy differences. First, differences in theK D values of agonist-stimulated [35S]GTPγS binding between high efficacy agonists (DAMGO, fentanyl, and morphine) and classic partial agonists (buprenorphine and levallorphan) were observed in all three systems. Second, differences in the B max value of agonist-stimulated [35S]GTPγS binding were observed between DAMGO and morphine or fentanyl in rat thalamus and SK-N-SH cells and between the high efficacy agonists and buprenorphine or levallorphan in all three systems. These results suggest that μ-opioid agonist efficacy is determined by the magnitude of the receptor-mediated affinity shift in the binding of GTP (or [35S]GTPγS) versus GDP to the G protein and by the number of G proteins activated per occupied receptor.
Opioid agonists differ in their maximal ability to produce biological responses. As with other drugs that act by binding to specific receptors, agonists that produce maximal efficacy are termed full agonists, whereas those that produce less-than-maximal efficacy (at full receptor occupancy) are called partial agonists. For the superfamily of G protein-coupled receptors, of which opioid receptors are members (1-4), efficacy is primarily determined by the interaction between receptors and the G protein transducers (5). Opioid receptors produce biological responses by selectively activating G proteins of the pertussis toxin-sensitive Gi/Go family (6), which couple to effectors, including inhibition of adenylyl cyclase (7, 8), stimulation of potassium channel conductance (9, 10), and inhibition of calcium channel conductance (11, 12). In the cycle of G protein activation, the receptor interacts with the G protein and decreases the affinity of the GDP-bound α subunit for GDP and increases its affinity for GTP, thus promoting guanine nucleotide exchange (13). The receptor catalytically activates G proteins, so that each receptor can activate multiple G proteins (14-16).
Recently, a method was developed to examine receptor activation of G proteins in isolated membranes by assaying agonist-stimulated binding of the hydrolysis-resistant GTP analog [35S]GTPγS in the presence of excess GDP (17-19). Traynor and Nahorski (19) have shown that different μ-selective opioid agonists have different efficacies in stimulating [35S]GTPγS binding to SH-SY5Y cell membranes, as measured by different maximal stimulation of [35S]GTPγS binding in agonist concentration-effect curves. Differences were also found among agonists that have been previously reported to produce “full” agonist responses in isolated organ preparations [e.g., the guinea pig ileum (20)] at different levels of receptor occupancy (21). These results correlate with the results of behavioral studies, which suggest that opioid agonists of different intrinsic efficacies have the ability to produce analgesic, discriminative stimulus, and reinforcing effects at different levels of receptor occupancy (22-26). Studies using G protein antisense oligonucleotides have indicated that analgesic efficacy differences may be related to the ability of these agonists to activate G proteins (27,28). Thus, there is reasonable evidence to suggest that various opioid analgesics have different intrinsic efficacies and that these differences are likely due to the different abilities of these agonists to activate G proteins through μ-opioid receptors.
Recent studies in rat striatal membranes have determined that different types of receptors (i.e., μ-opioid, δ-opioid, and cannabinoid) exhibit different abilities to stimulate [35S]GTPγS binding under conditions of maximal receptor occupancy by full agonists (16). Comparison of the B max value of receptor binding with the B max value of agonist-stimulated [35S]GTPγS binding revealed that these differences were due to the overall catalytic amplification by receptors under these conditions (i.e., the number of G proteins activated by each receptor depended on the receptor type). On the other hand, the KD value of agonist-stimulated [35S]GTPγS binding, a measure of the inherent ability of the agonist to change the conformation of the G protein α subunit to a high affinity GTP-preferring state, was not different among the different receptor types. Because these findings were determined for full agonists in three different receptor systems, the current study was designed to use the same technique to explore whether full and partial agonists of the same receptor system (in this case, μ-opioid receptors) produce similar differences in receptor-mediated G protein activation. These experiments were performed in rat thalamus, a brain region enriched in μ-opioid receptors (29, 30); human neuroblastoma SK-N-SH cells, which are also enriched in μ-opioid receptors (31-33); and CHO cells transfected with cDNA encoding the mouse μ-opioid receptor, which express high levels of the μ receptor (mMOR-CHO cells) (34a). These experiments reveal that differences in agonist efficacy may be the result of differential abilities of these agonists both to induce a receptor-mediated high affinity GTP-binding state in receptor-coupled G proteins and to stimulate different levels of receptor-mediated catalytic activation of G proteins. These studies help to elucidate the signal transduction mechanisms underlying agonist efficacy for μ-opioid receptors.
Experimental Procedures
Materials.
[35S]GTPγS (1150–1300 Ci/mmol) was purchased from New England Nuclear (Boston, MA). SK-N-SH cells were obtained from American Type Culture Collection (Rockville, MD). mMOR-CHO cells were generously provided by Drs. Lawrence Toll (SRI International, Menlo Park, CA) and Christopher Evans (University of California, Los Angeles, CA). Ecolite scintillation fluid was obtained from ICN Biomedicals (Irvine, CA). DAMGO, naloxone, and penicillin/streptomycin were purchased from Sigma Chemical (St. Louis, MO). All nonpeptide opioid agonists were obtained from the National Institute for Drug Abuse drug supply program (Research Triangle Institute, Research Triangle Park, NC). FBS and geneticin (G-418) were purchased from GIBCO (Grand Island, NY). GTPγS and guanosine-5′-diphosphate were purchased from Boehringer-Mannheim Biochemicals (New York, NY). All other chemicals (reagent grade) were obtained from Sigma or Fisher Scientific (Fair Lawn, NJ).
Cell culture.
Cells were cultured at 37° in a humidified atmosphere of 5% CO2/95% air. SK-N-SH cells were cultured in Dulbecco’s modified Eagle’s medium containing 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS. mMOR-CHO cells were cultured in 50% Dulbecco’s modified Eagle’s medium and 50% Ham’s F-12 Nutrient Mixture containing 100 units/ml penicillin, 100 μg/ml streptomycin, and 5% FBS. Cells were harvested by replacing the media with cold phosphate-buffered saline containing 0.04% EDTA for 5 min, followed by agitation. Cells were collected by centrifugation at 345 × g for 10 min.
Membrane preparation.
Rats were killed by decapitation, and the thalamus was dissected on ice. Rat thalamic tissue, SK-N-SH cells, or mMOR-CHO cells were homogenized in 20 volumes of ice-cold 50 mm Tris·HCl, 3 mm MgCl2, 1 mm EGTA, pH 7.4 (membrane buffer) with a Tissumizer (Tekmar, Cinncinnati, OH). The homogenate was centrifuged at 48,000 × g at 4° for 10 min, resuspended in membrane buffer, centrifuged again at 48,000 × g at 4° for 10 min, and finally resuspended in the assay buffer (50 mmTris·HCl, 3 mm MgCl2, 0.2 mmEGTA, 100 mm NaCl, pH 7.4). For SK-N-SH cell membranes, this procedure was preceded by a low-speed centrifugation at 500 × g, from which the supernatant was kept and the pellet was discarded. Membrane protein levels were determined according to the method of Bradford (35).
[35S]GTPγS binding assays.
Agonist-stimulated [35S]GTPγS binding was examined by a modification of previously published methods (19, 30). Rat thalamic (10 μg of protein), SK-N-SH cell (50 μg of protein), or mMOR-CHO cell (25 μg of protein) membranes were incubated for 1 hr at 30°, with and without various drugs, in assay buffer containing 0.05 nm [35S]GTPγS and 10–30 μmGDP. Basal binding was assessed in the presence of GDP and absence of drug, whereas nonspecific binding was measured in the presence of 10 μm GTPγS. In some experiments, 0.03–50 μm GDP was used. For Scatchard analysis, 0.1 nm [35S]GTPγS was incubated with 0.05–2000 nm GTPγS in the presence of 50 μm (rat thalamus), 30 μm (SK-N-SH), or 10 μm GDP (mMOR-CHO) with and without various drugs for 1 hr (SK-N-SH and mMOR-CHO) or 2 hr (rat thalamus) at 30°. The incubation was terminated by rapid filtration under vacuum through Whatman GF/B glass fiber filters, followed by three washes with 3 ml of ice-cold 50 mm Tris·HCl, pH 7.4. Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency after overnight extraction in 5 ml of Ecolite scintillation fluid.
[35S]GTPγS binding autoradiography.
[35S]GTPγS autoradiography was performed in vitro as previously described (30). Briefly, coronal sections from male Sprague-Dawley rats were cut at the level of the thalamus on a cryostat at −20° and thaw-mounted onto gelatin-coated slides. Sections were rinsed in assay buffer at 25° and then preincubated with 2 mm GDP in assay buffer at 25° for 15 min. Sections were incubated with and without various drugs in the presence of 0.04 nm [35S]GTPγS and 2 mm GDP in assay buffer at 25° for 2 hr. Basal binding was assessed in the presence of GDP and absence of drug. Slides were then rinsed twice in ice-cold 50 mm Tris·HCl, pH 7.0, at 25° and rinsed once briefly in deionized water. Slides were dried overnight and exposed to Reflections film (New England Nuclear) for 6 days. Films were digitized with a Sony XC-77 video camera and analyzed using the NIH Image program for Macintosh computers. Images were quantified by comparison with 14C standards, and values were adjusted to 35S as previously described (36).
Data analysis.
Unless otherwise indicated, data are reported as mean ± standard error of at least three separate experiments that were each performed in triplicate. Net stimulated [35S]GTPγS binding is defined as stimulated binding minus basal binding. Percent stimulation is defined as (net stimulated binding/basal binding) × 100%. Percent maximal stimulation is defined as (net stimulated binding by agonist/by 10 μm DAMGO) × 100%. This parameter was determined within each individual experiment by inclusion of 10 μm DAMGO in each assay, and the normalized data were subjected to nonlinear regression analysis to determine ED50 and efficacy (E max) values. Statistical significance of the data was determined by analysis of variance followed by the nonpaired two-tailed Student’st test, using JMP (SAS Institute, Cary, NY). Nonlinear regression analysis of concentration-effect curves was also performed with JMP using an iterative model. Scatchard analyses were conducted using EBDA and LIGAND.
Results
Concentration-effect relationship of opioid stimulation of [35S]GTPγS binding in membranes.
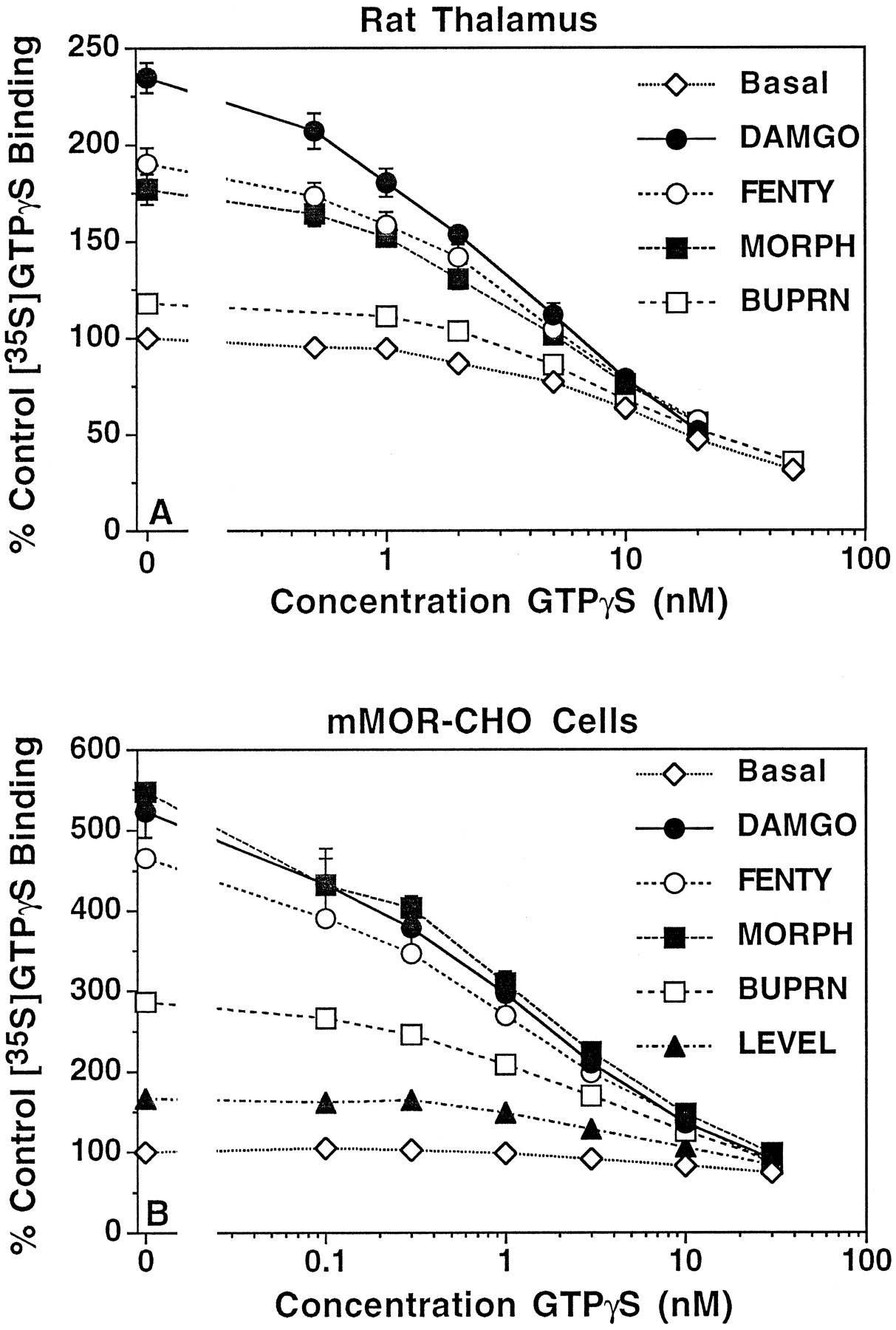
To determine the E max and ED50 values of opioid agonists for stimulation of [35S]GTPγS binding in membranes, concentration-effect curves were constructed using opioid agonists that have been reported to have different efficacies for G protein activation (19): DAMGO, fentanyl, morphine, buprenorphine, and levallorphan. Because agonist stimulation of [35S]GTPγS binding in membranes requires the presence of excess GDP, a GDP concentration (30 μm) was chosen that has previously been found to allow maximal stimulation of [35S]GTPγS binding by DAMGO in rat thalamic membranes (30). The concentration-effect curves for opioid agonist stimulation of [35S]GTPγS binding to rat thalamic membranes are shown in Fig. 1, A and B. Because DAMGO was the most efficacious agonist examined, producing an average stimulation of >165% above base-line, all agonists are presented as a percentage of the maximal stimulation produced by DAMGO (Fig. 1B). This calculation of the data was based on the inclusion of an internal “full efficacy” standard of 10 μm DAMGO in each individual experiment to control for interassy variability in the absolute stimulation. The results of these experiments showed that morphine and fentanyl were approximately equally efficacious and produced ∼55-60% of the maximal stimulation observed with DAMGO. Buprenorphine was much less efficacious and produced only ∼10% of the maximal stimulation obtained with DAMGO. Levallorphan acted as a pure antagonist in this system and produced no significant effect. The ED50 andE max values of the opioid agonists are summarized in Table 1. There was no correlation between efficacy and potency: the order of decreasing efficacy was DAMGO > fentanyl = morphine ≫ buprenorphine, whereas the order of decreasing potency (increasing ED50) was buprenorphine ≫ fentanyl > DAMGO > morphine.

Concentration-effect relationship of opioid stimulation of [35S]GTPγS binding in rat thalamic and mMOR-CHO cell membranes. Membranes prepared from (A and B) rat thalamus or (C and D) mMOR-CHO cells were incubated with 0.05 nm[35S]GTPγS, 30 μm (rat thalamus), or 10 μm (mMOR-CHO) GDP and various concentrations of DAMGO, fentanyl, morphine, or buprenorphine. Data are mean ± standard error of (A and C) net agonist-stimulated [35S]GTPγS binding or (B and D) percent of stimulation produced by 10 μm DAMGO. Basal [35S]GTPγS binding values and ED50 and E max values from curve-fitting of these data are shown in Table 1.
Efficacy and potency of opioid agonists for stimulation of [35S]GTPγS binding to rat thalamus, SK-N-SH, and mMOR-CHO membranes
Similar results were obtained in SK-N-SH cell membranes (Table 1), in which the full agonist DAMGO stimulated [35S]GTPγS binding by ∼220% above basal. Again, DAMGO was the most efficacious agonist, with fentanyl and morphine producing ∼65-75% and buprenorphine producing only ∼15% of the maximal stimulation obtained with DAMGO. The orders of efficacies and potencies of the various agonists were the same as observed in rat thalamic membranes (Table 1). Thus, although there were some quantitative differences among the potencies and the absolute and relative efficacies of these agonists in rat thalamic membranes compared with SK-N-SH membranes, the qualitative relationship among the agonists was similar in both of the μ-opioid receptor-containing preparations.
Somewhat different results were obtained in mMOR-CHO cell membranes (Fig. 1, C and D). In this system, there was no distinguishable difference among DAMGO, fentanyl, and morphine, all of which stimulated [35S]GTPγS binding by ∼400% above basal. Buprenorphine, however, still acted as a partial agonist, producing ∼40% of the stimulation obtained with DAMGO. Levallorphan was also a partial agonist in these cells, producing ∼12% of the stimulation observed with DAMGO. Potency and efficacy values of the agonists (Table1) revealed that significant quantitative differences in relative agonist efficacy were observed between mMOR-CHO cell membranes and the other two systems examined because all agonists were more efficacious relative to DAMGO than in the rat thalamic or SK-N-SH membranes. Furthermore, morphine, fentanyl, and DAMGO were more potent in SK-N-SH and mMOR-CHO cell membranes than in rat thalamic membranes.
Autoradiographic examination of opioid agonist-stimulated [35S]GTPγS binding in rat brain sections.
To confirm that membranes prepared from dissected rat thalamus actually contained the μ receptor-enriched region, and to examine the efficacy relationships among the opioid agonists in a more intact system, in vitro autoradiography of opioid agonist-stimulated [35S]GTPγS binding was examined in rat brain sections at the level of the thalamus, using maximally effective concentrations of agonists as determined from the concentration-effect curves. The results (Fig. 2) showed that all four agonists stimulated [35S]GTPγS binding with a distribution consistent with that of μ receptors (29, 36). This is best shown by DAMGO, which produced high levels of stimulation throughout the medial thalamus, medial hypothalamus, and amygdala, and produced low levels of stimulation in hippocampus and cortex. Comparison of labeling stimulated by all four agonists revealed similar efficacy relationships as observed in thalamic membranes: DAMGO > fentanyl = morphine ≫ buprenorphine. When agonist-stimulated binding in the medial thalamus was quantified by densitometry (Table2), some significant quantitative differences emerged between the results obtained in membranes and brain sections. Although buprenorphine produced approximately the same efficacy (13%) relative to DAMGO as that observed in thalamus membranes, the relative efficacies of fentanyl and morphine (37%) were much less than expected based on the results obtained in membranes.

In vitro autoradiography of basal and opioid agonist-stimulated [35S]GTPγS binding in rat brain sections at the level of the thalamus. Sections were incubated with 0.04 nm [35S]GTPγS, 2 mmGDP, and 5 μm DAMGO, 5 μm fentanyl, 10 μm morphine, or 0.03 μm buprenorphine. Images shown are from a typical experiment that was performed on triplicate (adjacent) sections and replicated three times. Values from densitometric analysis of this data are given in Table 2.
Effect of opioid agonists on [35S]GTPγS binding:in vitro autoradiographic analysis in rat thalamus
Effect of GDP on opioid agonist efficacy for stimulation of [35S]GTPγS binding.
The quantitative differences observed in the relative efficacies of the opioid agonists between rat thalamic membranes and sections suggested that a difference in assay conditions may produce different relative efficacies. The main difference in assay conditions was the GDP concentration: 30 μm GDP was used in rat thalamic membranes and 2 mm GDP was used in the autoradiography. Furthermore, similar quantitative differences were observed between relative agonist efficacies in SK-N-SH cell membranes in the current study and those previously reported in SH-SY5Y cell membranes, where 3 μm GDP was used in the assay (19). Therefore, the effect of GDP concentration on relative opioid agonist efficacy was investigated with maximal stimulatory concentrations of agonists (determined by the concentration-effect curves). The results in rat thalamic membranes showed that [35S]GTPγS binding was inhibited by GDP in both the absence and presence of opioid agonists (Fig. 3A), with 50 μm GDP inhibiting >90% of total [35S]GTPγS binding. However, the potency of GDP in inhibiting agonist-stimulated [35S]GTPγS binding was inversely proportional to the efficacy of the agonist. Net buprenorphine-stimulated [35S]GTPγS binding (not shown) was decreased half-maximally by ∼3 μm GDP, whereas DAMGO-, fentanyl-, and morphine-stimulated binding was reduced half-maximally by ∼30 μm GDP, and naloxone did not significantly stimulate [35S]GTPγS binding at any concentration of GDP. In Fig.3B, the the data are expressed as percent stimulation of [35S]GTPγS binding above basal. These results shows that for DAMGO, fentanyl, and morphine, the percent stimulation of binding increased with increasing GDP concentration. However, the percent stimulation by buprenorphine was maximal at 0.2 μm GDP. Perhaps most importantly, the relative differences in efficacy among the four agonists apparently increased with increasing GDP concentration. Similar results were obtained in SK-N-SH cell membranes (not shown), in which half-maximal inhibition of net buprenorphine-stimulated [35S]GTPγS binding was observed at 4 μm GDP and half-maximal inhibition of net-stimulated [35S]GTPγS binding with the higher efficacy agonists was obtained at ∼20 μm GDP. Again, relative efficacy differences among the agonists were magnified by increasing GDP concentrations.

Effect of GDP on basal and agonist-stimulated [35S]GTPγS binding in rat thalamic and mMOR-CHO cell membranes. Membranes prepared from (A and B) rat thalamus or (C and D) mMOR-CHO cells were incubated with 0.05 nm[35S]GTPγS and various concentrations of GDP in the presence and absence of opioid agonists. Agonist concentrations were 5 μm DAMGO, 5 μm fentanyl, 10 μm morphine, and 0.03 μm buprenorphine (rat thalamus) and 1 μm DAMGO, 1 μm fentanyl, 5 μm morphine, 0.03 μm buprenorphine, and 0.1 μm levallorphan (mMOR-CHO). Data shown are mean ± standard error of (A and C) percent total [35S]GTPγS binding in the absence of GDP or agonist and (B and D) percent stimulation by agonist over basal binding measured at each GDP concentration. Total [35S]GTPγS binding in the absence of GDP was 2174 ± 56 and 460 ± 17 fmol/mg of protein in rat thalamic and mMOR-CHO cell membranes, respectively.
The same experiment was conducted in mMOR-CHO cell membranes with similar results (Fig. 3, C and D). Again, the potency of GDP in inhibiting net agonist-stimulated binding was inversely correlated with agonist efficacy. Half-maximal inhibition of net agonist-stimulated [35S]GTPγS binding was observed at ∼2, ∼8, and ∼20 μm GDP with levallorphan, buprenorphine, and DAMGO/fentanyl/morphine, respectively. The GDP concentration also directly affected the relative agonist efficacies, measured as percent stimulation of [35S]GTPγS binding, with efficacy differences among the agonists apparently increasing with increasing GDP concentration. However, it was necessary to use lower GDP concentrations to eliminate the differences in relative agonist efficacy in this system. At 0.1 μm GDP, all agonists produced similar levels of stimulation. At GDP concentrations of >0.1 μm, significantly less stimulation was observed with levallorphan than with the other agonists. DAMGO, fentanyl, and morphine produced more stimulation than buprenorphine at GDP concentrations of >1 μm, reaching a maximum at 10–30 μm GDP. Thus, the effects of GDP on agonist efficacy were qualitatively similar among rat thalamus, SK-N-SH, and mMOR-CHO cell membranes, although some quantitative differences were evident.
Scatchard analysis of agonist-stimulated [35S]GTPγS binding.
The primary mechanism of G protein activation is the receptor-induced shift in the affinity of the G protein α subunit for guanine nucleotides: the GDP affinity is decreased, and the GTP affinity is increased. Thus, it is possible to determine whether differences in agonist efficacy are due to differences in the degree to which the agonist-occupied receptor can induce a high affinity GTP binding site on the G protein (measured as the KD value of [35S]GTPγS binding) and/or the number of G proteins activated by the agonist-occupied receptor (measured as theB max value of [35S]GTPγS binding). To examine these parameters, Scatchard analysis of basal and agonist-stimulated [35S]GTPγS binding to membranes was performed in the presence of GDP, as previously described (15).1 These experiments revealed little high affinity [35S]GTPγS binding in the absence of agonist, as demonstrated by both the homologous displacement of [35S]GTPγS binding by increasing concentrations of GTPγS (Fig. 4A) and Scatchard analysis of these data (Fig. 4B). The addition of the full agonist DAMGO produced significant stimulation of [35S]GTPγS binding above basal levels (Fig. 4A). This stimulation resulted in a biphasic Scatchard plot (Fig.4B) with a large increase in high affinity binding and no significant change in low affinity binding (Table 3), as previously observed for agonist-stimulated [35S]GTPγS binding in both SK-N-SH1 and HL-60 (15) cell membranes. Scatchard analysis of net receptor-stimulated [35S]GTPγS binding was obtained by subtraction of basal [35S]GTPγS binding from agonist-stimulated binding, according to previously published calculations (15, 16, 19).1 The resulting Scatchard plot of net-stimulated binding consisted of a single class of high affinity [35S]GTPγS binding sites (Fig. 4B, inset, and Table 3). This type of analysis is not only simpler than biphasic Scatchard modeling of the data but also statistically valid because there was no significant difference betweenKD and B maxvalues of agonist-stimulated high affinity [35S]GTPγS binding obtained from this monophasic Scatchard analysis of net agonist-stimulated [35S]GTPγS binding and those obtained by biphasic Scatchard analysis of absolute agonist-stimulated [35S]GTPγS binding (Table 3).1
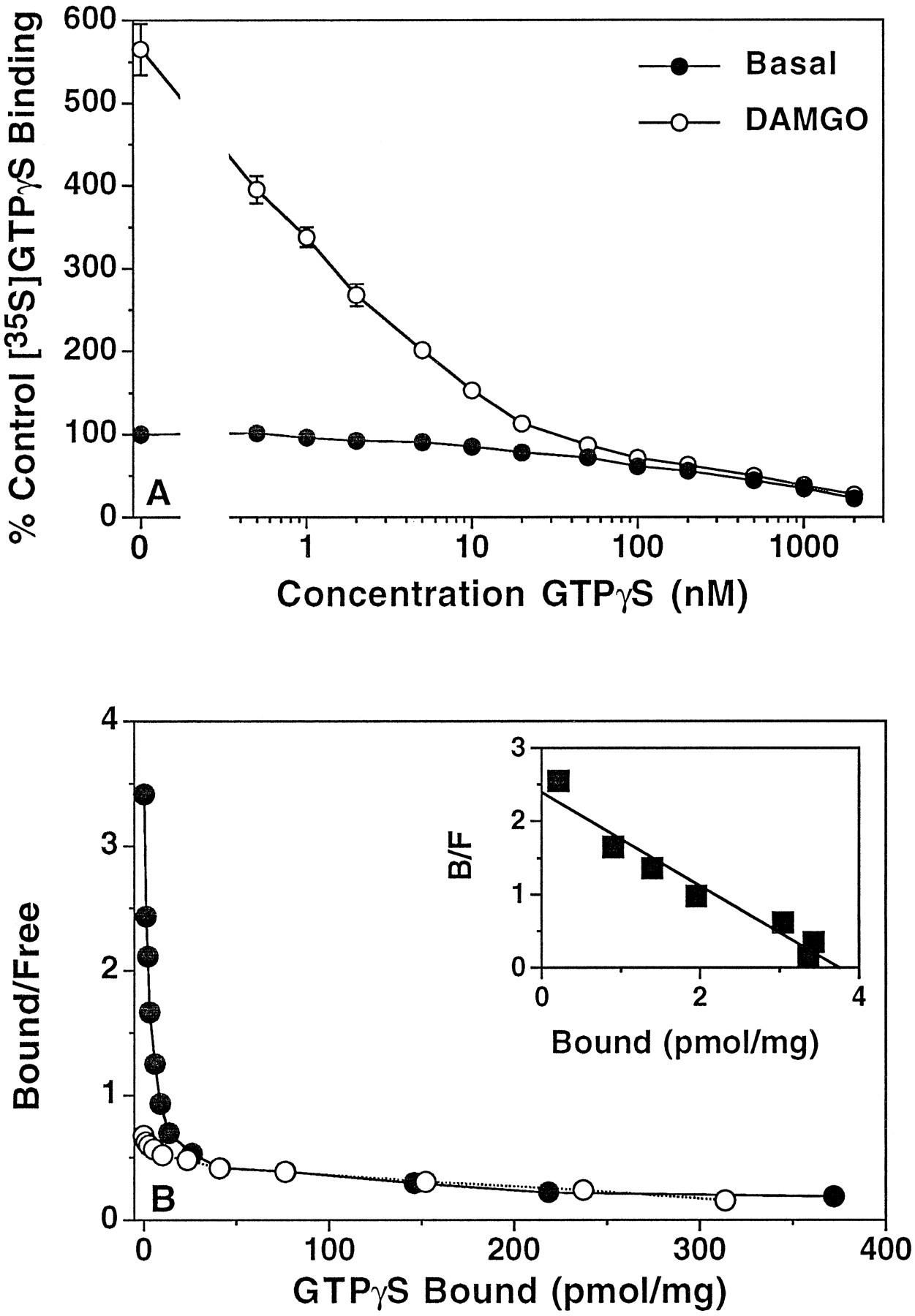
Homologous displacement and Scatchard analysis of [35S]GTPγS binding in mMOR-CHO cell membranes. Membranes were incubated with 0.1 nm[35S]GTPγS, 10 μm GDP, and 0.5–2000 nm unlabeled GTPγS in the presence and absence of 5 μm DAMGO. Data shown are (A) mean percent of control [35S]GTPγS binding (binding measured in the absence of agonist or unlabeled GTPγS) ± standard error and (B) Scatchard analysis of basal and agonist-stimulated [35S]GTPγS binding calculated from a typical experiment that was performed in triplicate and replicated four times. Inset, Scatchard analysis of net DAMGO-stimulated [35S]GTPγS (stimulated minus basal binding measured at each concentration of GTPγS) calculated from a typical experiment that was performed in triplicate and replicated four times. Control [35S]GTPγS binding was 61.1 ± 1.4 fmol/mg of protein.K D andB max values from Scatchard analyses are given in Table 3.
Scatchard analysis of basal and DAMGO-stimulated [35S]GTPγS binding to mMOR-CHO cell membranes
Scatchard analysis of net agonist-stimulated [35S]GTPγS binding was used to compare opioid agonists of different efficacies in all three systems. In rat thalamic membranes, homologous displacement of [35S]GTPγS binding by GTPγS (Fig.5A) showed that DAMGO produced greater stimulation of [35S]GTPγS binding than fentanyl or morphine at GTPγS concentrations of ≤1 nm and ≤2 nm(p < 0.05), respectively. All three of the high efficacy agonists produced greater stimulation than buprenorphine at GTPγS concentrations of ≤5 nm(p < 0.05). Scatchard analysis of net agonist-stimulated binding (Table 4) showed thatKD values produced by DAMGO, fentanyl, and morphine were significantly different from that produced by buprenorphine (p < 0.05). TheKD value obtained in the presence of morphine was also significantly, but very slightly, different from that observed in the presence of DAMGO. Analysis ofB max values (Table 4) showed that buprenorphine stimulated a significantly lower number of [35S]GTPγS binding sites than all three of the higher efficacy agonists, DAMGO, fentanyl, and morphine (p < 0.05). DAMGO apparently stimulated a higher number of [35S]GTPγS binding sites than morphine (p < 0.05), although the fentanyl-stimulated B max value was not statistically different from that for DAMGO.
Homologous displacement of [35S]GTPγS binding in rat thalamic and mMOR-CHO cell membranes. Membranes prepared from (A) rat thalamus or (B) mMOR-CHO cells were incubated with 0.1 nm[35S]GTPγS and 50 μm (rat thalamus) or 10 μm (mMOR-CHO) GDP and 0.1–50 nm unlabeled GTPγS in the presence and absence of opioid agonists. Agonist concentrations were 5 μmDAMGO, 5 μm fentanyl, 10 μmmorphine, and 0.03 μm buprenorphine (rat thalamus) and 1 μm DAMGO, 1 μm fentanyl, 5 μm morphine, 0.03 μmbuprenorphine, and 0.1 μm levallorphan (mMOR-CHO). Data are mean percent of control [35S]GTPγS binding (binding measured in the absence of agonist or unlabeled GTPγS) ± standard error. Control [35S]GTPγS binding was 313 ± 30 and 68.9 ± 3.8 fmol/mg of protein in rat thalamic and mMOR-CHO cell membranes, respectively.K D andB max values from Scatchard analyses of these data are shown in Table 4.
Scatchard analysis of net agonist-stimulated [35S]GTPγS binding to rat thalamus, SK-N-SH, and mMOR-CHO membranes
Scatchard analysis of net agonist-stimulated binding data in SK-N-SH cell membranes (Table 4) also revealed that buprenorphine-stimulated [35S]GTPγS binding displayed significantly higherKD and lowerB max values (p < 0.05) than those stimulated by DAMGO, fentanyl, and morphine. In contrast, the three high efficacy agonists (DAMGO, fentanyl and morphine) all produced similar KD values but differed only in their B max values. Although theB max values produced by DAMGO and morphine were not statistically different, there was a significant difference between the B max values of [35S]GTPγS binding stimulated by DAMGO and that obtained with fentanyl (p < 0.05). Thus, differences in μ-opioid agonist efficacy for G protein activation in rat thalamic and SK-N-SH cell membranes seemed to be due to differences in both the increase in the apparent [35S]GTPγS affinity produced by the agonist-occupied receptor and in the catalytic activation of G proteins (B max) by the agonist-occupied receptor.
In mMOR-CHO cell membranes, homologous displacement of [35S]GTPγS binding by unlabeled GTPγS revealed that DAMGO-, fentanyl-, and morphine-stimulated binding was significantly greater than that stimulated by buprenorphine at GTPγS concentrations of ≤3 nm (p < 0.05) and greater than levallorphan-stimulated binding at GTPγS concentrations of ≤10 nm (p < 0.05). Buprenorphine-stimulated [35S]GTPγS binding was also greater than levallorphan-stimulated binding at GTPγS concentrations of ≤3 nm (p < 0.05). Scatchard analysis (Table 4) showed that KD values for net-stimulated [35S]GTPγS binding with DAMGO, fentanyl, and morphine were significantly lower than those obtained with either buprenorphine or levallorphan, whereas buprenorphine produced a significantly lowerKD value than levallorphan. Similarly, B max values obtained with DAMGO, morphine, and buprenorphine were significantly higher than that obtained with levallorphan. The B max value produced by fentanyl was intermediate between that obtained with levallorphan and those obtained with the other higher efficacy agonists but was not statistically different from that for either DAMGO or levallorphan. Taken together with the results obtained in rat thalamus and SK-N-SH cells, these results indicate that the relative differences in affinity of agonist-stimulated [35S]GTPγS binding observed between low and high efficacy agonists are tissue independent, whereas the B max differences observed among the various agonists are dependent on both the agonist and the system in which it is acting. Nevertheless, differences in agonist efficacy in all systems examined seemed to be dependent on both the affinity with which activated G proteins bind [35S]GTPγS and the number of G proteins catalytically activated by the agonist-occupied receptor.
Discussion
The results of the current study confirm those of Traynor and Nahorski (19), demonstrating that opioid agonists of various reported intrinsic efficacies have different abilities to maximally stimulate [35S]GTPγS binding to μ receptor-containing cell membranes. The results of the current study also show that similar efficacy differences for stimulation of [35S]GTPγS binding are observed both in membranes prepared from rat thalamus and (by [35S]GTPγS autoradiography) in rat brain sections at the level of the thalamus. The finding that qualitatively similar results have been obtained in three different tissues, SH-SY5Y (19) and SK-N-SH neuroblastoma cells, as well as rat thalamus, indicates that these efficacy differences are a property of the agonist/receptor interaction and not of the system under investigation. However, the quantitative differences observed among the different systems indicate that the tissue and/or conditions under which the experiment is conducted will determine how these efficacy differences are expressed in terms of a functional response. For example, the effect of GDP concentration on the magnitude of the differences in agonist efficacy that were observed in the [35S]GTPγS binding assays are quite profound. The finding that efficacy differences were magnified by increasing GDP concentrations was also an indication that there was a guanine nucleotide affinity component involved in the determination of agonist efficacy. These results demonstrated that agonists of higher efficacy were better able to overcome the driving force of excess GDP to “turn off,” or prevent the “turn on,” of receptor-activated G proteins. The catalytic activation component of agonist efficacy may also be affected by GDP concentration: Because GDP stabilizes G proteins in the inactive, holotrimeric state (37, 38), it is possible that the catalytic rate of receptor/G protein activation is slowed by excess GDP. Agonists of higher efficacy may be less affected by this stabilizing effect of GDP in their ability to catalytically activate G proteins. It is also clear that the receptor density ratio is an important factor in the determination of both absolute and relative agonist efficacy, as demonstrated by the results obtained in mMOR-CHO cell membranes, which contain an overabundance of μ-opioid receptors compared with neural tissues (34).
The mechanisms of μ-opioid agonist efficacy were explored in this study using Scatchard analysis of agonist-stimulated [35S]GTPγS binding. However, there are both advantages and disadvantages to this type of analysis. It is important to note that Scatchard analysis of [35S]GTPγS binding actually measures the competition of a radiolabeled ligand ([35S]GTPγS) for a nonlabeled ligand (GDP) under nonequilibrium conditions. These conditions are necessary to measure agonist-stimulated binding; at equilibrium, the [35S]GTPγS would displace as much of the GDP as it is capable of displacing, and no agonist-stimulated binding could then be observed. Therefore, this type of analysis is not quantitatively accurate in the sense that a given B maxrepresents the exact maximal number of G proteins that are capable of being activated by the agonist-occupied receptor. However, this type of analysis is useful for relative comparisons. Previous studies have demonstrated the usefulness of Scatchard analysis of [35S]GTPγS binding to examine catalytic activation of G proteins (15), differences in the ability of different receptor types to catalytically activate G proteins (16), and changes in the catalytic activation of G proteins during agonist-induced receptor desensitization.1 Thus, Scatchard analysis of [35S]GTPγS binding is useful for examining relative measurements of agonist activity, such as with agonists of different efficacies, as in the current study. One problem with using this type of analysis to compare agonists of different efficacies is that the Scatchard analysis was performed at high GDP concentrations, which maximizes efficacy differences among the different agonists but considerably reduces [35S]GTPγS binding and potentially decreases the accuracy of B max determinations. Nevertheless, significant differences betweenB max values were obtained with DAMGO versus morphine or fentanyl in either rat thalamus or SK-N-SH cell membranes, which validates the conclusion that differences in catalytic activation of G proteins contribute to the determination of agonist efficacy (see below).
Scatchard analysis of basal and agonist-stimulated [35S]GTPγS binding confirmed that agonists of higher efficacy produced a higher affinity GTP-binding state, presumably in the guanine nucleotide binding site of μ receptor-coupled G protein α subunits, than agonists of lower efficacy. This was most evident when comparing the agonist of lowest efficacy, buprenorphine, with the agonist of highest efficacy, DAMGO, for which significant differences in the agonist-stimulated [35S]GTPγS binding affinity were observed in both SK-N-SH and rat thalamus membranes. Among the agonists of higher efficacy, DAMGO, fentanyl, and morphine, differences in the agonist-induced guanine nucleotide affinity shifts were less evident. However, differences in catalytic activation of G proteins, as measured by the agonist-stimulated B max of [35S]GTPγS binding, were apparent among the higher efficacy agonists. Although the differences betweenB max values obtained with fentanyl and morphine versus those obtained with DAMGO were relatively small (15–30% lower than DAMGO) compared with the differences between DAMGO and buprenorphine (50–60% lower), statistical significance was achieved with at least one of the two agonists in both SK-N-SH and rat thalamus membranes. Thus, opioid agonist efficacy seems to be determined by at least two signal transduction components: differences in the ability to promote receptor-stimulated guanine nucleotide exchange on the G protein α subunits (KD ) and differences in the number of G proteins activated per occupied receptor (B max). These data suggest that there are actually three classes of agonists: 1) full agonists (e.g., DAMGO), which induce a maximally high affinity GTP binding state and also catalytically activate a maximal number of G proteins; 2) classic partial agonists (e.g., buprenorphine and levallorphan), which induce a lower affinity GTP binding state and (possibly by virtue of the lower probability that a given G protein will bind GTP and thus become activated) also catalytically activate a lower number of G proteins; and 3) mixed full/partial agonists (high efficacy partial agonists; e.g., fentanyl and morphine), which induce the same high affinity GTP binding state as full agonists but may catalytically activate a lower number of G proteins. This hypothesis predicts that classic partial agonists, such as buprenorphine and levallorphan, would be partial agonists in any cell type but that the efficacy of the mixed full/partial agonist, such as morphine, would depend on additional tissue-specific factors, such as the ratio of receptors to G proteins. In systems where the receptor number is high relative to the pool of G proteins available for activation by the receptor, such agonists may seem to be full agonists.
This hypothesis was confirmed by the experiments performed in mMOR-CHO cell membranes. These cells have been reported to contain μ receptors at a density of 7.5 pmol/mg of membrane protein (34) compared with 0.15 pmol/mg in SK-N-SH cell membranes1 and ∼0.1 pmol/mg in rat thalamic membranes (39). In membranes from mMOR-CHO cells, DAMGO stimulated [35S]GTPγS binding by >400% above basal, as did morphine and fentanyl. The apparent lack of an efficacy difference for G protein activation by the high efficacy agonists in this cell line is likely due to the high receptor density compared with the density of G proteins available for activation because theB max of agonist-stimulated [35S]GTPγS binding was in the 3–5 pmol/mg range for the “full” agonists. Only those agonists that did not fully shift the affinity of the G proteins into a GTP-preferring state (i.e., buprenorphine and levallorphan) showed decreased efficacy in this system, although they did show increased relative efficacy compared with rat thalamus or SK-N-SH cells. These results suggest that relative agonist efficacy depends on the receptor/G protein ratio as well as the activity state of the G proteins and the intrinsic ability of the agonist to activate the receptor. The findings of the present study may help to explain the differences in μ-opioid agonist intrinsic efficacy observed at the level of effectors (40, 41) and in behavioral and analgesic studies (22-26).
Acknowledgements
The authors thank Christopher S. Breivogel for developing the [35S]GTPγS Scatchard assays in SK-N-SH cell membranes, Dr. Christopher Evans and Duane Keith for development of the mMOR-CHO cell line, and Dr. Lawrence Toll for helpful discussions regarding the mechanisms of opioid agonist efficacy.
Footnotes
- Received May 28, 1996.
- Accepted September 27, 1996.
-
Send reprint requests to: Dr. Steven R. Childers, Department of Physiology and Pharmacology, Bowman Gray School of Medicine, Wake Forest University, Medical Center Boulevard, Winston-Salem, NC 27157. E-mail: childers{at}bgsm.edu
-
↵1 C. Breivogel, D. E. Selley, and S. R. Childers. Acute and chronic affects of opioids on delta and mureceptor activation of G proteins in NG108-15 and SK-N-SH cell membranes, submitted for publication.
-
This work was partially supported by United States Public Health Service Grant DA02904 from the National Institute on Drug Abuse and a Young Investigator Award (D.E.S.) from the North Carolina Governor’s Institute on Alcohol and Substance Abuse.
Abbreviations
- GTPγS
- guanosine-5′-O-(γ-thio)-triphosphate
- DAMGO
- [d-Ala2,N-MePhe4,Gly5-ol]enkephalin
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- FBS
- fetal bovine serum
- CHO
- Chinese hamster ovary
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}