Article Text

Abstract
Objective Sorafenib is effective in hepatocellular carcinoma (HCC), but patients ultimately present disease progression. Molecular mechanisms underlying acquired resistance are still unknown. Herein, we characterise the role of tumour-initiating cells (T-ICs) and signalling pathways involved in sorafenib resistance.
Design HCC xenograft mice treated with sorafenib (n=22) were explored for responsiveness (n=5) and acquired resistance (n=17). Mechanism of acquired resistance were assessed by: (1) role of T-ICs by in vitro sphere formation and in vivo tumourigenesis assays using NOD/SCID mice, (2) activation of alternative signalling pathways and (3) efficacy of anti-FGF and anti-IGF drugs in experimental models. Gene expression (microarray, quantitative real-time PCR (qRT-PCR)) and protein analyses (immunohistochemistry, western blot) were conducted. A novel gene signature of sorafenib resistance was generated and tested in two independent cohorts.
Results Sorafenib-acquired resistant tumours showed significant enrichment of T-ICs (164 cells needed to create a tumour) versus sorafenib-sensitive tumours (13 400 cells) and non-treated tumours (1292 cells), p<0.001. Tumours with sorafenib-acquired resistance were enriched with insulin-like growth factor (IGF) and fibroblast growth factor (FGF) signalling cascades (false discovery rate (FDR)<0.05). In vitro, cells derived from sorafenib-acquired resistant tumours and two sorafenib-resistant HCC cell lines were responsive to IGF or FGF inhibition. In vivo, FGF blockade delayed tumour growth and improved survival in sorafenib-resistant tumours. A sorafenib-resistance 175 gene signature was characterised by enrichment of progenitor cell features, aggressive tumorous traits and predicted poor survival in two cohorts (n=442 patients with HCC).
Conclusions Acquired resistance to sorafenib is driven by T-ICs with enrichment of progenitor markers and activation of IGF and FGF signalling. Inhibition of these pathways would benefit a subset of patients after sorafenib progression.
- HEPATOCELLULAR CARCINOMA
- DRUG RESISTANCE
- MOLECULAR MECHANISMS
- STEM CELLS
- MOLECULAR GENETICS
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The multitarget tyrosine kinase inhibitor (TKI) sorafenib is the unique Food and Drug Administration (FDA)-approved therapy for patients with advanced hepatocellular carcinoma (HCC). However, despite favourable initial response, most patients develop disease progression. There is no effective second-line therapy approved for these patients.
The mechanisms underlying resistance to sorafenib are complex and not clearly elucidated. They may include compensatory activation of signalling pathways, acquired mutations and epithelial to mesenchymal transition events among others.
Cancer stem cells have been proposed as key mediators of resistance to antitumour therapies in solid malignancies.
What are the new findings?
Long-term exposure to sorafenib induces HCC tumours with enrichment of tumour-initiating cells (T-ICs) with progenitor/stem cell features.
Acquired resistance to sorafenib is led by the activation of two major signalling axes essential for tumour growth and survival, FGF and IGF signalling. Specific inhibition of these pathways can overcome resistance in vivo and reduce viability of cells derived from acquired resistant tumours and sorafenib-resistant cell lines.
Genomic characterisation of tumours that develop acquired resistance allowed us to propose a gene signature, which is linked to poor prognosis.
How might it impact on clinical practice in the foreseeable future?
Our study provides experimental evidence of potential mechanisms underlying resistance to sorafenib. Eventually, these findings will contribute to the optimal design of clinical trials to evaluate new drugs blocking IGF and FGF pathways in HCC in second-line treatment, a current unmet medical need.
Introduction
Hepatocellular carcinoma (HCC) is a major health problem, being currently the third cause of cancer-related death worldwide.1 Most patients are diagnosed when the metastatic process is already present. In these cases, the multitarget tyrosine kinase inhibitor (TKI) sorafenib is the only Food and Drug Administration (FDA)-approved systemic therapy, expanding patient median survival from 7.9 to 10.7 months.2 Despite initial response, most patients develop disease progression. In the case of HCC, radiological progression under sorafenib occurs after 4–5 months of treatment.2 As sorafenib targets several signalling pathways, acquisition of resistance might involve different mechanisms, including the activation of compensatory signalling cascades, rather than specific DNA aberrations, as was described with BCR-ABL and imatinib3 and BRAF mutations in melanomas resistant to vemurafenib.4 As the precise molecular mechanisms underlying resistance to sorafenib are still barely understood,5 ,6 there is an urgent need to characterise drivers of resistance to identify ideal targets for second-line therapies.
Many solid tumours, including HCC, contain a small subpopulation of cells bearing progenitor cell-like features, termed cancer stem cells (CSC) or tumour-initiating cells (T-ICs).7 A growing number of studies using human samples and preclinical models suggest that T-ICs are responsible for tumour relapse, metastasis and chemoresistance to antitumour drugs leading to disease progression and mortality.7 Thus, therapeutic strategies aimed to target T-ICs are particularly attractive as they could circumvent, at least partially, the development of resistance.
In the present study, we explored the mechanisms underlying acquisition of resistance to sorafenib in an animal model of HCC. Resistant tumours had enrichment of T-ICs, which showed enhanced tumourigenicity when transplanted in NOD/SCID mice. Transcriptomic analysis revealed that activation of IGF and FGF pathways contributes to the development of this resistance and that it could be overcome with selective inhibitors. Finally, we proposed a gene signature derived from sorafenib-resistant tumours with prognostic value in patients with HCC.
Material and methods
Establishment of a HCC xenograft model of acquired resistance to sorafenib
Subcutaneous Huh7 cells-derived tumours treated with sorafenib (30 mg/kg/day)8 for 4 weeks were excised in small pieces and engrafted in nude Balb/C mice (n=35). When tumours reached 100 mm3 volume, mice were treated with sorafenib (n=26) or placebo (n=5). Upon development of acquired resistance (see online supplementary material and methods), animals were used to explore T-ICs enrichment (n=5) or randomised to receive either anti-FGF therapy (brivanib, 100 mg/kg/day, n=6)9 or be maintained on sorafenib (n=6). Tumours exhibiting slow growth rate (ratio <1.3; tumour volume day 3/tumour volume day 1) or regression were considered sorafenib-sensitive. Four mice were excluded due to lack of response to sorafenib. We defined survival as the time comprised between randomisation and euthanasia. According to institutional ethical guidelines, mice were euthanised when tumours reached 10% body weight (∼2000 mm3) or mice showed discomfort, as shown by significant body weight loss. One hour after the last dose of treatment, animals were euthanised, and tumours collected and cut into portions to isolate cells, fixed for immunohistochemical analysis or frozen for mRNA and protein analysis.
Supplementary materials
Sphere formation assay
At least three subcutaneous Huh7-derived tumours corresponding to each experimental group, sorafenib-resistant, sorafenib-sensitive and non-treated control group, were excised and subjected to isolation and spheroid assays as previously described.10 For more details see online supplementary materials and methods section.
Tumour-initiating capacity
Tumour-initiating capacity was performed as previously described.7 Briefly, limiting dilutions (102, 103 and 104) of cells were injected subcutaneously into the flanks of NOD.Cg-Prkdcscid IL2rgtm1Wjl (NSG) mice in a mixture (1:1) of 1×PBS and Matrigel (BD Biosciences). Tumour incidence (number of tumours per number of injections) and tumour latency (time from injection to first tumour palpability) were assessed weekly. Tumours were confirmed on histology. If a tumour was palpable at a single injection site, it was removed surgically to allow continued evaluation of other injection sites. Mice were assessed for up to 36 weeks, and animals with no sign of tumour formation were examined at necropsy for confirmation.
Reagents, cell culture conditions, siRNA transfection, immunohistochemical analysis, and western blot analysis
See online supplementary materials and methods section.
Human samples
A total of 442 HCCs (87.5% early stages, Barcelona clinic liver cancer (BCLC) 0/A) collected from patients undergoing resection at three hospitals of the HCC Genomic Consortium (Hospital Clínic, Barcelona; Mount Sinai, New York and National Cancer Institute, Milan) were included in the study. These samples, with complete annotated clinical data and follow-up, have been used in previous studies conducted by the HCC Genomic Consortium.11–15 A first set, Cohort A, included 223 HCCs (45% hepatitis C virus (HCV), 20% HBV and 35% other aetiologies) and a second independent set, Cohort B, included 219 HCCs (61% HCV, 21% HBV and 18 other aetiologies). Online supplementary table S1 presents the main clinicopathological features of the patients included in the study.
Gene expression analysis profiling
For microarray profiling of mice xenografts and tumour-derived spheres, RNA isolation, cDNA transcription and mRNA processing were done as previously described.12 ,15 For more details, see online supplementary materials and methods section.
Bioinformatic and statistical analysis
See online supplementary materials and methods section.
Results
Acquired resistance developed after long-term exposure to sorafenib
To study acquired resistance to sorafenib, we generated an in vivo xenograft mouse model (figure 1A). Tumour monitoring revealed that 65% of mice (17/26) developed acquired resistance after a median of 42 days (p25–p75, percentiles: 33–49), defined by ≥30% increase in tumour volume in 3 days in tumours that had showed response to sorafenib for >15 days (figure 1B). Among the remaining nine animals, four (15%) were excluded from the study, since no response was observed during the first 15 days of treatment, and five animals (19%) were defined as long-term responders harbouring sensitive tumours. Tumour growth slopes of acquired resistant and sensitive tumours for the 4 weeks prior to development of resistance show a sharp increase in tumour volume (figure 1C).

Acquired resistance to sorafenib therapy in an in vivo model of hepatocellular carcinoma (HCC). (A) Experimental design of a HCC xenograft model of acquired resistance to sorafenib. (B) tumour growth reveals three patterns of growth in 26 animals: primary resistance, acquired resistance and long-term responsiveness. Acquired resistance developed after a 30% increase in tumour volume in animals responsive to sorafenib for at least 15 days. Significant statistical differences (*) were observed in sorafenib-resistant and sensitive tumour volumes in all time points in between. (C) Tumour growth evolution for 28 days prior to development of acquired resistance. Significant statistical differences (*) were observed in all time. Plotted data are expressed as mean and SDs of tumour volume.
Genomic profiling reveals a role of IGF and FGF signalling in sorafenib-acquired resistance
Microarray gene expression analysis was used to unravel molecular mechanisms underlying acquired resistance to sorafenib in the tumours of the experimental mouse model. We found 528 genes differentially expressed between acquired resistant and sensitive tumours (>1.5-fold, p<0.05, figure 2A, see online supplementary tables S3 and S4). Gene ontology and ingenuity pathways analysis (IPA) software mapped these genes to several functions and regulatory networks; 170 overexpressed genes in acquired resistant tumours were mostly involved in cell cycle progression, proliferation, migration and apoptosis (ie, IGF1R, FGFR1, FGF18, MYC, PDGFD, TGFBR2, PIK3C2G, MMP16) and mediators of cellular adhesion (ie, VCAN, FN1, SLC7A11, LAMA4). Conversely, 358 downregulated genes were mainly implicated in liver development and differentiation. The main networks associated with acquired resistance were those involving IGF1R, PI3K, MYC and FGFR1 activation (figure 2B).

Identification of mechanisms underlying acquired resistance to sorafenib. (A) Heat map showing 528 differentially expressed genes in sorafenib-acquired resistant (n=4) and sensitive tumours (n=3; >1.5-fold, p<0.05). The highest upregulated (red) and downregulated (green) genes in sorafenib-acquired resistant tumours are listed. (B) Top two rated networks generated by Ingenuity pathway analysis reflecting transcriptomic changes after acquired resistance involving upregulated genes in red (FGFR1, IGF1R, PI3K and MYC) and down regulated genes (green). (C) gene set enrichment analysis (GSEA) (FDR<0.05) revealed enrichment of gene sets of proliferation,12 IGF1R activation17 and progenitor features in acquired resistant tumours.
Sorafenib-acquired resistant tumours were enriched with gene signatures of poor prognosis in liver cancer,16 activation of proliferative pathways,12 a signature of IGF1R activation in HCC,17 mTOR complex 1 (mTORC1) activation, induction of HIF-1α target genes and embryonic stem/progenitor cell origin (FDR<0.05) reported in MSigDatabase (see figure 2C and online supplementary table S5). Interestingly, they were also associated with gene sets of acquired resistance to antitumour therapies in multiple tumour types, including chemotherapy and tyrosine kinase inhibitors (TKIs). By contrast, sorafenib-sensitive tumours were enriched in gene signatures of hepatic differentiation,18 cell adhesion, inflammatory response and better outcome, suggesting preservation of the liver function (see online supplementary table S6).
Sorafenib-acquired resistant tumours are enriched in T-ICs
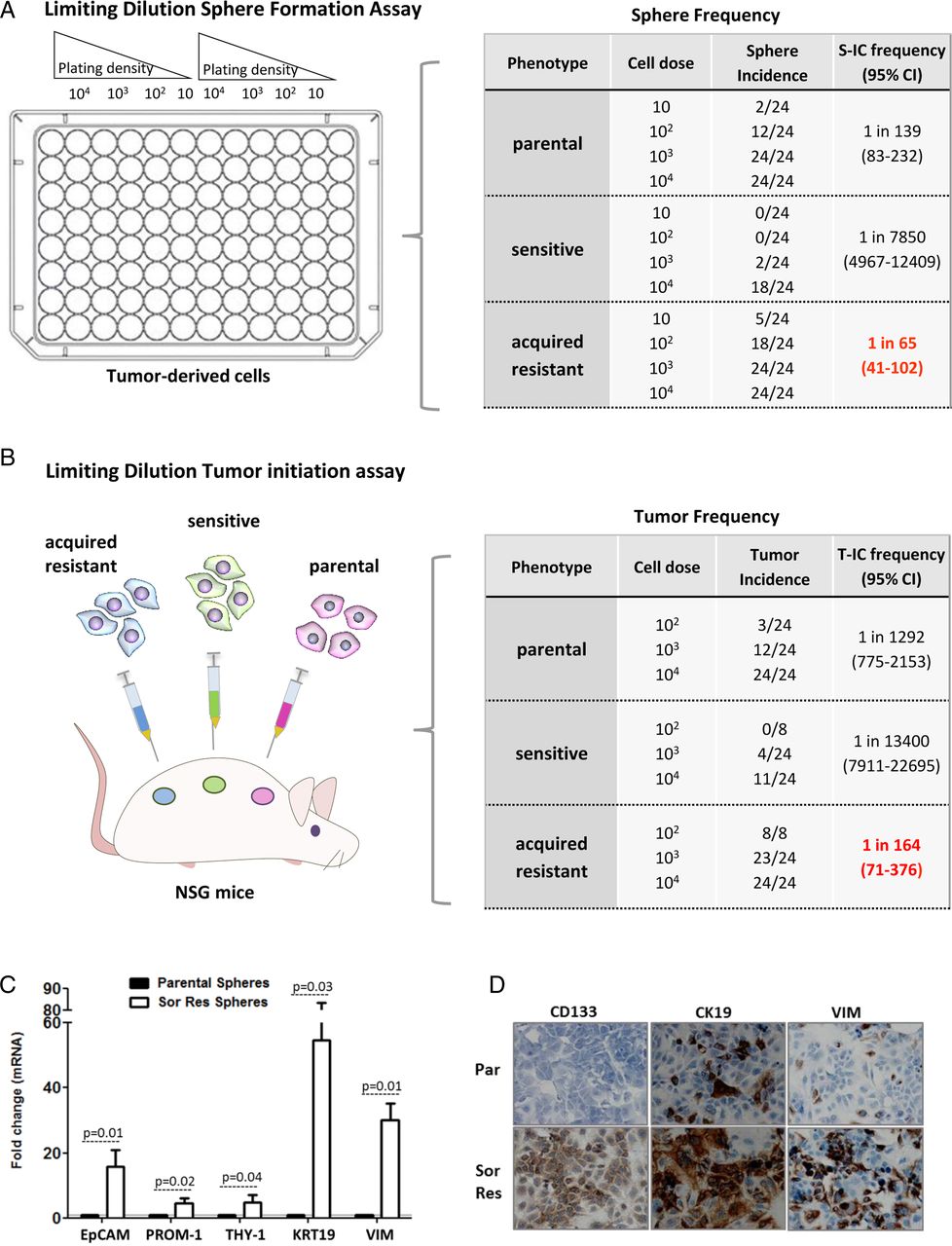
Transcriptomic analysis revealed an enrichment of progenitor cell features in tumours resistant to sorafenib (see figure 2C and online supplementary table S5). Thus, we hypothesised that these tumours could contain features of CSC or T-ICs. These cells are mainly responsible of tumour progression as described in other malignancies,7 being characterised by having self-renewal potential and the ability to initiate tumours once transplanted into immunodeficient mice. For this purpose, we first evaluated the capacity of cells isolated from xenograft tumours to generate spheroids in limiting dilution assays under non-attached culture conditions. Results showed that cells from sorafenib-acquired resistant tumours generated spheres at higher frequency (1 sphere for 65 cells (95% CI 41 to 102)) than those derived from sensitive (1 for 7850 (95% CI 4967 to 12 409)) or non-treated tumours (1 for 139 (95% CI 83 to 232)) p<0.05 (figure 3A). After dissociation of these spheres into single suspension, sphere-forming cells from acquired resistant tumours gave rise to secondary spheroids with high efficiency (data not shown).

Sorafenib-resistant tumours exhibit enrichment of cells with cancer stem-like properties. (A) Sphere formation capacity for three groups of tumours: parental non-treated, sorafenib-sensitive and sorafenib-resistant tumours. Ratio (ie, 1 in 65 refer to number of cells forming spheres out of all cells plated). (B) Limiting dilution tumour initiation assay after subcutaneous cell transplantation in NOD.Cg-Prkdcscid IL2rgtm1Wjl (NSG). Ratio (ie, 1 in 164 refer to number of cells inoculated needed to develop a tumour in vivo). Differences between number of cells needed in acquired resistance group versus others are significant (p<0.001). (C) Expression of liver progenitor cell markers and vimentin in spheres derived from sorafenib-acquired resistant tumours (empty bars) and non-treated tumours (dark bars). Data represent the mean expression value for a gene normalised to 1 (mean expression value of its corresponding parental non-treated cell line). Expression level is relative to the GAPDH gene. Bars indicate SD. (D) Immunocytochemical analysis of progenitor cell markers in sorafenib-resistant tumour cells and parental. T-ICs, tumour-initiating cells.
Next, we confirmed that sorafenib-acquired resistant tumours were enriched with T-ICs in vivo by performing serial transplant limiting dilutional tumourigenicity assays into NOD/SCID mice. Subcutaneous injection of as few as 102 sorafenib-acquired resistant tumour cells induced tumours in 100% of the mice, whereas only 12.5% of mice developed tumours from non-treated tumour cells, and no tumours developed from sensitive tumour cells. The frequency of T-ICs in acquired resistance, parental and sensitive to sorafenib was 1/164 (95% CI 71 to 376), 1/1292 (95% CI 775 to 2153) and 1/3400 cells (95% CI 7911 to 22 695), respectively (figure 3B). Taken together, these data suggest a significant enrichment of T-ICs in HCCs with acquired resistance to sorafenib.
We further confirmed that spheres from sorafenib-acquired resistant tumours had enhanced expression of several liver progenitor markers19 epithelial cell adhesion molecule (EpCAM), CD133/PROM-1, CD90/THY-1, the epithelial progenitor marker CK19 and the epithelial–mesenchymal transition (EMT) protein vimentin compared with spheres from non-treated tumours (see figure 3C and online supplementary figure S1A). Growing in adherent conditions, the expression of some of these markers was still higher in cells from sorafenib-acquired resistant tumors than in parental non-treated cells (see figure 3D and online supplementary figure S1A).
Cells isolated from sorafenib-acquired resistant tumours have activation of pathways involved in tumour progression and survival
To better understand the signalling pathways deregulated in T-ICs in sorafenib-acquired resistant tumours, we conducted a microarray gene expression profile analysis. A total of 342 well-annotated genes were differentially expressed (≥1.5-fold, FDR <0.05) between sorafenib-acquired resistant and parental non-treated cells growing under sphere formation conditions (see figure 4A and online supplementary tables S7 and S8). GSEA showed that spheres derived from sorafenib-acquired resistant tumours were particularly enriched in gene signatures related to stem cell/progenitor features in the liver (ie, CK19 signature,14 EpCAM20 and hepatoblastoma-C2,21 http://www.broadinstitute.org/msigdb, FDR<0.05, figure 4A). Moreover, there was a clear enrichment for gene sets related to pathways essential for tumour development and cell survival (ie, IGF1R signalling,17 mTOR signalling) and biological functions involved in cell death and survival, cellular movement or embryonic development (see figure 4A and online supplementary table S8).

Cells from sorafenib-acquired resistant tumours are sensitive to IGF and FGF inhibition. (A) Heat map showing 342 differentially expressed genes between sorafenib-acquired resistant spheres and parental non-treated spheres (≥1.5-fold, FDR<0.05). GSEA shows significant enrichment of signatures of progenitor features, proliferation and IGF1R signalling in sorafenib-acquired resistant spheres (FDR<0.05). (B) Parental cells present significant decrease of cell viability on sorafenib treatment compared with sorafenib-acquired resistant cells. (C) Upregulation of IGF1R, FGFR1 and downstream protein kinase B (AKT) signalling in sorafenib-acquired resistant cells and Huh7 parental cells. (D) Cell viability in sorafenib-acquired resistant and parental cells, after transient transfection with siRNA against IGF1R or FGFR1 alone or in combination with sorafenib. (E) Cell viability of sorafenib-acquired resistant cells and parental cells treated with sorafenib, IGF1R inhibitor (linsitinib (5 µM)) FGFR inhibitors (brivanib (5 µM) and BGJ398 (5 µM)) and combinations. Results are the mean of at least three independent experiments assayed in triplicate. Bars depict the SDs. Statistical significance is set at p<0.05.
Considering the genomic data obtained in our xenograft tumour model (figure 2B, C), increased expression/activation of IGF1R and FGFR1 was confirmed in cells derived from sorafenib-acquired resistant tumours in comparison with parental non-treated cells (figure 4C), as well as increased activity of downstream protein kinase B (AKT) signalling. We also confirmed that cells from sorafenib-resistant tumours had increased cell viability with respect to parental cells when cultured with sorafenib (5 µM) for 72 h (64% vs 48%, p=0.01; figure 4B).
Next, we tested the effects of specific IGF and FGF inhibition in spheres derived from resistant tumours. Using siRNA, IGF1R and FGFR1 expressions were downregulated in cells derived from sorafenib-acquired resistant tumour and parental cells (see online supplementary figure S2A, B). However, siRNA against IGF1R or FGFR1 did not significantly decreased cell viability when used as single agents and only achieved significant decrease in viability when combined with sorafenib (3 µM; figure 4D). Conversely, when using linsitinib (5 µM), a dual TKI of IGF1R/IR, and the pan-FGFR inhibitor BGJ398, a significant decrease in cell viability was obtained (figure 4E). We speculate that these suboptimal outcomes when using siRNA are, in part, the result of the modest knockdown efficiency obtained in acquired resistant cells compared with parental cells (see online supplementary figure S2A). Then, we explored the mechanism of resistance in two sorafenib-resistant cell lines derived from Hep3B and Huh6 cells (figure 5). Western blot analysis showed that Huh6 had activation of IGF1R and downstream AKT (figure 5A left panel), while Hep3B, that was previously reported to mediate resistance to sorafenib through epidermal growth factor receptor (EGFR) activation,22 showed increased upregulation of FGFR1 expression/activation but not of IGF1R signalling (figure 5B, left panel). In consequence, IGF1R inhibitor linsitinib had stronger efficacy in reducing cell viability in Huh6-resistant cells than in parental cells (78% vs 92%, p=0.04) (figure 5A). Finally, in agreement with our observations in cells from sorafenib-resistant tumours, Hep3B-resistant and Huh6-resistant cell lines had increased expression of several progenitor cell markers (see online supplementary figure S1A and B).

Assessment of IGF1R and/or FGFR1 signalling status in hepatocellular carcinoma (HCC)-resistant cell lines. Expression/activation of IGF1R and FGFR1 and downstream signalling pathways (left panel), cell viability in the presence of sorafenib (central panel) and treatment with linsitinib, brivanib, or BGJ398 as single agents or in combination with sorafenib (right panel) were evaluated in (A) Huh6 parental and resistant cells lines and (B) Hep3B parental and resistant cells lines. Results are represented as the mean of at least three independent experiments assayed in triplicate. Bars depict the SDs. Statistical significance is set at p<0.05.
Other previously described mechanisms of acquired resistance to sorafenib, such as increased Mitogen-Activated Protein Kinase 14 (MAPK14) activation5 (as measured by pMAPK14), where not observed in neither our in vivo and in vitro models nor the two additional cell lines resistant to sorafenib (see online supplementary figure S2C). Furthermore, GSEA analysis comparing the gene expression profile of sorafenib-acquired resistant and sensitive tumours showed no enrichment of p38/MAPK14 pathway gene set (data not shown).
Anti-FGF therapy reverts resistance and induces liver-specific functions in sorafenib-acquired resistant tumours
Upon development of resistance, mice were randomly assigned to continue to receive a FGFR inhibitor (brivanib, 100 mg/kg/day, figure 1A) or continue on sorafenib. Brivanib induced a significant delay of tumour growth (day 12: 1141±424 vs 1659±264 mm3 in sorafenib-treated animals, p=0.03, figure 6A), also showing a trend to increased survival, from a median of 14 days (95% CI 12 to 16) in sorafenib-treated animals up to 32 days (95% CI 5 to 59) in mice receiving brivanib (p=0.07, figure 6B). No significant treatment-related side effects were observed.

An FGF inhibitor delayed tumour growth after development of acquired resistance and inhibited FGFR1 activation and downstream signalling. (A) Tumour growth in acquired resistant tumours of mice randomised to receive brivanib or sorafenib therapy. Data represent mean±SD. (B) Kaplan–Meier survival curves comparing the two groups. (C) qRT-PCR analysis of FGFR1 and IGFR1 mRNA levels in each experimental group. Acquired resistant tumours present significant upregulation of IGF1R and FGFR1. (D) Representative immunostaining assessing the activation status of FGFR1, IGF-1R, Extracellular Signal-regulated Kinase-1 (ERK) and RPS6 in formalin-fixed paraffin-embedded (FFPE) sections from xenograft tumours of each experimental group.
Quantitative real-time PCR analysis confirmed significant higher expression of IGF1R (2.1-fold, p=0.021) and FGFR1 (4.1-fold, p=0.015) in sorafenib-acquired resistant tumours compared with sensitive tumours (figure 6C), as detected by microarray analysis, whereas brivanib treatment reduced FGFR1 expression (2.4-fold, p=0.007). On the other hand, no changes of other proangiogenic factors, such as vascular endothelial growth factor (VEGFA), FGF1, FGF2, IL8 and ANGT2 (see online supplementary figure S3) were found. Immunohistochemical analysis further confirmed increased FGFR1 and IGF1R protein expression in sorafenib-acquired resistance (see online supplementary figure S4) and, importantly, a notable enhanced activation of these receptors and downstream activation of p-Extracellular Signal-regulated Kinase-1 (ERK) and p-RPS6, surrogates of Ras-MAPK and PI3K-Akt-mTOR signalling activation, respectively (figure 6D). Furthermore, brivanib treatment decreased FGFR1 activation, as well as ERK and p-RPS6 effectors (figure 6D), supporting that the development of acquired resistance involves signals triggered by IGF1R and FGFR1 activation.
Moreover, transcriptomic analysis of tumours treated with the FGFR inhibitor versus those following on sorafenib after randomisation revealed that brivanib induced differential expression of 180 genes, including inhibitors of proliferation, as well as regulators of FGF signalling (ie, FGF18, FGFR1OP FRLT3, HRASLS, SPRY2; greater than twofold, p<0.05, see online supplementary table S9). GSEA further showed reversion of genomic traits of proliferation and tumour progression and enrichment of liver-specific functions after treatment with brivanib (see online supplementary table S10).
A gene signature of resistance to sorafenib is associated with poor outcome in patients with HCC
Genomic profiling of sorafenib resistance and sensitive mice xenografts enabled us to generate a 175-gene signature of sorafenib resistance (FDR<0.20) (see figure 7A and online supplementary table S11). Due to the lack of genomic information from biopsies derived from patients treated with sorafenib, prediction class was performed in two independent cohorts of surgically resected patients, cohort A (n=223) and cohort B (n=219), whose gene expression profiles had been previously analysed.11–15 Prediction analysis showed that 16% and 12% of patients harboured the gene signature in cohort A and cohort B, respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gene signature of acquired resistance to sorafenib predicts poor survival of patients with resected hepatocellular carcinoma (HCC; n=442), including training (n=223) and validation sets (n=219). (A) Generation of a 175 gene signature based on the transcriptomic analysis of mice tumours resistant to sorafenib. (B) and (C) Around 15% of patients in the training and validation set harbour the 175 gene signature of sorafenib resistance, which is significantly associated to poor outcome.
Next, we evaluated the prognostic performance of the gene signature of resistance and showed that it was able to identify a subset of patients with HCC with worse prognosis after resection (median survival: 4.1 vs 5.6 years (p=0.046), and 3.1 vs 9.5 years (p=0.002; figure 7B, C), in both independent cohorts. Patients harbouring the signature had significantly more advanced tumours (BCLC-B/C stage, p=0.043), vascular invasion (p<0.001), tumour satellites (p=0.02) and enrichment with signatures of poor prognosis (EpCAM, CK19), proliferation class (FDR<0.05) and those identifying patients with HCC with IGF1R and Notch signalling activation (p<0.05; see online supplementary table S12).
Discussion
In the present study, we demonstrated that acquired resistance to sorafenib, the unique effective therapy for advanced HCC, is characterised by novel molecular features and observations including: (1) clear enrichment in T-ICs with progenitor cell features, (2) activation of signalling cascades essential for growth and survival, such as IGF, FGF, Myc and PI3K, not activated in sensitive tumours, (3) resistance can be overcome with inhibitors of these pathways and (4) the genomic characterisation of acquired resistant tumours identify a gene signature linked to poor prognosis.
During the last decade, a particular emphasis has been made on resistance to kinase inhibitors in the context of tumour dependency or oncogenic addiction. In this scenario, the most common traits of acquired resistance mechanisms are the persistent activation of the oncogenic target itself or its downstream signaling pathways due to secondary mutations in the target kinase (e.g. EGFR (T790M) in gefitinib/erlotinib-resistant non small-cell lung carcinoma (NSCLC) patients,23 or in TSC2 in a tumor treated with everolimus in anaplastic thyroid) or to increased gene dosage (e.g. BCR-ABL amplifications in imatinib-resistant CML patients).24 Alternatively, deregulation of feedback loops, such as the case of PI3K/AKT activation by mTOR or MEK inhibitors,25 ,26 has been proposed as mediators to escape from drug pressure. In addition to these well-established mechanisms, pathway-independent resistance mechanisms, such as acquisition of stem/progenitor cell phenotype, within the heterogeneous tumour cell population have also been suggested.7 This later model constitutes an attractive framework to understand the onset of acquired resistance, considering that treatment-resistant initiating cells present self-renewal capacities required for disease survival and dissemination.
The identification of key molecular events responsible for the acquisition of resistance to sorafenib is particularly challenging due to: (1) the high molecular heterogeneity of HCC, (2) the multiple potential drivers but no clear oncogenic addiction loops and (3) the limited understanding of the mechanisms of action of sorafenib. Recent reports have attempted to unveil the complex mechanisms that underlie the development of resistance to sorafenib, the unique effective therapy for advanced HCC, including crosstalks of PI3 K/Akt27 and JAK-STAT signalling, hypoxia-inducible pathways6 and epithelial to mesenchymal transition events28 or Mapk14-dependent activation of Mek-Erk and Atf2 signalling.5 In line with our findings, a previous study suggested that in vitro long-term exposure to sorafenib induced the generation of a population of cells with increased expression of progenitor cell markers and enhanced migratory and invasive abilities.28
The broad spectrum of activity of sorafenib suggests a complex plethora of events contributing to the development of acquired resistance. In our mouse model, the mechanisms that ultimately leads to upregulation of IGF1R and/or FGFR1 could involve, among others, miRNA or epigenetic regulation followed by clonal selection.29 Nonetheless, no evidence of other recently reported mechanism of resistance was observed (ie, EGFR signalling upregulation or MAPK14 activation.5 ,22 ,30 This highlights the functional redundancy of diverse TK signalling pathways involved in the acquisition of resistance.
We previously reported that IGF signalling is activated in 20% of early HCC, and can be efficiently inhibited in preclinical models.17 Similarly to our data, increasing evidence strongly suggests an important role of IGF and FGF signalling in conferring drug resistance in human malignancies,29 ,31 including a study that highlighted the role of IGF1R in CSC biology within the liver.32 This signalling cascade has been identified as a promoter of cell proliferation and survival in the context of chronic exposure to EGFR and BRAF inhibitors and chemotherapy in several malignancies.33 ,34 Our data support exploring IGF TKI in HCC as second-line therapy. The fact that monoclonal antibodies against IGF1R, where not effective in front line, does not undermine the potential therapeutic use of TKI after progression to sorafenib, since there is a relevant activation of IGF pathway in this setting and the mechanism of action of small molecules simultaneously block both IGF1R and insulin receptor (IR).
FGF signalling has a central role in tumour progression, angiogenesis and has emerged as a driver of resistance.31 In the current study, we observed a positive effect of FGF inhibition in experimental models of acquired resistance to sorafenib results that were consistent with pancreatic neuroendocrine tumours with acquired resistance to sorafenib.35 Unfortunately, this effect has not been confirmed in a phase III trial in second line in HCC.36
In conclusion, the experimental study reported herein points to IGF and FGF signalling as relevant activated axis partially responsible of acquired resistance to sorafenib therapy in HCC. Further understanding of these mechanisms will allow the design of phase II–III studies using biomarker-based trial enrichment where samples can be obtained before and after treatment. Collecting tissue samples in all HCC clinical trials has emerged as a critical recommendation in the recent Guidelines of Management of HCC.37 This approach would allow us to further understand the mechanisms of sorafenib resistance and also guide trial enrichment and personalised/stratified oncotherapeutics.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Acknowledgements The authors thank Silvia Secanella for technical assistance. The authors also thank Corina Buta (INSERM UMR_S938) for establishing the sorafenib-resistant cells lines derived from Hep3B and Huh6 cells.
Contributors Study concept and design: VT, HC, JDD, JML; Acquisition of data: VT, HC, AM, SV, DS, JP, LC, CA, ST, IMQ, JDD; Analysis and interpretation of data: VT, HC, AM, SV, YH, DS, JP, CA, JJL, CDM, MS, JDD, AV, JML; Drafting of the manuscript: VT, HC, AM, SV, JML; Critical revision of the manuscript for important intellectual content: VT, HC, AM, SV, YH, DS, JP, LC, CA, IMQ, CDM, MS, JDD, AV, JML.
Funding Bristol-Myers Squibb partially supported the study with a research grant. JML is supported by grants from The European Commission Framework Programme 7 (HEP-CAR grant, Number 667273-2 and HEPTROMIC, Proposal No:259744), the Samuel Waxman Cancer Research Foundation, the Spanish National Health Institute (SAF-2013-41027), and the Asociación Española Contra el Cáncer (AECC). Victoria Tovar is supported by a grant from the AECC. Helena Cornella and Clara Alsinet are recipients of grants from the Instituto de Salud Carlos III (PFIS programme). Agrin Moeini is recipient of a grant from Spanish National Health Institute (FPI programme). Daniela Sia is supported by the Italian Association for Cancer Research and the Italian National Ministry of Health. Yujin Hoshida is supported by grants from the U.S. National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK099558). Christèle Desbois-Mouthon is supported by French institute of health and medical research (INSERM) and French Institute of Cancer (INCA, grant No: INCa-DGOS_5790).
Competing interests Prof. Josep M. Llovet has been a consultant, advisory board member and received research funding from Bayer and Bristol-Myers Squibb.
Ethics approval These samples were collected upon IRB approval at three hospitals of the HCC Genomic Consortium (Hospital Clínic-Barcelona, Mount Sinai-New York and National Cancer Institute-Milan) and have been used in previous studies (Villanueva, Hepatology, 2015; Chiang, Cancer Res, 2008; Villanueva, Gastroenterology, 2008; Wurmbach, Hepatology, 2007).
Provenance and peer review Not commissioned; externally peer reviewed.