Article Text

Abstract
Objective Epigenetic mechanisms are potential targets to relieve somatic pain. However, little is known whether epigenetic regulation interferes with visceral pain. Previous studies show that oestrogen facilitates visceral pain. This study aimed to determine whether histone hyperacetylation in the spinal cord could attenuate oestrogen-facilitated visceral pain.
Design The effect of the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) on the magnitude of the visceromotor response (VMR) to colorectal distention was examined in ovariectomised rats with/without oestrogen replacement. An additional interaction with the metabotropic glutamate receptor 2/3 (mGluR2/3) antagonist LY341495 was tested. The levels of acetylated histone and mGluR2 mRNA and protein were analysed. The binding of acetylated H3 and oestrogen receptor α (ERα) to the GRM2 promoter was measured by chromatin immunoprecipitation coupled with qPCR.
Results In ovariectomised rats, 17β-estradiol (E2), but not safflower oil, increased the magnitude of the VMR to colorectal distention. SAHA attenuated the E2-facilitated VMR, but had no effect in safflower oil-treated rats. Subsequent spinal administration of LY341495 reversed the antinociceptive effect of SAHA in E2 rats. In addition, SAHA increased mGluR2 mRNA and protein in the spinal dorsal horn following E2, but not vehicle, treatment. In contrast, neither E2 nor SAHA alone altered mGluR2 mRNA. SAHA increased binding of H3K9ac and ERα to the same regions of the GRM2 promoter in E2-SAHA-treated animals.
Conclusions Histone hyperacetylation in the spinal cord attenuates the pronociceptive effects of oestrogen on visceral sensitivity, suggesting that epigenetic regulation may be a potential approach to relieve visceral pain.
- VISCERAL NOCICEPTION
- VISCERAL HYPERSENSITIVITY
- SEX STEROIDS
- IRRITABLE BOWEL SYNDROME
- NEUROPHARMACOLOGY
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Women are more susceptible to functional pain syndromes including IBS.
Oestrogen facilitates visceral pain.
Epigenetic mechanisms are potential targets to relieve somatic pain.
What are the new findings?
Histone deacetylase (HDAC) inhibitors attenuate oestrogen-induced visceral hypersensitivity in the rat.
HDAC inhibitors upregulate metabotropic glutamate receptor 2 mRNA and protein in the spinal cord.
HDAC inhibitors increase binding of histone 3 hyperacetylated at lysine 9 and ligand-bound oestrogen receptor α to the same regions of the GRM2 promoter.
An mGlu2/3 receptor antagonist reversed the inhibitory effect of HDAC inhibitors.
How might it impact on clinical practice in the foreseeable future?
Inhibition of HDAC may be a potential approach to relieve visceral pain, a defining characteristic of IBS.
Introduction
Visceral pain is one of the most common reasons for clinical visits and it impacts the quality of life of millions of people. Currently, visceral pain is poorly managed because the underlying mechanisms remain largely unclear. In general, women exhibit a higher morbidity of functional pain syndromes such as IBS than men and oestrogen is thought to be a critical player in IBS severity.1 ,2 Our previous studies in animal models have demonstrated that oestrogen facilitates colorectal distention-evoked visceral pain through activation of oestrogen receptor α (ERα) in the spinal cord.3 ,4 ERα is a nuclear receptor that primarily functions as a transcription factor to regulate promoter activity.5 Therefore, regulation of gene expression is critical to mediate oestrogen's effects on visceral sensitivity, although little is known of the underlying mechanism.
Recently, it has been reported that gene transcription or expression is regulated by genetic and by epigenetic mechanisms that are independent of genomic DNA sequences and impacted largely by environmental changes.6 ,7 Three molecular mechanisms, that is, DNA methylation, chromatin remodelling and non-coding RNAs, are involved in epigenetic regulation.6–8 Histones are basic structures of chromatin, and post-translational modification of histones remodels chromatin structure regulating transcriptional activity.7 Acetylation of lysine residues in the N-terminal of histones is a well-studied histone modification. Increase in histone acetylation following pharmacological inhibition of histone deacetylase (HDAC) has been found to attenuate inflammatory and neuropathic pain in animal studies.9–13 However, it remains largely unknown whether histone acetylation is critical to modulate visceral sensitivity.
In this study, we tested our working hypothesis that epigenetic mechanisms are critical to modulate visceral pain at the spinal level. We applied HDAC inhibitors to the spinal cord in ovariectomised rats receiving 17β-estradiol (E2) and found that HDAC inhibition increased histone acetylation in the spinal dorsal horn and attenuated E2-induced visceral hypersensitivity. We also observed that HDAC inhibition significantly increased binding of acetylated histone 3 and ERα to selective regions of the promoter of the metabotropic glutamate receptor 2 (mGluR2) gene (GRM2) and upregulated mGluR2 mRNA and protein. Blocking mGluR2 activity disrupted the antinociceptive effect of HDAC inhibition. These data suggest that HDAC inhibition interacts with E2-activated ERs to modulate visceral sensitivity.
Materials and methods
Animals
Experimental protocols were approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee and adhered to guidelines for experimental pain in animals published by the International Association for the Study of Pain. Female Sprague-Dawley rats weighing 225–250 g were obtained from Harlan. Rats were housed in pairs with free access to food and water at 25°C with 12 h/12 h alternating light/dark cycle.
Surgery
Rats were anaesthetised with 55 mg/kg ketamine, 5.5 mg/kg xylazine and 1.1 mg/kg acepromazine. Ovariectomies (OVx) were performed by a dorsolateral approach. In order to direct drugs to the spinal cord, the atlantooccipital membrane was slit and a polyethylene catheter (32 g, ReCathCo, Allison Park, Pennsylvania, USA) was inserted in the subdural space to the level of the lumbosacral spinal cord (L6–S2). After catheter placement, electromyogram (EMG) electrodes made from Teflon-coated 32 gauge stainless steel wire (Cooner Wire Company, Chatsworth, California, USA) were stitched into the ventrolateral abdominal wall. The electrode leads were tunnelled subcutaneously and exteriorised at the back of the neck with the catheter. Rats were individually housed and allowed 10–14 days to recover from surgery.
Visceromotor response
Rats were fasted for 18–24 h prior to recording the visceromotor response (VMR). Water was available ad libitum. On the day of testing, rats were briefly sedated with isoflurane and a 5–6 cm balloon attached to Tygon tubing was inserted into descending colon and rectum through the anus. The secured end of the balloon was at least 1 cm proximal to the external anal sphincter and the tubing was taped to the tail. Rats were loosely restrained in acrylic tubes and given 30 min to recover from sedation. The EMG signals were recorded with a CED 1401 and analysed using Spike 2 for Windows software (Cambridge Electronic Design, Cambridge, UK). Colorectal distention trials (CRD; each trial was five distentions to 60 mm Hg CRD, 20 s duration, 3 min interstimulus interval) were produced by inflating the distention balloon with air under computer control. The recorded EMG signal was rectified in Spike 2 and the area under the curve (AUC) calculated. The response for each distention stimulus was the AUC of the EMG for the 20 s before distention (background) subtracted from the AUC during the 20 s distention. The responses of the five distentions during a trial were averaged for the response for that trial.
Tissue collection
Rats were euthanised with CO2 and decapitated at selected time points after treatment. The spinal cord was removed by pressure ejection with ice-cold saline as described previously.14 Using the lumbar enlargement as a landmark, the L6 to S2 section of the spinal cord was isolated. The dorsal part of isolated spinal cord was dissected, frozen on dry ice and saved at −80°C until use.
Western blots analysis
Tissues were homogenised in radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate (SDS)) supplemented with 1 mM Na3VO4 and protease inhibitor cocktail (Roche, Branford, Connecticut, USA). The homogenates were centrifuged at 14 000×g for 10 min at 4°C, and the supernatant was collected. Protein contents in supernatants were measured using the Bradford method. After denaturing, protein samples were fractionated 25 µg per lane on 4%–12% SDS-NuPAGE gel and blotted to nitrocellulose membrane. The membranes were incubated with primary antibody (1:1000) directed against acetylated lysine 9 on histone 3 (H3K9ac, Cell Signaling Technology, Danvers, Massachusetts, USA) or mGluR2 (Abcam, Cambridge, Massachusetts, USA) at 4°C overnight. The membranes were further incubated for 1 h in blocking buffer with IRDye 800CW goat antirabbit secondary antibody (Li-Cor Biosciences, Lincoln, Nebraska, USA). Washed membranes were scanned and analysed with the Li-Cor Odyssey System (Li-Cor Biosciences). Blots were then stripped for 30 min and reprobed with antibodies for pan-histone 3 (1:1000, Cell Signaling Technology) or β-actin primary antibody (1:5000, Sigma, St. Louis, Missouri, USA) and IRDye 800CW goat antirabbit secondary antibody or IRDye 680LT goat antimouse secondary antibody (1:20 000, Li-Cor Biosciences) with same processes as above.
Quantitative reverse transcription PCR
Total RNA was extracted from the dissected L6–S2 dorsal spinal cord using absolutely RNA miniprep kit (Stratgene, La Jolla, California, USA) and reverse transcribed into cDNAs using SuperScript II Reverse Transcriptase kit with random primers (Invitrogen, Grand Island, New York, USA). Quantification of rat mGluR2 and mGluR3 mRNAs was completed by SYBR green-based real-time PCR (qPCR). PCR primers were designed based on mRNA sequences to cross at least one exon/exon junction (GenBank accession number NM_001105711 for mGluR2 and NM_001105712 for mGlu3) using Primer3 software. Three pairs of primers per mRNA were evaluated and one each (mGluR2, forward, 5′-CGTGAGTTCTGGGAGGAGAG, reverse, 5′- GCGGACCTCA TCGTCAGTAT; mGluR3, forward, 5′-GTGGTCTTGGGCTGTT TGTT, reverse, 5′-GCAGCATGTGAGCACTTTGT) with comparable PCR efficiency to that of the glyceraldehyde-3-phospate dehydrogenase (GAPDH) mRNA (forward, 5′-TCACCACCATG GAGAAGGC; reverse, 5′-GCTAAGCAGTTGGTGGTGCA) was chosen. qPCR was completed in Maxima SYBR Green/Rox qPCR Master Mix (Thermo Scientific, Waltham, Massachusetts, USA) on the Eppendorf Mastercycler Real-plex system (Eppendorf, Hauppauge, New York, USA), and CT values were obtained from the system. The efficiency of PCR was calculated from the slope of the standard curve of serially diluted cDNA and was found within the range of 90%–110%. Relative level of mRNA was calculated using the ΔΔCT method after normalisation to GAPDH mRNA as described previously.15
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using a previously described method with the following modifications.16 Half of the dissected spinal dorsal horn was minced in 1× phosphate buffered saline (pH 7.4) supplemented with protease inhibitors and subjected to DNA–protein cross-link by incubation with 1.5% paraformaldehyde for 20 min at room temperature. After homogenisation on ice, the homogenate was centrifuged and the resulting pellet incubated in swelling buffer (25 mM Hepes, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5% NP-40) on ice for 15 min and subjected to a second homogenisation. Nuclei were isolated by centrifugation, broken in lysis buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 1% SDS, protease inhibitor cocktail, 100 µg/mL valproic acid) and sonicated in the Covaris M2 system to yield DNA fragments around 500 bp. For immunoprecipitation, 3 µg of polyclonal antibodies specific to H3K9ac or H3K18ac (Cell Signaling Technology, Danvers, Massachusetts, USA) or to ERα (Millipore, Temecula, California, USA) were incubated overnight with 200 µg protein at 4°C. The antibody–antigen complexes were pulled down by protein A/G-agarose followed by successive washes once each with low-salt buffer (20 mM Tris-HCl, pH 8.0, 150 mM KCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100), high-salt buffer (the same as low-salt buffer except 500 mM KCl), LiCl buffer (10 mM Tris-HCl, pH 8.0, 250 mM LiCl, 1 mM EDTA, 1% NP-40, 1% sodium dexoycholate) and twice in Tris-EDTA buffer (pH 8.0). The complex was eluted into solution of 100 mM NaHCO3–1% SDS. After reverse cross-link in 250 mM NaCl at 65°C for 6–12 h, DNA was purified by a Qiaquick column (Qiagen, Frederick, Maryland, USA). Lysate equal to 5% of the immunoprecipitate was directly subjected to reverse cross-link and Qiaquick column purification without antibody immunoprecipitation and served as input. Two per cent of eluted DNA was subjected to qPCR with SYBR green (Thermo Scientific, Waltham, Massachusetts, USA) in duplicate using primers covering five different regions in the GRM2 promoter (figure 4A). qPCR was completed on the Eppendorf RealPlex2 system with a programme of initial denaturing at 95°C for 4 min followed by cycling at 95°C for 15 s and 57°C –64°C for 30 s.
Two negative controls were performed. First, in a mock ChIP, immunoglobulin (IgG) purified from non-immunised rabbit serum (VWR, Philadelphia, Pennsylvania, USA) was added to replace antibody during immunoprecipitation for initial studies of ChIP specificity. Second, a 187 bp intergenic locus in rat chromosome 4 was examined by qPCR from ChIP elutes and inputs of every sample, which was then used for calculation. Primers used for this negative locus are forward, 5′-TGCAAACTTCACACGTCCTC, and reverse, 5′-GGGGTGGAAATTTCTGGTTT. Antibody-enriched experimental DNA over the negative control was calculated using the ΔΔCT method with a normalisation of immunoprecipitation data to relevant input of each sample for the promoter regions and the intergenic locus17 (see online supplementary figure S2). Six rats per group were tested for each pair of primers in ChIP.
Hormones and drugs
E2 was dissolved in safflower oil to a concentration of 50 µg/100 µL. E2 (100 µL) or safflower oil was injected subcutaneously. Suberoylanilide hydroxamic acid (SAHA) (Cayman Chemical, Ann Arbor, Michigan, USA), made fresh, was dissolved in 20% dimethyl sulfoxide (DMSO) to a final concentration of 5–80 µg/20 µL. The HDAC inhibitor trichostatin A (TSA) and mGluR2/3 antagonist LY341495 (Tocris Bioscience, Bristol, UK) were also dissolved in 20% DMSO. For intrathecal injection, 20 µl volume of drug was injected followed by a 10 µL saline flush.
Data analysis
All data are expressed as mean±SEM. Data were analysed by one-way or two-way analysis of variance (ANOVA) as appropriate. Significant ANOVA results were further analysed using Bonferroni's multiple comparison test and reported in the figures. t Test was used for two-group comparison. p<0.05 was considered significant.
Results
HDAC inhibitors attenuate the E2-faciliated VMR and increase histone acetylation
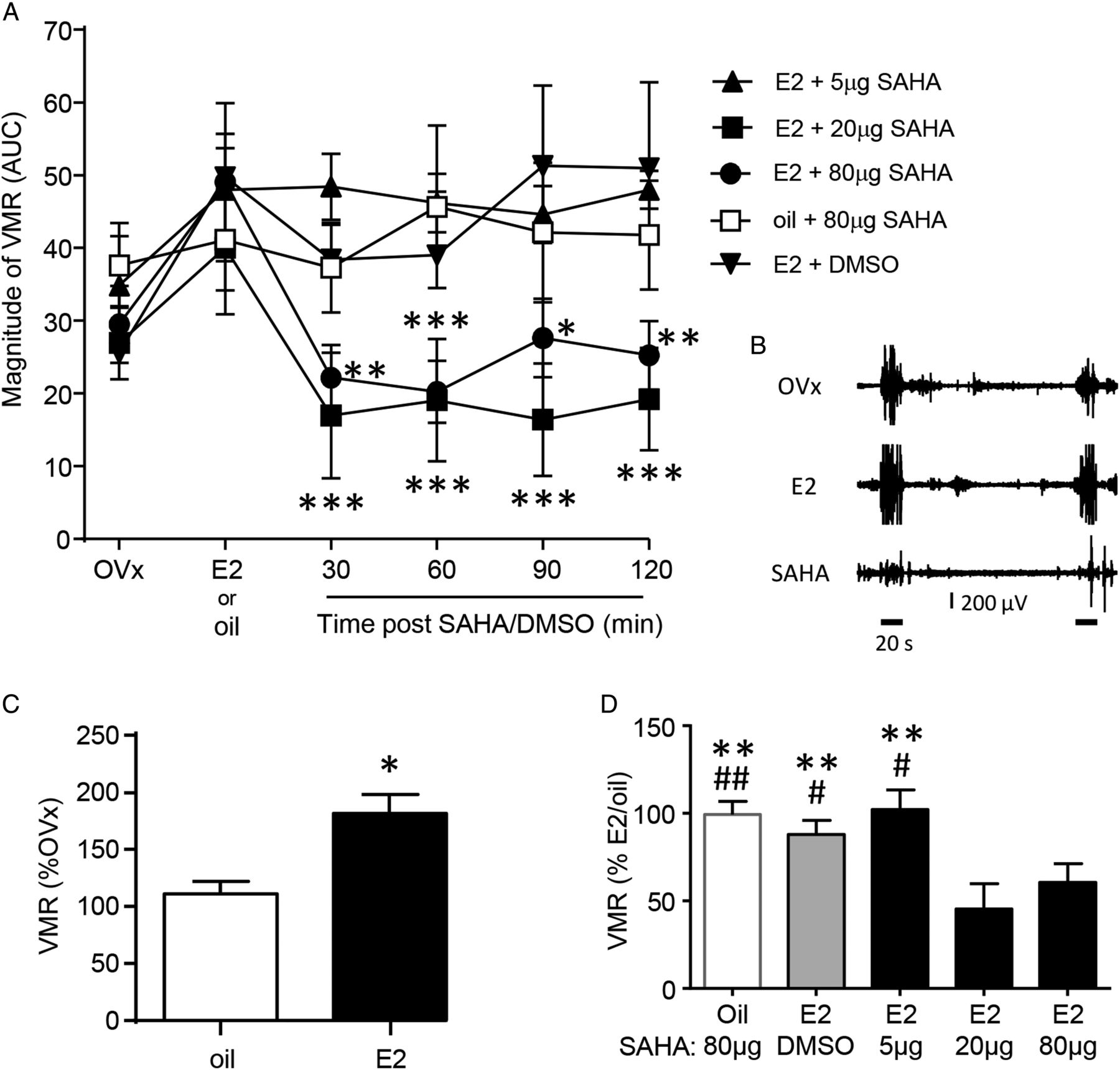
Ten to fourteen days following ovariectomy, the baseline VMR was recorded. Rats were subsequently injected with E2 or safflower oil and the VMR recorded again 4 h later. Rats were then injected with SAHA or vehicle and the VMR recorded over the next 2 h. There was no difference in the baseline response (labelled OVx in figure 1A, one-way ANOVA, p>0.05) between treatment groups. Following E2, but not oil injection, there was a significant increase in the magnitude of the VMR as previously reported (t test, p<0.05; figure 1C).3 ,4

Magnitude of the VMR following intrathecal injection of the HDAC inhibitor SAHA or vehicle in E2-treated or oil-treated rats. (A) SAHA (20 and 80 μg) attenuated the E2-facilitated VMR. *p<0.05, **p<0.01, ***p<0.001 versus E2 response in each treatment group. n=5–10 in each treatment group. (B) Example electromyogram recordings from a rat in the 20 µg SAHA group showing the baseline response (top; OVx), 4 h post-E2 (middle; E2) and 30 min post-SAHA injection (bottom; SAHA). Two distentions are indicated by the 20 s horizontal lines under the bottom trace. (C) Average magnitude of the VMR in oil-treated or E2-treated rats normalised to their OVx (baseline) response. Data were derived from the OVx and E2/oil points in (A), the E2 data were pooled from all E2-treated groups. E2, but not oil, increased the magnitude of the VMR. *p<0.05 versus oil. (D) Average magnitude of the VMR from 30 min to 2 h post-SAHA or DMSO (vehicle) injection normalised to E2/oil response. **p<0.01 versus E2+20 μg SAHA; #p<0.05,##p<0.01 versus E2+80 μg SAHA. VMR, visceromotor response; HDAC, histone deacetylase; SAHA, suberoylanilide hydroxamic acid; OVx, ovariectomies; DMSO, dimethyl sulfoxide; AUC, area under the curve.
The HDAC inhibitor SAHA or its vehicle DMSO was then intrathecally injected. The VMR trial was recorded starting 30, 60, 90 and 120 min after injection of SAHA/vehicle. SAHA at doses of 20 and 80 μg reversed the E2-facilitated VMR for at least 2 h (one-way repeated measures (RM) ANOVA for each treatment group, p<0.0001 and 0.01, respectively; figure 1A). A lower dose of SAHA (5 μg) or vehicle (20% DMSO) had no effect on the E2-facilitated VMR (one-way RM ANOVA, p>0.05 for both). In the absence of E2 (safflower oil-treated ovariectomised rats), SAHA showed no effect on the magnitude of the VMR compared with the OVx or post-oil-injected state (one-way RM ANOVA, p>0.05).
Within each treatment group, the response post-SAHA remained stable over the 2 h recording period so the response was averaged and normalised to the E2 or oil response prior to SAHA/DMSO injection. Comparison between treatment groups shows that 20 and 80 µg SAHA significantly attenuated the magnitude of the VMR compared with 5 µg SAHA or DMSO in E2-treated rats or 80 µg SAHA in oil-treated rats (one-way ANOVA, p<0.005, figure 1D).
The reversal of the E2-faciliated VMR by HDAC inhibition was confirmed by intrathecal administration of another HDAC inhibitor, TSA (20 µg, one-way RM ANOVA, p<0.005, see online supplementary figure S1). A lower dose of TSA (5 µg) had no effect (one-way RM ANOVA, p>0.05). Similar to SAHA, TSA had no effect on the VMR in the absence of E2 (one-way RM ANOVA, p>0.05).
Western blots were used to confirm that SAHA inhibited HDAC and increased global histone acetylation in the spinal cord. Rats were treated with E2 or safflower oil for 4 h followed by SAHA (80 µg) or DMSO treatment for 30 min or 2 h before tissues were collected for western blot analysis. SAHA significantly increased H3K9ac in E2-treated animals compared with the E2+DMSO (two-way ANOVA, p<0.001 at 30 min post-SAHA; p<0.01 at 2 h post-SAHA, figure 2). SAHA had no effect on histone acetylation in safflower oil-treated animals compared with the oil+DMSO (p>0.05 at both 30 min and 2 h post-SAHA). Interestingly, E2 alone did not alter the level of histone acetylation (E2+DMSO group vs oil+DMSO group, p>0.05 at both 30 min and 2 h post-DMSO).

Intrathecal injection of SAHA, but not its vehicle DMSO, increased the acetylation level of H3K9ac in E2-treated rats at 30 min (A) and 2 h (B) post-SAHA injection. **p<0.01, ***p<0.001 versus E2+DMSO. # p<0.05, ##p<0.01 versus oil+SAHA. n=2 for oil+DMSO group and n=4 for other groups at 30 min post-SAHA injection. n=4 for oil+DMSO group and n=3 for other groups at 2 h post-SAHA injection. SAHA, suberoylanilide hydroxamic acid; DMSO, dimethyl sulfoxide.
These data show that E2-induced visceral hypersensitivity is attenuated by spinal administration of HDAC inhibitors. In addition, the HDAC inhibitor increased global histone acetylation in the presence of E2.
HDAC inhibitor attenuation of the E2-facilitated VMR is mGluR2 mediated
The simplest explanation for the inhibitory effect of SAHA on visceral sensitivity is an increase in spinal inhibitory processing. Considering that HDAC inhibitors attenuated nociceptive responses in the formalin test through upregulation of mGluR2,11 we tested the hypothesis that SAHA upregulates mGluR2 to strengthen inhibitory processing and in turn attenuate visceral hypersensitivity. Results of quantitative reverse transcription PCR (qRT-PCR) revealed that the mGluR2 mRNA level significantly increased 2 h after SAHA (80 µg) injection in E2-treated rats (two-way ANOVA, p<0.05; figure 3A). In comparison, injection of SAHA following oil treatment at that time point had no effect on mGluR2 mRNA. There were no significant differences in mGluR2 mRNA among groups at 30 min and 1 h after SAHA injection (two-way ANOVA, p>0.05). Consistent with the mRNA change, mGluR2 protein expression also significantly increased 2 h after SAHA injection in E2-treated rats in comparison with DMSO-treated rats (two-way ANOVA, p<0.05, figure 3B). However, there was no difference in the expression level of mGluR3 mRNA at any time point (figure 3C) or protein 2 h (not shown) after SAHA administration (two-way ANOVA, p>0.05).

Intrathecal injection of SAHA increased mGluR2 mRNA (A) and mGluR2 protein expression (B), but not mGluR3 mRNA (C), in E2-treated rats 2 h post-SAHA. *p <0.05 (n=3–5/group). SAHA, suberoylanilide hydroxamic acid; mGluR2, metabotropic glutamate receptor 2; DMSO, dimethyl sulfoxide.
Since the transcription rate is sensitive to histone acetylation and directly impacts mRNA levels, we applied ChIP to examine the interaction between acetylated histones to the GRM2 promoter following E2+SAHA treatment. The rat GRM2 promoter was defined for the region upstream of and surrounding the transcriptional start site (TSS) or the first base of exon 1, which has 397 bp sequences as provided by the University of California Santa Cruz genome database (genome.ucsc.edu). We searched putative cis-elements within ±2 kb of the TSS using TRANSFAC software (BIOBASE Biological Database, Qiagen, Frederick, Maryland, USA) and designed primers to cover five regions containing motifs for potential positive transcription factors (figure 4A). After optimising the PCR programme, we were able to obtain a single band of the expected size from each pair of primers from gDNA precipitated with an antibody against H3K9ac in ChIP of naive animals (figure 4C). The specificity of these primers was also confirmed by the single peak in melting curves of SYBR green qPCR (see online supplementary figure S3). In comparison, these primers were unable to generate signal or band from negative control of IgG mock ChIP elute (figure 4C). We also examined an intergenic locus as negative control for calculation of ChIP results. This locus does not contain binding sites for oestrogen receptors and exhibited a low binding to acetylated histone 3 (see online supplementary figure S4). Our results demonstrate that E2+80 µg SAHA treatment significantly increased binding of H3K9ac to three regions (A1, A4, A5) of the GRM2 promoter (t test, p<0.05) while binding of H3K9ac to the other two regions and binding of H3K18ac to all five regions revealed no significant change compared with E2+DMSO (figure 4D).

HDAC inhibition on binding of acetylated histone 3 or oestrogen receptor α (ERα) to the GRM2 promoter. (A) PCR primers designed to cover the proximal transcriptional regulatory region are shown schematically. (B) Primer sequences, relation to TSS, amplicon size and relevant cis-elements. (C) Sample of PCR amplicons fractionated on agarose gel. (D) Following immunoprecipitation with an antibody to H3K9ac, binding to five regions of the GRM2 promoter was quantified. SAHA increased H3K9ac enriched amplicons 1, 4 and 5 compared with DMSO in E2-treated rats. (E) Following immunoprecipitation with an antibody to ERα, binding to three regions of the GRM2 promoter was quantified. SAHA increased ERα binding to amplicons 4 and 5 compared with DMSO in E2-treated rats. *p<0.05, **p<0.01, ***p<0.001 for E2+SAHA compared with E2+DMSO, n=6 for each group. HDAC, histone deacetylase; SAHA, suberoylanilide hydroxamic acid; DMSO, dimethyl sulfoxide; IgG, immunoglobulin G; ERα, oestrogen receptor α; TSS, transcriptional start site; SRF, serum response factor.
Interestingly, atypical or half oestrogen response elements (aERE) were found in the A2 (TGACC, −1106/−1102), A4 (GTGAC, −503/−499) and A5 (GGGTCA, 1133/1138) regions of the GRM2 promoter. It has been reported that more than 50% of oestrogen receptor-bound sequences are atypical sites.18 To examine whether these aEREs are functional, we performed ChIP with a polyclonal antibody against ERα and found that regions A2, A4 and A5 were precipitated by this antibody (figure 4E). After normalisation to the input, A5 had the highest binding capacity. Furthermore, similar to the increase in H3K9ac binding, SAHA significantly increased ERα binding to A4 and A5 (t test, p<0.05). In comparison, the low binding level of ERα to A2 was not altered by SAHA, consistent with no change in H3K9ac binding to this region. These data indicate that hyperacetylation of K9 in histone 3 is associated with increased binding of ligand-bound ERα to the GRM2 promoter and with upregulation of GRM2 transcription.
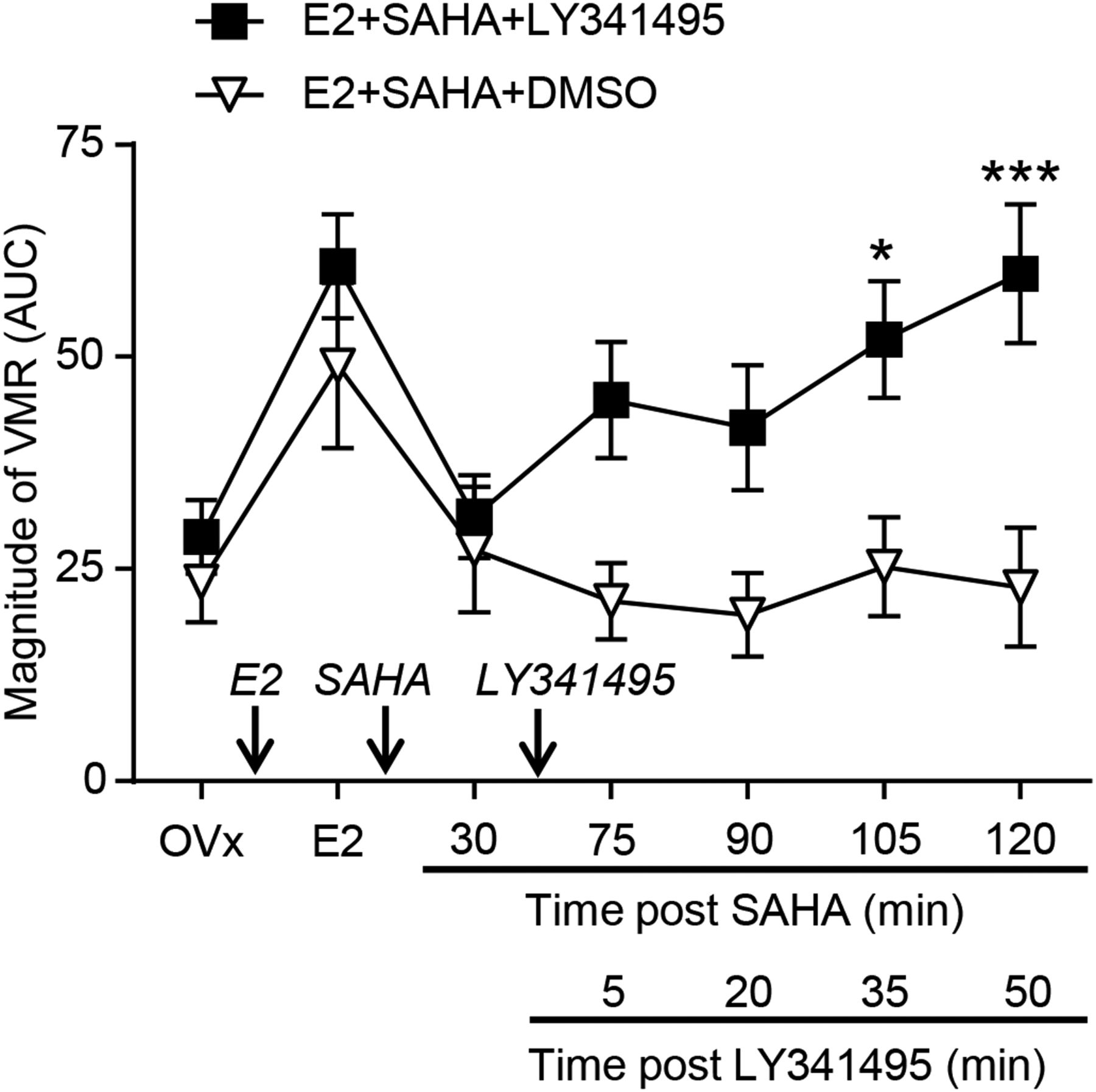
Due to the observed increase in mGluR2 expression and the binding of H3K9ac and ERα to the GRM2 promoter in E2-SAHA-treated animals, we next determined whether there was any functional significance of these changes. Thirty minutes following SAHA (80 µg) injection and establishing attenuation of the E2-faciliated VMR, the mGluR2/3 antagonist LY341495 (20 nmol, intrathecal) was injected (figure 5). Starting 5 min later the VMR was recorded over the next hour. LY341495 gradually reversed the attenuation of the VMR evoked by SAHA, and the magnitude of the VMR was significantly increased at 35 and 50 min after LY341495 application (ie, 105 and 120 min after SAHA application) compared with the VMR at same time point in E2 plus SAHA-treated animals receiving DMSO (vehicle for LY341495) (two-way RM ANOVA, p<0.01 for treatment; figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Magnitude of VMR following intrathecal injection of the mGlu2/3 receptor antagonist LY341495 (n=9) or vehicle (n=8) in E2+SAHA-treated rats. *p<0.05, ***p<0.001 versus E2+SAHA+DMSO. AUC, area under the curve; SAHA, suberoylanilide hydroxamic acid; mGluR2/3, metabotropic glutamate receptor 2/3; DMSO, dimethyl sulfoxide; OVx, ovariectomies; VMR, visceromotor response.
Discussion
In this study, we show that increasing histone acetylation by spinal administration of HDAC inhibitors attenuated E2-induced visceral hypersensitivity. We further demonstrated that SAHA reversed this E2-induced hypersensitivity by increasing mGluR2 expression in the spinal cord and that SAHA treatment significantly increased binding of H3K9ac to three of five tested regions in the GRM2 promoter. More importantly, SAHA also significantly increased binding of E2-activated ERα specifically to the GRM2 promoter regions, which contain aERE and exhibited increased binding to H3K9ac. Spinal administration of the mGluR2/3 antagonist LY341495 reversed the effects of SAHA, confirming that epigenetic regulation of mGluR2 by HDAC inhibitors may contribute to attenuation of visceral pain. Previous reports have shown that the inhibitory effect of HDAC inhibitors on somatic pain may be due to epigenetic regulation of mGluR2 in the spinal cord11 ,19 and activation of oestrogen receptors triggers mGluR2/3 signalling to decrease cAMP response element-binding protein.20 ,21 Our data indicate that oestrogen receptors interact with mGluR2 in the spinal cord to regulate visceral pain through epigenetic mechanisms. To the best of our knowledge, this is the first report to show that an epigenetic mechanism modifies visceral pain at the spinal cord level and that the access of ligand-bound ERα to the promoter of an antinociceptive gene is enhanced by hyperacetylated histone interacting with the same promoter regions. Our findings provide a new approach to relieve existing visceral pain involving oestrogen function.
Our previous studies found that oestrogen increases sensitivity of the VMR to colorectal distention through activation of spinal ERα.3 ,4 Furthermore, E2 increased expression of the N-methyl-D-aspartate (NMDA) receptor subunit GluN1 and phosphorylation of GluN1 in the spinal cord, suggesting genomic and non-genomic actions of ER in facilitating visceral pain.14 In the nucleus, ERα bound and activated by oestrogen is generally considered a regulator (activator or repressor) of transcription or promoter activity via two different mechanisms.5 First, activated ERα directly binds an ERE on a promoter22 and regulates transcription. Second, ERα binds to a half ERE, termed aERE, and further interacts with nearby DNA bound-transcription factors including members of the nuclear factor-κB (NF-κB), Sp, serum response factor and Ap families.5 ,18 ,23 Consistent with this concept, the GluN1 gene Grin1 promoter contains an aERE and the elements for Sp and Ap factors.24
In this study, we identified several aERE elements from the promoter or regulatory region of the GRM2 gene. We found that ligand-bound ERα could not alter GRM2 transcription until histone 3 became hyperacetylated by SAHA and then binding of activated ERα to this promoter increased (figure 4E). As indicated by our ChIP data, ERα binds only to these aERE elements that are located in the region(s) whose binding to acetylated H3 is enhanced by E2+SAHA (A4, A5 in figure 4D, E). An interaction between ERα and hyperacetylated histones is further supported by the observation that histone hyperacetylation by SAHA did not alter the nociceptive response in control animals in the absence of E2 (figure 1). It has been observed previously that binding of transcription factors such as NF-κB to the promoter may trigger histone acetylation surrounding ERE sites, resulting in recruitment of ER.23 ,25 On the other hand, in most cases, binding of ER to the promoter recruits factors with histone acetylase activity increasing histone acetylation on the promoter and recruiting other transcription factors for transcription regulation.25 ,26 Here we provide a novel mechanism in which access of ligand-bound or activated ERα to aERE on the promoter depends first on acetylation of K9 on histone 3 binding to sequences encompassing these elements and this mechanism is important for the expression of the antinociceptive gene, GRM2. This novel mechanism of ERα action supports the notion that histone acetylation allows transcription factors to access promoters and regulate transcription.6 ,7 In addition, this regulation may explain why E2 is antinociceptive under some conditions (eg, histone hyperacetylation increasing mGluR2) while it is pronociceptive under other conditions (eg, increasing NMDA receptor function).
It has been reported that acetylated histones bind transcribed genes in a bell-shape centring on the TSS.27 Our ChIP data showed acetylated histone 3 binding starting approximately 1 kb upstream of the GRM2 TSS without a robust increase closer to the GRM2 TSS. There are three possible reasons to account for this different pattern. First, relatively large amplicons were examined in our qPCR, which differs from small tiled amplicons used for mapping acetylated histone binding to the genome. Thus, the large amplicon may mask any sudden change in acetylated histone binding. Second, this acetylated histone 3 binding distal to the GRM2 TSS might suggest multiple TSS, which may be either an alternative TSS of the GRM2 gene or a TSS for an unknown gene.28 Lastly, the spinal cord is composed of a heterologous population of cells. It is known that GRM2 is expressed only in a limited portion of dorsal horn neurons (Allen Institute.org) while our samples of spinal cord consist of a large number of multiple types of cells including neurons and glia. This may mask the pattern of acetylated histone binding reported in homologous populations of cultured cells.27 Nevertheless, our ChIP data combined with the pharmacological results support the notion that SAHA plus E2 treatment altered binding of acetylated histone to the GRM2 promoter and thus regulated mGluR2 expression and impacted E2-related visceral hypersensitivity.
Further, our finding that spinal administration of HDAC inhibitors attenuated oestrogen-induced facilitation of visceral pain is consistent with recent reports that increasing spinal histone acetylation attenuates hyperalgesia in neuropathic and inflammatory pain models.10 ,12 Intrathecal application of HDAC inhibitors does not increase histone acetylation in dorsal root ganglion,12 suggesting that the spinal cord instead of the dorsal root ganglion may be the main epigenetic regulation region in this study.
In this study, we showed that spinal mGluR2 is the downstream target of the inhibition of HDAC for attenuation of visceral pain. We also recognise possible roles of other genes as targets mediating the effects of histone hyperacetylation on the pain circuitry through epigenetic modulation. For example, HDAC inhibition in the nucleus raphe magnus enhanced gamma-amino butyric acid activity and thus relieved persistent pain.9 While the studies mentioned above focused on somatic pain, one recent study showed the contribution of epigenetic regulation to visceral pain. Tran et al29 reported that application of the HDAC inhibitor TSA into the cerebral ventricles attenuated chronic psychological stress-induced visceral hypersensitivity. In addition, this study focused on a noxious distention pressure as determined in normal rats. Future studies should also evaluate the effect of HDAC inhibitors on less intense distention pressures experienced during normal digestive functioning.
HDAC inhibitors have shown potential therapeutic efficacy in many rodent models of neurodegenerative diseases for neuroprotective function, preventing or delaying neuronal dysfunction. In addition, HDAC inhibitors including SAHA have been approved or are under evaluation in clinical trials for several neurological disorders and cancer.30–32 This study together with other studies9–12 provides evidence that HDAC inhibitors may be used for analgesia on both somatic and visceral pain. Spinal administration of HDAC inhibitors did not affect visceral pain in normal rats, suggesting that increasing histone acetylation may have specific function on pathophysiological condition and HDAC inhibitors may have few side effects when they are used for treatment.10
Acknowledgments
The authors thank Ms. Jane Karpowicz and Sangeeta Pandya for technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Contributors D-YC: data acquisition, data analysis and drafting of manuscript. GB: data acquisition, data analysis, study design and critical reading of manuscript. YJ: data acquisition, data analysis and critical reading of manuscript. RJT: study concept and design, obtained funding, study supervision and critical reading of manuscript. All authors read and agreed on the final manuscript.
Funding This work was supported by the National Institutes of Health (NIH) grant R01 NS 37424 to RJT.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.