

Abstract
Menthol and many of its derivatives produce profound sensory and mental effects. The receptor for menthol has been cloned and named cold- and menthol-sensitive receptor-1 (CMR1) or transient receptor potential channel M8 (TRPM8) receptor. Using a dorsal root ganglion (DRG) and dorsal horn (DH) coculture system as a model for the first sensory synapse in the CNS, we studied menthol effects on sensory synaptic transmission and the underlying mechanisms. We found that menthol increased the frequency of miniature EPSCs (mEPSCs). The effects persisted under an extracellular Ca2+-free condition but were abolished by intracellular BAPTA and pretreatment with thapsigargin. Menthol-induced increases of mEPSC frequency were blocked by 2-aminoethoxydiphenylborane (2-APB) but not affected by the phospholipase C inhibitor U73122 or by the cADP receptor inhibitor 8-bromo-cADPR (8Br-cADPR). Double-patch recordings from DRG-DH pairs showed that menthol could potentiate evoked EPSCs (eEPSCs) and change the paired-pulse ratio of eEPSCs. A Ca2+ imaging study on DRG neurons demonstrated that menthol could directly release Ca2+ from intracellular Ca2+ stores. Menthol-induced Ca2+ release was abolished by 2-APB but not affected by U73122 or 8Br-cADPR. Taken together, our results indicate that menthol can act directly on presynaptic Ca2+ stores of sensory neurons to release Ca2+, resulting in a facilitation of glutamate release and a modulation of neuronal transmission at sensory synapses. Expression of TRPM8 receptor on presynaptic Ca2+ stores, a novel localization for this ligand-gated ion channel, is also strongly suggested.
Introduction
TRPM8, also known as cold- and menthol-sensitive receptor-1 (CMR1), has recently been cloned from primary sensory neurons (McKemy et al., 2002; Peier at al., 2002). This new receptor belongs to a transient receptor potential (TRP) family, a large family of membrane proteins with conserved molecular domains that may interact with intracellular signaling systems (Clapham et al., 2001; Minke and Cook, 2002). Some members in the TRP family are molecules that sense physical and chemical stimuli, including light, mechanical force, heat, odors, nerve growth factor, pheromones, pH, osmolarity, and metabolic stress (Minke and Cook, 2002). Similarly, TRPM8 is also a sensory molecule expressed on a subpopulation of primary afferent neurons. One identified function of TRPM8 is to serve as a sensor for cool temperature at peripheral nerve endings (McKemy et al., 2002; Peier at al., 2002). TRPM8, however, can also be activated by menthol, an active ingredient of peppermint (McKemy et al., 2002; Peier at al., 2002; Reid et al., 2002). In addition, several other chilling compounds can activate TRPM8 as well (McKemy et al., 2002). Electrophysiological studies have indicated that TRPM8 is a ligand-gated nonselective cation channel permeable to Ca2+ (McKemy et al., 2002; Peier at al., 2002; Reid et al., 2002). Activation of TRPM8 can result in an increase of intracellular Ca2+ levels (Okazawa et al., 2000; McKemy et al., 2002; Peier at al., 2002; Reid et al., 2002), which may signal multiple intracellular responses.
Menthol is a naturally occurring ligand for TRPM8 receptor. It is one of the most widely used compounds in food, cigarette, and pharmaceutical industries (Eccles, 2000). Menthol-containing substances and medicines are administered through different routes (Eccles, 2000; Umezu et al., 2001). Behavioral studies have shown that menthol has neuronal actions independent of its effects at peripheral nerve endings (Umezu et al., 2001). Although not identified yet, the potential presence of endogenous ligands for TRPM8 has been proposed; however, it is currently unknown whether TRPM8 has any other functions in addition to its role in sensing cold at peripheral nerve endings.
We have hypothesized that one action site of TRPM8 ligands is at the presynaptic terminals of the first somatic sensory synapses. These synapses are formed between primary afferent fibers and dorsal horn (DH) neurons in the spinal cord or the equivalent brain regions. Sensory transmission at these synapses is highly regulated by a number of mechanisms (MacDermott et al., 1999), including presynaptic inhibition (e.g., activation of presynaptic GABA-A receptors) and presynaptic facilitation (e.g., activation of presynaptic NMDA receptors). These presynaptic regulations affect sensory information flow from the periphery to the CNS. If TRPM8 is expressed at the presynaptic terminals of primary afferent fibers, its activation may modulate sensory transmission to the CNS. To test this hypothesis, we studied the effects of menthol and cold on synaptic transmission in a dorsal root ganglion (DRG)-DH coculture system, a model that has been used to study synaptic transmission and modulation at the first sensory synapses (Gu and MacDermott, 1997). We further studied the intracellular mechanisms by which menthol induced modulation of sensory synaptic transmission.
Materials and Methods
Preparation of DRG-DH coculture. Sprague Dawley rats were used according to the Institutional Animal Care and Use Committee guidelines of the University of Florida. DH-DRG cocultures were prepared as described previously (Gu and MacDermott, 1997). In brief, bilateral DRGs and spinal cord dorsal horns were dissected out from rat embryos at the age of 16 d in utero (E16). DRGs and DHs were incubated separately for 25 min at 37°C in S-MEM medium (Invitrogen, Grand Island, NY) with 2.5% trypsin (Invitrogen) and then triturated to dissociate neurons. The neurons were plated on glass coverslips prepared previously with a monolayer of rat cortical astrocytes. In some dishes, only DH neurons were plated to obtain DH neuron monocultures. Neurons were maintained in MEM (Invitrogen) culture medium that also contained nerve growth factor (2.5S NGF; 10 ng/ml; Roche Molecular Biochemicals, Indianapolis, IN), 5% heat-inactivated horse serum (JRH Biosciences, Lenexa, KS), uridine/5-fluoro-2′-deoxyuridine (10 μm; Sigma, St. Louis, MO), 8 mg/ml glucose, and 1% vitamin solution (Invitrogen). The cultures were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2 and fed weekly with fresh culture medium.
Patch-clamp recordings from DH neurons in DRG-DH cocultures. Coverslips with the cocultured neurons were mounted in a 0.5 ml recording chamber and placed on the stage of an Olympus IX70 microscope (Olympus, Lake Success, NY). Cells were perfused continuously with standard extracellular bath solution flowing at 1 ml/min. The bath solution contained (in mm): 150 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES, pH 7.2; osmolarity was adjusted to 320 mOsm with sucrose. Most experiments were performed at room temperature (24.0 ± 0.2°C).
For patch-clamp recordings from DH neurons, electrode intracellular solution contained (in mm): 110 Cs2SO4, 2 MgCl2, 0.5 CaCl2, 5 TEA-Cl, 5 EGTA, 5 HEPES, pH 7.3. Recording electrode resistance was ∼5 MΩ. DH neurons were voltage clamped at -70 mV in whole-cell configuration. Signals were amplified with Axopatch 200B (Axon Instruments, Union City, CA), filtered at 2 kHz, and sampled at 5 kHz using pCLAMP 8.1 (Axon Instruments). Unless indicated otherwise, the miniature EPSCs (mEPSCs) were recorded from DH neurons in the presence of lidocaine (10 mm), strychnine (2 μm), and bicuculline (20 μm). The high concentration of lidocaine was used to block both TTX-sensitive and TTX-resistant Na+ channels (Gu and MacDermott, 1997). In some experiments, the mEPSCs were recorded in the presence of lanthanum chloride (30 μm LaCl3) to block voltage-gated Ca2+ channels. The effects of menthol on mEPSCs were tested by bath application of menthol (1 and 100 μm) for 2 min. The effects of cold stimulation on mEPSCs were tested by bath application of cold bath solution, which yielded a temperature drop from 24 to ∼18°C in 20 sec. In all of the above experiments, mEPSCs were recorded for 1 min in normal bath solution and recorded continuously for 4 min after the application of menthol or cold bath solution.
For experiments performed in Ca2+-free (0 [Ca2+]o) bath solution, Ca2+ was omitted from normal bath solution and 5 mm EGTA was included. All experiments in 0 [Ca2+]o were performed using the perforated-patch technique. The electrode internal solution for the perforated-patch recordings was Cs2SO4 intracellular solution with 0.2 mg/ml amphotericin B (Sigma). In some 0 [Ca2+]o experiments, DRG-DH cocultures were preincubated for 45-60 min with 20 μm BAPTA-AM or 10 μm thapsigargin. Recordings were then performed in the continuous presence of BAPTA-AM and thapsigargin. To test the effects of 2-aminoethoxydiphenylborane (2-APB) and the cADP receptor inhibitor 8-bromo (8Br-cADPR) on menthol-induced responses in 0 [Ca2+]o bath solution, coculture dishes were preincubated with 100 μm 2-APB or 100 μm 8Br-cADPR for 30 min; 100 μm menthol was then co-applied with 2-APB. In experiments testing U73122, U73122 (2 μm) was pre-applied for 5 min, and then 100 μm menthol was co-applied with it.
Patch-clamp recordings from DRG neurons in the cocultures. Menthol- and cold-evoked whole-cell currents were recorded from DRG neurons with diameters in a range of 12-25 μm. Cell size was determined by calculating the average of shortest and longest diameters. Unless indicated otherwise, DRG neurons were held at -60 mV. All of the recording conditions for DRG neurons were the same as those for DH neurons. Menthol (100 μm) was applied for 4 or 20 sec to the recorded neurons through a glass tube (500 μm in diameter) positioned 1 mm away. Cold bath solution was applied to the recorded neurons, which yielded a temperature drop from 24 to ∼18°C in 20 sec at the recording site. The gravity-driven solution flow was controlled electronically by solenoid valves and triggered from a computer. Changes of temperature around the recording area were measured by an ultra-sensitive digital thermometer during the application of cold bath solution (Electro-therm Digital Thermometer TM99A; Cooper Instrument Corporation, Middlefield, CT). To test the effect of 2-APB on menthol-evoked whole-cell currents, 100 μm 2-APB was pre-applied for 3 min and then it was co-applied with 100 μm menthol; recovery was tested further after washout of 2-ABP for >10 min.
Double patch-clamp recordings from DRG-DH pairs in the cocultures. In experiments to determine the effects of menthol on monosynaptic EPSCs, double patch-clamp recordings were performed on DRG-DH pairs. Experiments were performed in a bath solution that contained (in mm): 150 NaCl, 5 KCl, 2 MgCl2, 0.5 CaCl2, 10 glucose, 10 HEPES, pH 7.2; osmolarity was adjusted to 320 mOsm with sucrose. DRG neurons were under current-clamp configuration with electrodes that had internal solution containing (in mm): 140 KCl, 10 HEPES, 2 MgCl2, pH 7.2. DH neurons were under voltage-clamp configuration held at -70 mm with electrodes containing CsSO4 internal solution. Monosynaptic connection was judged by the constant latency of eEPSCs responding to repetitive stimulation. Paired-pulse stimuli were applied to DRG neurons by injecting two brief depolarizing currents at an interpulse interval of 200 msec; each depolarizing current was applied for 10 msec. Both action potentials on DRG neurons and monosynaptic EPSCs on DH neurons were recorded simultaneously. The intensity of stimulus was pretested for each experiment and ranged from 100 to 550 pA for different cells. In each experiment, trials were repeated 10 times (10 sweeps) every 10 sec, and averaged values of paired-pulse eEPSCs were presented in the results. Unless indicated otherwise, 1 μm menthol and 5 μm thapsigargin were tested. In all electrophysiological recordings, membrane capacitance (Cm), membrane resistance (Rm), and access resistance (Ra) were monitored continuously; only stable recordings were included.
Ca2+ imaging experiments on DRG neurons. DRG neurons were dissociated from rats (Sprague Dawley; 100-250 gm) and plated on coverslips precoated with poly-d-lysine and laminin as described previously (Tsuzuki et al., 2003). Neurons were maintained in culture medium as described above for DRG-DH cocultures. Ca2+ imaging experiments were performed on neurons 2-5 d in culture. To load the Ca2+ indicator Fluo-3, the DRG neurons on coverslips were incubated with 5 μm Fluo-3-AM in 20% pluronic acid (Molecular Probes, Eugene, OR) for 30 min at 37°C. After dye loading, the cells were perfused with normal bath solution in a 0.5 ml chamber on a microscope stage (Olympus IX70). Fluo-3 fluorescence in the cells was detected with a Peltier-cooled charge-coupled device camera (PentaMAX-III System; Roper Scientific, Trenton, NJ) under a 20× objective. Excitation and emission were achieved by a fluorescence filter set at 450 and 550 nm, respectively. Images were taken at one frame per second and digitized using WinView software (Roper Scientific). The effects of menthol (100 μm) on intracellular Ca2+ levels were tested by bath application of 100 μm menthol. Unless indicated otherwise, menthol was applied for 70 sec in each test. The experiments were performed in both normal and 0 [Ca2+]o bath solution. Normal bath solution and 0 [Ca2+]o bath solution were the same as those used for electrophysiological experiments, and experiments were performed at room temperature. Unless indicated otherwise, a new dish was used, and cells were exposed to menthol only once in each experiment. In experiments to test the effects of 2-APB, 8Br-cADPR, and U73122 on menthol-induced responses in 0 [Ca2+]o bath solution, these compounds were applied in the same way as described in “Patch-clamp recordings from DH in DRG-DH cocultures.”
Drugs and data analysis. Menthol, EGTA, LaCl3, lidocaine, bicuculline, strychnine, BAPTA-AM, thapsigargin, 8Br-cADPR, and U73122 were purchased from Sigma. 2-APB was from Tocris (Ellisville, MO).
Synaptic events of the mEPSCs were analyzed using the Mini Analysis Program (Synaptosoft, Decatur, GA) with the same criteria as reported previously (Gu and MacDermott, 1997). The time course of mEPSC events was constructed with a time bin of 10 sec. The averages of frequency and amplitude in three sequential bins (30 sec) around the peaks were used for calculating the changes of mEPSC frequency and amplitude. For paired-pulse eEPSCs, averages of 10 sweeps were used in calculating paired-pulse eEPSC and first-pulse EPSC ratios. The first-pulse EPSC ratio of >1.2 was considered to be potentiation and the ratio of <0.8 to be depression. For Ca2+-imaging experiments, relative fluorescence intensity ΔF/F0 was used. Neurons with ΔF/F0 values of >0.1 (i.e., 10% increase) were arbitrarily designated responsive cells. All of the data were represented as mean ± SEM. Paired Student's t test was used for statistical comparison, and significance was considered at the p < 0.05 level.
Results
Presynaptic effects of menthol and cold at DRG-DH synapses
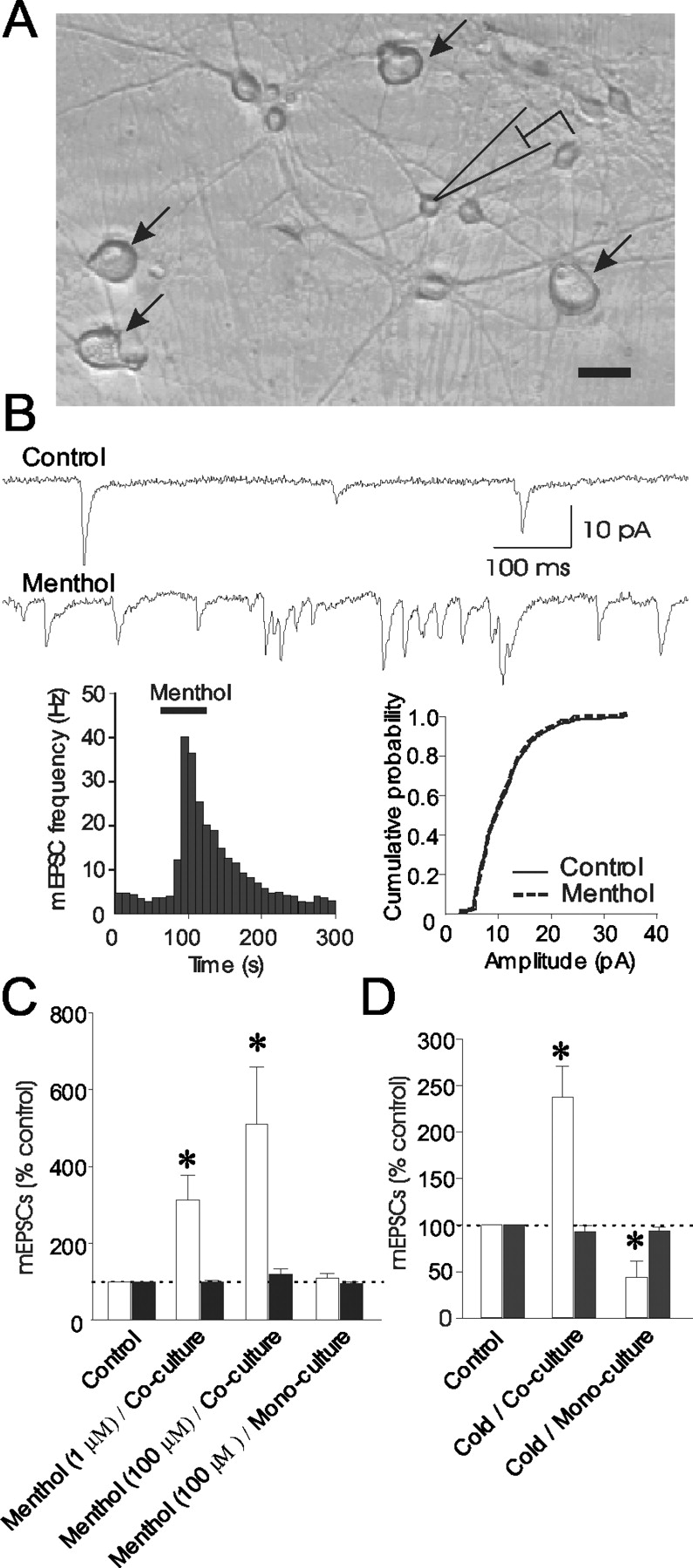
To determine whether menthol and cold may have actions at presynaptic terminals of sensory synapses, we examined the effects of menthol and cold stimulation on mEPSCs recorded from DH neurons. A DRG-DH coculture system was used (Fig. 1A). Similar to sensory synapses in vivo, glutamatergic synapses are formed between DRG and DH neurons in the coculture system that represent the first somatic sensory synapses in the CNS (Gu and MacDermott, 1997; Kerchner et al., 2002).

Increases of spontaneous glutamate release at sensory neuron synapses by menthol and cold stimulation. A, Micrograph shows a coculture of DRG neurons (arrow) and DH neurons (rest of cells). Scale bar, 25 μm. B, Sample traces represent mEPSCs recorded from a DH neuron in DRG-DH coculture before (control; top) and after (bottom) application of 100 μm menthol. Two graphs show that menthol increased mEPSC frequency (left) without affecting mEPSC amplitude (right). C, Graph is a summary of changes in mEPSC frequency (white bars) and amplitude (black bars) induced by menthol. Experiments were performed in DRG-DH coculture (second and third set of bars) and DH mono-culture (fourth set of bars). Control (first set of bars) was mEPSC frequency and amplitude before menthol application and is scaled to 100%. Menthol significantly increased mEPSC frequency in DRG-DH coculture (1 μm, n = 9; 100 μm, n = 11) but did not affect mEPSCs in DH mono-culture (n = 9). D, Graph summarizes changes in mEPSC frequency (white bars) and amplitude (black bars) induced by cold bath solution. Cold stimulation, ramp from 24 to 18°C, increased mEPSC frequency but not amplitude in DRG-DH coculture (second set of bars; n = 5). In DH mono-culture, cold stimulation decreased mEPSC frequency without affecting mEPSC amplitude (third set of bars; n = 7). In all experiments, menthol or cold bath solution was applied in the presence of strychnine (2 μm), bicuculline (20 μm), and lidocaine (10 mm). Data represent mean ± SEM, *p < 0.05.
Bath application of menthol (100 μm) for 1 min increased spontaneous glutamate release as evidenced by an increase of mEPSC frequency in the recorded DH neurons (Fig. 1B,C). Menthol-induced increases of mEPSC frequency occurred in all neurons recorded. Overall, the mEPSC frequency was increased significantly to 510.9 ± 148.3% of control in response to 100 μm menthol (n = 11; p < 0.05) (Fig. 1B,C). There was no significant difference between mEPSC amplitude in control and mEPSC amplitude after menthol application (120.4 ± 13.5% of control; n = 11) (Fig. 1B,C). Menthol at a concentration of 1 μm (Fig. 1C) still increased mEPSC frequency (311.8 ± 66.4% of control; n = 9; p < 0.05) without affecting mEPSC amplitude (99.6 ± 3.3% of control; n = 9). The increases of mEPSC frequency, not mEPSC amplitude, suggest an action of menthol at presynaptic sites. To test whether the effects of menthol occurred only at synapses formed between DRG and DH neurons, we examined the effect of menthol at synapses formed between DH neurons. This was achieved by using DH neuron monoculture in which DRG neurons were not present. In DH neuron monoculture, application of menthol (100 μm) for 2 min did not have significant effects on mEPSC frequency (110.0 ± 10.9% of control; n = 9) (Fig. 1C) and amplitude (94.3 ± 4.5% of control; n = 9) (Fig. 1C). These results indicate that menthol-induced spontaneous glutamate release is mediated by its action on the presynaptic terminals of DRG neurons. It should be noted that the high incidence of menthol-induced increases of mEPSC frequency in the above experiments did not mean that every DRG neuron in our coculture system was sensitive to menthol. This is because a single DH neuron received multiple synaptic inputs from many DRG neurons in the massive neuron coculture system.
We next tested the effects of cold stimulation on mEPSCs recorded from DH neurons in the DRG-DH coculture system. Cold stimulation was achieved through the application of cold bath solution, which had a temperature ramp from 24 to 18°C within 20 sec at recording sites. Cold stimulation significantly increased mEPSC frequency (237.3 ± 33.2% of control; n = 5; p < 0.05) (Fig. 1D) without affecting mEPSC amplitude (92.9 ± 7.0% of control; n = 5) (Fig. 1D). In contrast, in DH neuron monoculture, cold stimulation did not induce an increase of mEPSC frequency; there was only a decrease of mEPSC frequency (43.9 ± 17.2% of control; n = 7; p < 0.05) (Fig. 1D) and no change in mEPSC amplitude (93.5 ± 4.3% of control; n = 7) (Fig. 1D). Thus, cold stimulation had effects similar to menthol at sensory synapses formed between DRG and DH neurons.
Menthol- and cold-evoked currents in DRG neurons
To provide direct evidence that the effects of menthol and cold on mEPSCs in our coculture system are involved in their actions on DRG neurons, we recorded whole-cell currents evoked by menthol (100 μm) and cold stimulation in DRG neurons. In a total of 121 small-diameter (≤25 μm) DRG neurons, application of 100 μm menthol (4 or 20 sec) evoked inward currents in 68 neurons when the cells were held at -60 mV (Fig. 2A). Menthol-evoked inward currents showed partial desensitization when menthol was applied for 4 sec (n = 52; data not shown). The desensitization became substantial when menthol was applied for 20 sec (n = 16) (Fig. 2A); recovery from desensitization occurred after several minutes (Fig. 2A). The averaged peak amplitude was 199.4 ± 27.4 pA (n = 68) (Fig. 2E). The relationship between menthol-evoked currents and holding potentials showed a reversal potential near 0 mV (1.4 ± 16.8 pA at 0 mV; n = 10) (Fig. 2B).

Menthol- and cold-evoked currents from the soma of DRG neurons. A, Whole-cell currents recorded from a DRG neuron after three applications of menthol (100 μm). The duration of each application was 20 sec. The time intervals between each application are indicated. Holding potential was -60 mV. B, Current-voltage relationship of menthol-evoked currents (n = 10). Currents were scaled in reference to the values obtained at -60 mV. C, A whole-cell current recorded from a DRG neuron after the application of cold bath solution for 20 sec. Holding potential was -60 mV. Similar results were obtained in nine other cells. D, Temperature ramp measured at the recording sites during the applications of cold bath solution for 20 sec (n = 25). E, Pooled results of the peak amplitude of inward currents evoked by menthol (n = 68) and cold (n = 10).
We tested the responses of DRG neurons to cold stimulation after the application of cold bath solution. The temperature ramp at the recording sites was from 24 to 18°C during a 20 sec cold bath application (Fig. 2D), which preferentially activates TRPM8 receptors with little effect on ANKTMI, a lower temperature receptor (Story et al., 2003). Similar to menthol, inward currents were evoked in some DRG neurons after the application of cold bath solution for 20 sec when the cells were held at -60 mV. In a total of 37 small DRG neurons tested, cold stimulation evoked inward currents in 10 neurons (Fig. 2C,E). The inward current evoked by cold stimulation had a slower rising phase compared with the menthol-evoked current. This was most likely caused by the slow ramp of temperature change. Similar to menthol-evoked inward currents, cold-induced current also showed desensitization during a 20 sec cold bath application. The amplitude of cold-evoked inward currents was 156.4 ± 47.8 pA (n = 10) (Fig. 2E).
Role of Ca2+ in menthol-induced spontaneous glutamate release
We examined the mechanisms by which menthol acts on presynaptic terminals to increase mEPSC frequency, i.e., the spontaneous glutamate release. In the presence of LaCl3 (30 μm) to completely block voltage-gated Ca2+ channels (Gu and MacDermott, 1997), menthol (100 μm) still produced increases of mEPSC frequency (446.6 ± 139.4% of control; n = 6; p < 0.05) (Fig. 3A,E) in normal extracellular Ca2+ (2 mm [Ca2+]o) bath solution. Although mEPSC frequency increased and mEPSC inter-event intervals decreased, no significant changes in mEPSC amplitude were observed (91.1 ± 4.1%; n = 6) (Fig. 3A,E). The above result suggests that menthol can increase glutamate release when voltage-gated Ca2+ channels are blocked.

Ca2+ dependence of menthol-induced spontaneous glutamate release. A, Traces of mEPSCs before (top) and after (bottom) application of 100 μm menthol in the presence of LaCl3 (30 μm). Graph below the traces shows cumulative probability of mEPSC inter-event intervals before (control) and after menthol application. Menthol significantly increased spontaneous glutamate release, i.e., mEPSC frequency, in the presence of La3+. B, Traces of mEPSCs before (top) and after (bottom) application of 100 μm menthol in 0 [Ca2+]o bath solution. Graph below the traces shows cumulative probability of inter-event intervals, indicating that menthol still increased mEPSC frequency under 0 [Ca2+]o condition. C, Sample traces of mEPSCs recorded from a neuron preincubated with BAPTA-AM (20 μm). The histogram below shows cumulative probability of mEPSC inter-event interval. The mEPSCs show no difference before and after 100 μm menthol application under 0 [Ca2+]o condition. D, Sample traces recorded from a neuron pretreated with thapsigargin (10 μm) show mEPSCs before (top) and after (bottom) application of 100 μm menthol under 0 [Ca2+]o condition. The histogram below shows cumulative probability of mEPSC inter-event interval. The cumulative probability histograms in A-D were constructed from recordings before (solid line) and during (dashed line) menthol application. E, Summary of menthol-induced changes of mEPSC frequency (white bars) and amplitude (black bars) in experiments illustrated in A (n = 6), B (n = 29), C (n = 9), and D (n = 8). Control (first set of bars) was mEPSC frequency and amplitude before menthol application and is scaled to 100%. In all experiments, menthol (100 μm) was applied for 2 min. Experiments in A were in normal Ca2+ bath solution (2 mm [Ca2+]o). Experiments in B-D were in 0 [Ca2+]o bath solution in which Ca2+ was omitted and 5 mm EGTA was included.
Menthol-induced increase of spontaneous glutamate release may be caused by a direct Ca2+ entry through menthol-activated cation channels, presumably the TRPM8 receptors expressed on the plasma membranes of DRG presynaptic terminals. To test this idea, we studied the effects of menthol on mEPSCs in a Ca2+-free bath solution (0 [Ca2+]o). If menthol-induced spontaneous glutamate release is entirely dependent on extracellular Ca2+ influx, the effects of menthol should be abolished completely in 0 [Ca2+]o. Menthol (100 μm) still produced a significant increase in spontaneous glutamate release, however, in 0 [Ca2+]o as evidenced by the increase of mEPSC frequency to 273.1 ± 32.4% of control (n = 29; p < 0.05) (Fig. 3B,E); the mEPSC amplitude was not affected (103.3 ± 2.7% of control; n = 29) (Fig. 3E). The degree of menthol-induced increase of mEPSC frequency in 0 [Ca2+]o appeared to be lower than that in normal [Ca2+]o bath solution (Fig. 3E, compare 1C), suggesting that extracellular Ca2+ entry might be one mechanism of menthol-induced spontaneous glutamate release. Extracellular Ca2+ entry could not fully account for the effects of menthol, however, and other mechanisms could be involved.
One possibility of menthol effect in 0 [Ca2+]o might be attributable to Ca2+ release from presynaptic Ca2+ stores. To test this hypothesis, DRG-DH cocultures were preincubated with BAPTA-AM (20 μm, 60 min) to load the fast Ca2+ chelator BAPTA into all cells in the dishes. By using these cells and in 0 [Ca2+]o bath solution, menthol (100 μm) did not produce any significant change of mEPSCs in the DH neurons tested (Fig. 3C). Overall, mEPSC frequency was 114.5 ± 7.3% of control and mEPSC amplitude was 98.9 ± 4.5% (n = 9) (Fig. 3E). If Ca2+ release from presynaptic Ca2+ stores was indeed involved in menthol-induced spontaneous glutamate release, the effect of menthol should also have been abolished when presynaptic Ca2+ stores were depleted. We preincubated cultures with thapsigargin (10 μm, 60 min) to deplete intracellular Ca2+ stores. Thapsigargin itself increased mEPSC frequency in 0 [Ca2+]o bath solution, which returned to basal level within 30 min (data not shown). We then examined the effect of menthol on mEPSCs in 0 [Ca2+]o bath solution. Under this condition, menthol (100 μm) did not have any significant effect on mEPSCs in the cells tested (Fig. 3D,E). The mEPSC frequency was 124.4 ± 14.7% of control (n = 8) (Fig. 3E), and the mEPSC amplitude was 94.4 ± 3.8% (n = 8). These results suggest that menthol may cause a mobilization of intracellular Ca2+ stores at presynaptic terminals, which in turn induce spontaneous glutamate release.
Release of Ca2+ from intracellular Ca2+ stores by menthol in DRG neurons
We examined the effect of menthol on the dynamic changes of intracellular Ca2+ levels in DRG neurons, using Ca2+ imaging with the Ca2+ indicator Fluo-3. In a total of 27 DRG neurons tested, menthol (100 μm, 70 sec) produced increases of fluorescence intensities in 10 cells in normal [Ca2+]o bath solution (Fig. 4A, top panel, B), indicating that these cells were sensitive to menthol and intracellular Ca2+ was increased by menthol. The remaining 17 cells showed a continuous decrease of fluorescence intensities attributable to photobleach, and these cells were considered to be menthol-insensitive neurons (Fig. 4A, top panel, B). The relatively higher percentage of menthol-sensitive neurons in our study than others (McKemy et al., 2002; Peier at al., 2002) could be the result of sampling bias or receptor upregulation in NGF-containing culture medium. For the cells that were sensitive to menthol, the intracellular Ca2+ level was increased rapidly by the menthol (Fig. 4B), which was similar to the effect caused by depolarizing a concentration of KCl (50 mm; 70 sec; n = 7); however, the menthol-induced Ca2+ increase had slower decay than the KCl-induced effect (Fig. 4B).

Menthol-induced increases of intracellular Ca2+ level in DRG neurons. A, Top panel shows a series of real-time Ca2+ imaging in DRG neurons after the application of menthol in normal bath solution (2 mm [Ca2+]o). Menthol (100 μm) was applied from 15 to 84 sec, and fluorescence images were taken up to 180 sec. The last image was taken in bright field. The bottom panel shows a similar experiment in different DRG neurons in 0 [Ca2+]o bath solution. B, Summary of changes of fluorescence intensity (ΔF/F0) in DRG neurons over time under the following conditions: in normal Ca2+ bath solution after 100 μm menthol applications (•, n = 10), in normal Ca2+ bath solution after 50 mm KCl applications (▪, n = 7), in 0 [Ca2+]o bath solution after 100 μm menthol applications (▴, n = 14), and in 0 [Ca2+]o bath solution after 1 μm bradykinin applications (♦, n = 9). Fluorescence intensity of menthol-insensitive neurons in normal bath solution (ΔF/F0 < 0.1; □, n = 17) is shown for comparison. Each experiment was performed in DRG neurons that did not have previous exposure to menthol. Images were taken up to 100 sec for the bradykinin experiment and up to 180 sec for all other experiments. C, Top panel shows a Ca2+ imaging experiment similar to A. Bottom panel shows the same cells after a subsequent application of menthol in 0 [Ca2+]o bath solution. Menthol (100 μm) was applied from 15 to 84 sec. D, Summary of results from experiments illustrated in the top (•, n = 7) and bottom (▴, n = 7) panels of C. E, Experiments were similar to C except that both the first and second menthol applications were performed in 0 [Ca2+]o bath solution, and the duration of menthol applications was 35 sec. F, Summary of results from experiments illustrated in E (n = 6). Scale bars: A, C, E, 25 μm. Horizontal bars in B, D, and F indicate applications of testing compounds.
To directly test whether menthol caused a release of Ca2+ from intracellular stores, we determined the effect of menthol on intracellular Ca2+ in 0 [Ca2+]o bath solution. In each test, we used fresh dishes in which DRG neurons had not been exposed to menthol; i.e., each dish was tested only once with a menthol application. This was to avoid the potential depletion of intracellular Ca2+ stores by previous menthol application. Under this condition, menthol (100 μm; 70 sec) increased the intracellular Ca2+ level in 14 cells of 32 DRG neurons tested (Fig. 4A, bottom panel, B). Compared with the effect of menthol in normal bath solution, the menthol-induced increases of the intracellular Ca2+ level in 0 [Ca2+]o bath solution were much slower (Fig. 4B). On the other hand, in 0 [Ca2+]o bath solution, the kinetics showed fewer differences between menthol-induced and bradykinin-induced increases of intracellular Ca2+ (Fig. 4B). These results suggest that activation of menthol increases the intracellular Ca2+ level by two mechanisms: Ca2+ entry from extracellular sites and Ca2+ release from intracellular Ca2+ stores. These results are consistent with the effects of menthol on mEPSCs.
In previous studies from several groups, activation of TRPM8 receptors was not found to significantly affect intracellular Ca2+ level when pre-identified TRPM8-expressing cells were tested further in 0 [Ca2+]o bath solution (Okazawa et al., 2000; Peier at al., 2002; Reid et al., 2002). One possibility was the depletion of intracellular Ca2+ stores by a previous menthol application. To test this possibility, we first identified menthol-responsive neurons by the application of 100 μm menthol (70 sec) in normal bath solution (Fig. 4C, top panel, D) (n = 7 of 14 cells). After a 20 min wash with normal bath solution, we then tested the effects of menthol in 0 [Ca2+]o bath solution in these pre-identified menthol-responsive neurons. None of these cells showed a significant increase in the intracellular Ca2+ level (Fig. 4C, bottom panel, D) (n = 7). Similarly, neurons responding to the first application of menthol in 0 [Ca2+]o bathsolution (Fig. 4E, top panel) did not respond to a subsequent menthol application in 0 [Ca2+]o bath solution within 20 min (Fig. 4E, bottom panel, F). The lack of response to the second menthol application was unlikely caused by TRPM8 desensitization because menthol-inducedcurrents could fully recover within several minutes (Fig. 2A). We also tested Ca2+ release from intracellular stores by bradykinin in DRG neurons as a parallel experiment. Of 40 cells, the first application of bradykinin (1 μm, 40 sec) significantly increased fluorescence intensity in 20 cells (ΔF/F0 = 0.5 ± 0.1; n = 20), but there was no significant increase of fluorescence intensity after the second bradykinin application within 20 min (ΔF/F0 = 0.05 ± 0.02; n = 20; data not shown). A similar effect of bradykinin in DRG neurons has also been shown in a previous study (Eun et al., 2001).
Mechanisms of menthol-induced Ca2+ release
We explored the intracellular signaling pathways that might be involved in intracellular Ca2+ release induced by menthol. In this experiment, DRG neurons were pretreated with 2-APB (100 μm; 30 min), a membrane-permeable inhibitor of IP3 receptors. The effects of menthol were then tested in 0 [Ca2+]o bath solution in the continuous presence of 2-APB. Of 38 cells tested, menthol did not increase fluorescence intensity in any of these DRG neurons. Figure 5A shows the changes of fluorescence intensity (ΔF/F0 = -0.14 ± 0.03) in seven cells that were identified as menthol-sensitive neurons afterward; the negative value of ΔF/F0 represents the decrease of fluorescence intensity over time attributable to photobleach. After washout of 2-APB with normal bath solution for 20 min, cells were tested with menthol again; the seven cells noted above were shown to be menthol-sensitive neurons, with the fluorescence intensity (ΔF/F0) increasing to 0.31 ± 0.09 in response to 100 μm menthol. The response to a second menthol application after washout of 2-APB suggested that 2-APB prevented the depletion of Ca2+ stores by the first menthol application.

Pharmacology of menthol-induced Ca2+ release from intracellular stores and menthol-induced spontaneous glutamate release at sensory synapses. A, A summary of Ca2+ imaging experiments in DRG neurons in 0 [Ca2+]o bath solution under the following conditions: menthol alone (n = 14), menthol in the presence of 100 μm 2-APB (n = 7), menthol in the presence of 2 μm U73122 (n = 12), 1 μm bradykinin (n = 53), 1 μm bradykinin in the presence of 2 μm U73122 (n = 16), and menthol in the presence of 100 μm 8Br-cADPR (n = 10). Imaging acquisitions were the same as Figure 4A. The fluorescence intensity of each experiment was taken at the 100 sec time point. In all experiments, 100 μm menthol was applied for 2 min. B, Bar graph shows the pooled results of whole-cell currents recorded in DRG neurons (n = 10) after the applications of menthol (100 μm, 4 sec) in the absence of 2-ABP (first bar), in the presence of 100 μm 2-ABP (middle), and after washout of 2-ABP (last). C, Changes of mEPSC frequency (white bars) and amplitude (black bars) recorded from DH neurons in DRG-DH cocultures under the following conditions: application of menthol alone (n = 29), menthol in the presence of 100 μm 2-APB (n = 12), menthol in the presence of 2 μm U73122 (n = 9), and menthol in the presence of 100 μm 8Br-CADPR (n = 7). All experiments were performed in 0 [Ca2+]o bath solution, and 100 μm menthol was applied for 2 min.
We further tested whether the IP3/IP3 receptor pathway was involved in menthol-induced Ca2+ release. U73122 (2 μm), a potent phospholipase C (PLC) inhibitor, was used to block IP3 production. Unexpectedly, U73122 did not block menthol-induced increases of the intracellular Ca2+ level in 0 [Ca2+]o bath solution; the ΔF/F0 value was 0.37 ± 0.06 (n = 12) and was not significantly different from that in the absence of U73122 (Fig. 5A). On the other hand, 2 μm U73122 prevented bradykinin-induced increases of the intracellular Ca2+ level in 0 [Ca2+]o bath solution (Fig. 5A). These results suggest that the IP3/IP3 receptor pathway is not involved in menthol-induced Ca2+ release. Menthol could still increase intracellular Ca2+ levels (ΔF/F0 = 0.37 ± 0.07; n = 10) (Fig. 5A) in 0 [Ca2+]o bath solution when 8Br-cADPR (100 μm) was used to block cADPR receptors, discounting the involvement of cADPR receptors. The above results suggested that menthol-induced Ca2+ release was unlikely to be through the conventional intracellular signaling pathways.
The inhibitory effect of 2-APB on menthol-induced Ca2+ release may be caused by its direct inhibition of menthol-activated channels. To test whether 2-APB might directly inhibit menthol-activated channels, whole-cell inward currents evoked by 100 μm menthol were tested in the presence of 100 μm 2-APB. We found that menthol-induced inward currents (-99.8 ± 33.1 pA in control; n = 10) were abolished by 100 μm 2-APB (n = 10) (Fig. 5B).
We determined whether menthol-induced release of intracellular Ca2+ at presynaptic terminals had pharmacological profiles similar to those shown in DRG soma. All experiments were performed in 0 [Ca2+]o bath solution. Although 100 μm menthol significantly increased mEPSC frequency in 0 [Ca2+]o bath solution, there was no significant increase of mEPSC frequency in the presence of 100 μm 2-APB (104.7 ± 2.3% of control; n = 12) (Fig. 5C). On the other hand, inhibition of PLC by including 2 μm U73122 did not significantly affect menthol-induced increases of mEPSC frequency (216.8 ± 65.7%; n = 9) (Fig. 5C). Furthermore, 8Br-cADPR (100 μm) also did not attenuate menthol-induced increases of mEPSC frequency (221.0 ± 52.0%; n = 7) (Fig. 5C). Thus, menthol-induced presynaptic modulation, although involved in Ca2+ release at presynaptic terminals, is not coupled with conventional intracellular signaling pathways.
Effect of menthol on synaptic transmission at the first sensory synapses
To study whether menthol may modulate sensory synaptic plasticity, we examined the effect of menthol on stimulation-evoked monosynaptic transmission from small-sized DRG neurons to DH neurons using double-patch recordings from DRG-DH pairs (Fig. 6A). In control, paired-pulse stimulation of DRG neurons produced paired-pulse facilitation of eEPSCs in all DRG-DH pairs tested. After the application of 1 μm menthol, paired-pulse eEPSCs were examined again. Recovery was also examined 10 min after washout of menthol (Fig. 6B). Only those recordings that showed complete recovery were included in this study (n = 15). We compared first-pulse-evoked EPSCs before (P1) and during the application of menthol (P1′) to determine whether menthol affected the eEPSCs. The first-pulse eEPSCs during menthol (P1′) were found to be potentiated in eight DRG-DH pairs when P′/P1 of >1.2 is arbitrarily defined as potentiation (Fig. 6B,C). The P′/P1 values were between 0.8 and 1.2 for the remaining seven DRG-DH pairs (Fig. 6C). Overall, on average for the 15 DRG-DH cell pairs examined, the P′/P1 values were 1.49 ± 0.22 (n = 15).

Menthol-induced changes of synaptic transmission. A, Micrograph shows an example of double-patch recording from a DRG-DH neuron pair. Asterisks indicate two patch electrodes on a DRG neuron (arrow) and a DH neuron (arrowhead). Scale bar, 25 μm. B, Three sets of traces on the top panel show paired-pulse eEPSCs recorded from the DH neuron in the absence (left) and presence of 1 μm menthol (middle) and washout of menthol (right). Three sets of traces on the bottom panel show action potentials recorded from the DRG neuron after the paired-pulse stimulation. Paired-pulse interval was 200 msec. Each trace represents the average of 10 sweeps at time intervals of 10 sec. P1 and P2 represent the amplitudes of eEPSC pair in control, and P1′and P2′ represent those during menthol application. C, Changes of first-pulse eEPSCs during menthol applications in 15 DRG-DH pairs. The changes are relative to P1 and expressed as ratios of P1′/P1. Each line represents a DRG-DH pair. Two dashed lines indicate P1′/P1 values of 1.2 and 0.8. P1′/P1 > 1.2 was considered to be potentiation, P1′/P1 < 0.8 was considered to be depression, and P1′/P1 values within 0.8-1.2 were not considered to have effect. All 15 pairs had complete recovery after washout of menthol. D, Paired-pulse eEPSC ratio in control (P2/P1, open bar) and at different times after application of 1 μm menthol (P2′/P1′, solid bars). The data are pooled from all 15 DRG-DH pairs. E, The experiment was similar to B except 5 μm thapsigargin was tested in a DRG-DH pair. The two sets of paired-pulse eEPSCs in control (left) and after the application of thapsigargin for 15 min (right) are shown. The changes of paired-pulse eEPSCs over different times after thapsigargin are plotted on a right graph. F, Changes of first-pulse eEPSCs after 15 min of thapsigargin application. The changes are relative to P1 and expressed as ratios of P1′/P1. Each line represents a DRG-DH pair. The dashed line indicates a P1′/P1 value of 1.2. G, Changes of paired-pulse eEPSC ratios after 5, 10, 15, and 20 min of thapsigargin application (n = 6).
If the site of menthol action is located at presynaptic terminals of DRG neurons, it is expected that the paired-pulse eEPSC ratio would be affected by menthol. We compared the paired-pulse eEPSC ratio before (P2/P1) and during the application of menthol (P2′/P1′). As shown in Figure 6D, the paired-pulse ratio before menthol application (P2/P1) was 1.88 ± 0.13. The ratios (P2′/P1′) were 1.59 ± 0.19 at 1 min, 1.38 ± 0.15 at 3 min, and 1.50 ± 0.11 at 5 min during menthol application (n = 15). The ratios at 3 and 5 min during menthol applications were significantly lower than those of control (n = 15; p < 0.05) (Fig. 6D). The paired-pulse ratio fully recovered to the control value (1.83 ± 0.13; n = 15) after ∼10 min washout of menthol. These results suggest that menthol modulates sensory synaptic transmission through its action at the presynaptic sites of sensory synapses.
The modulation of synaptic transmission by menthol could be caused by menthol-induced Ca2+ release and Ca2+ entry. We tested whether release of intracellular Ca2+ was sufficient to modulate synaptic transmission at the first sensory synapses. We first tested thapsigargin because it releases Ca2+ from Ca2+ stores without directly inducing Ca2+ entry. eEPSCs were potentiated after the application of 5 μm thapsigargin (Fig. 6E,F). The ratio of P1′/P1 was increased to 2.0 ± 0.2 (n = 6; p < 0.05) (Fig. 6F) after the application of 5 μm thapsigargin for 15 min. The paired-pulse ratios were also changed in these six DRG-DH pairs. The paired-pulse ratio before thapsigargin applications (P2/P1, control) was 1.7 ± 0.3 (n = 6) (Fig. 6G). The paired-pulse ratios during thapsigargin application (P2′/P1′) were 1.5 ± 0.2 at 5 min, 1.2 ± 0.1 at 10 min, 0.8 ± 0.1 at 15 min, and 0.9 ± 0.1 at 20 min (n = 6); significant changes were found after thapsigargin applications for 15 and 20 min. The onset of thapsigargin action appears to be slower than that of menthol, probably because of its slower crossing of plasma membranes caused by much lower lipid solubility than that of menthol. The effects of thapsigargin on eEPSCs indicate that Ca2+ release from intracellular stores at presynaptic terminals alone could potentiate synaptic transmission at the first sensory synapses.
To evaluate whether menthol-induced Ca2+ release contributed to synaptic potentiation, we examined the changes of eEPSCs after repeated applications of menthol. As shown in Figure 7, menthol (1 μm) produced significant synaptic potentiation, and the amplitude of eEPSCs increased 203 ± 55% of control (n = 4) after a 3 min menthol application. After a 10 min wash of menthol, eEPSCs returned to control level (115 ± 38% of control). We then tested menthol again in the same manner as the first menthol test. It was found that the second menthol application could not potentiate eEPSCs (112 ± 39% of control; n = 4) on these DRG-DH pairs. The lack of synaptic potentiation by a second menthol application is consistent with the lack of effects on Ca2+ release by a second menthol application within 20 min after the first menthol application (Fig. 4). This suggests that menthol-induced Ca2+ release in presynaptic terminals is essential for its synaptic potentiation of evoked sensory transmission.

Effects on eEPSCs by first and second applications of menthol. A, Four traces show eEPSCs recorded from a DRG-DH neuron pair in the absence (control), after first application of 1 μm menthol, after washout of menthol (recovery), and after second application of 2 μm menthol. Each trace represents the average of 10 sweeps at time intervals of 10 sec. Action potentials recorded from the DRG neuron are not shown. B, Pooled results from four DRG-DH pairs. eEPSCs before menthol application (control) are scaled as 100%. In each experiment, menthol (1 μm) was applied for 3 min and eEPSCs were then recorded; recovery was recorded 10 min after wash off of menthol with normal bath solution.
Discussion
We have provided electrophysiological evidence showing that menthol can act on the presynaptic terminals of sensory neurons to facilitate spontaneous glutamate release as well as to potentiate evoked glutamate release at the first somatosensory synapses. The modulation of glutamate release is mediated by the increases of intracellular Ca2+ levels at presynaptic terminals. We have demonstrated that menthol not only induces Ca2+ entry, but also elicits a direct release of Ca2+ from intracellular Ca2+ stores. Menthol-induced mobilization of intracellular Ca2+ stores at presynaptic terminals plays a significant role in modulation of sensory synaptic transmission.
In our study, TRPM8 receptors are most likely to be involved in the effects of menthol. First, TRPM8 receptors are expressed at the somata of DRG neurons, and menthol is a naturally occurring ligand of these cation channels. Direct recordings from DRG neurons show that menthol evokes whole-cell currents with a reversal potential near 0 mV, indicating that menthol activates a nonselective cation channel on our DRG neurons. This menthol-activated nonselective cation current is consistent with TRPM8 current in a heterologous expression system and TRPM8-like current in sensory neurons recorded by others (Okazawa et al., 2000; McKemy et al., 2002; Peier at al., 2002; Reid et al., 2002). In our study, outward rectification of menthol-evoked currents (McKemy et al., 2002) was not clearly observed, a situation that has also been encountered for other ligand-gated ion channels studied in native tissues (Goldstein et al., 1995). Second, both menthol and cold induced increases of mEPSC frequency in DRG-DH cocultures but not in DH mono-cultures. This clearly indicates that the effect of menthol is not nonspecific action on cell membranes. The increase of mEPSC frequency indicates that the action of menthol and cold was at presynaptic sites. The specificity of menthol-induced response in DRG-DH cocultures indicated that the action of menthol and cold was at presynaptic sites of DRG neurons. It has been shown previously that K+ channels (Viana et al., 2002) and Na+-K+ ATPase (Pierau et al., 1975) also play roles in sensing cold. The lack of increases in mEPSC frequency in DH monocultures suggested that K+ channels and Na+-K+ ATPase were unlikely to be involved in menthol- and cold-induced synaptic effects observed in this study. Third, menthol- and cold-induced increases of mEPSCs were Ca2+ dependent, consistent with the involvement of TRPM8 in presynaptic modulation of glutamate release. Although ANKTM1, a cold receptor sensing lower temperature, has been identified very recently (Story et al., 2003), the results in our study were unlikely to be caused by the activation of ANKTM1 receptors because ANKTM1 receptors are not sensitive to menthol. The relationship between menthol responsiveness and TRPM8 mRNA has been examined in trigeminal neurons (Nealen et al., 2003). Nearly all TRPM8-positive cells tested were menthol responsive, and nearly all menthol-unresponsive neurons showed a lack of TRPM8 mRNA. Interestingly, some menthol-responsive trigerminal neurons did not have any detectable TRPM8 mRNA (Nealen et al., 2003), raising the issue of whether menthol-induced responses can be attributed entirely to TRPM8 receptor activation. Nevertheless, the involvement of TRPM8 receptors in menthol-induced responses appears to best fit the results of our study.
Menthol application results in the increase of intracellular Ca2+ level, and this increase is caused at least partially by the release of Ca2+ from intracellular Ca2+ stores. Ca2+ release was clearly evidenced by the menthol-induced increase of intracellular Ca2+ levels in 0 [Ca2+]o bath solution. Ca2+ release from intracellular Ca2+ stores could not be induced repeatedly by 100 μm menthol in 20 min, raising the possibility that menthol might cause a depletion of intracellular Ca2+ stores. This could be one of the reasons that previous studies did not observe menthol-induced Ca2+ release in TRPM8-expressing cells (Okazawa et al., 2000; Peier at al., 2002; Reid et al., 2002). Menthol-induced Ca2+ release was unlikely through the conventional intracellular signaling pathways such as through IP3/IP3 receptors. We initially used 2-APB to probe the potential involvement of IP3/IP3 receptors because 2-APB is known to be an IP3 receptor antagonist (Bootman et al., 2002). Menthol-induced Ca2+ release was indeed blocked by 2-APB; however, in the presence of the PLC inhibitor U73122 to prevent IP3 production, menthol still induced Ca2+ release. This does not support the involvement of the IP3/IP3 receptor intracellular signaling pathway.
After an examination of the effect of 2-APB on menthol-evoked whole-cell currents in DRG neurons, it turned out that 2-APB also strongly inhibited menthol-evoked inward currents. Because menthol-induced Ca2+ release did not appear to be associated with the conventional intracellular signaling pathways, the Ca2+ release in 0 [Ca2+]o was unlikely to be mediated by TRPM8 receptors on plasma membranes. The inhibitory effect of 2-APB on menthol-induced Ca2+ release was also unlikely because of its block on the TRPM8 receptors on plasma membranes. One possibility is that TRPM8 receptors are also expressed on intracellular Ca2+ stores and that menthol-induced Ca2+ release may result from activation of these receptors. 2-APB is known to be membrane permeable, and it may block TRPM8 receptors expressed on intracellular Ca2+ stores. Although the expression of ligand-gated cation channels on intracellular Ca2+ stores is uncommon, recent studies have shown that transient receptor potential channel V1 (TRPV1) is expressed on endoplasmic reticulum, and its activation also directly mobilizes intracellular Ca2+ stores (Olah et al., 2001; Liu et al., 2003).
Menthol-induced Ca2+ mobilization also occurs at presynaptic terminals and has significant effects on synaptic transmission at the first sensory synapses. In agreement with this suggestion, menthol increased mEPSC frequency in 0 [Ca2+]o bath solution. This effect was abolished by the Ca2+ chelator BAPTA or after thapsigargin treatment. The menthol-induced increase of mEPSC frequency was inhibited by 2-APB but not by U73122 or 8Br-cADPR, a result that is consistent with the pharmacological profile of menthol-induced Ca2+ release observed in DRG neurons. On the basis of these results, we propose that TRPM8 receptors (or TRPM8-like receptors) are expressed on Ca2+ stores within presynaptic terminals of sensory neurons and that activation of these receptors releases Ca2+ in presynaptic terminals. In addition to its localization on Ca2+ stores, TRPM8 receptors may also be expressed on the plasma membrane of presynaptic terminals. Activation of these receptors may mediate Ca2+ entry directly through TRPM8 receptors and indirectly through voltage-gated Ca2+ channels because of terminal membrane depolarization and Ca2+-induced Ca2+ release. Calcium entry through several ligand-gated Ca2+-permeable receptors (McGehee et al., 1995; Gu and MacDermott, 1997; Kerchner et al., 2001) plays a modulatory role in synaptic transmission. The increases of presynaptic Ca2+ levels through both Ca2+ release and Ca2+ entry may cause an increase in the probability of glutamate release and potentiate synaptic transmission. Consistent with this idea, the double-patch recordings from some DRG-DH pairs have shown synaptic potentiation of the eEPSCs during menthol application. This potentiation was caused by presynaptic modulation because the ratios of paired-pulse eEPSCs were significantly altered. Menthol-induced Ca2+ release at presynaptic terminals appears to be essential for the potentiation of eEPSCs because repeated application of menthol that failed to release Ca2+ also failed to produce synaptic potentiation. This result also indicates that activation of plasma membrane TRPM8 receptors alone may not be sufficient to mediate synaptic potentiation of eEPSCs under our experimental conditions.
The expression of menthol-gated receptors (TRPM8 or TRPM8-like receptors) at presynaptic terminals raises an issue about how these receptors may be activated. These receptors are unlikely to be temperature sensors because temperature within the spinal cord is constant (∼37°C). These presynaptic receptors, however, may be activated by exogenous ligands such as menthol. The presence of menthol-like endogenous ligands has been proposed. If present, endogenous ligands may also act on presynaptic terminals to modulate sensory synaptic transmission. In the TRP receptor family, another temperature-sensing molecule, TRPV1 (Caterina et al., 1997, 2000), is shown to have several endogenous ligands (Zygmunt et al., 1999; Huang et al., 2002) in addition to its naturally occurring ligand capsaicin. By increasing the gain of sensory inputs through presynaptic facilitation of glutamate release (Woolf and Salter, 2000), thermal sensation may be affected by menthol, its derivatives, and menthol-like endogenous ligands.
Footnotes
This work was supported by a National Institutes of Health Grant NS38254 to J.G.G. We thank A. MacDermott for providing suggestions in the early stage of this study.
Correspondence should be addressed to Jianguo G. Gu, Department of Oral and Maxillofacial Surgery, McKnight Brain Institute and College of Dentistry, University of Florida, Box 100416, Gainesville, FL 32610. E-mail: jgu{at}dental.ufl.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240762-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}