


Abstract
Schizophrenic patients are thought to have an impaired ability to process sensory information. This deficit leads to disrupted auditory gating measured electrophysiologically as a reduced suppression of the second of paired auditoryevoked responses (P50) and is proposed to be associated with decreased function and/or expression of the homomeric α7 nicotinic acetylcholine receptor (nAChR). Here, we provide evidence that N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride (PNU-282987), a novel selective agonist of the α7 nAChR, evoked whole-cell currents from cultured rat hippocampal neurons that were sensitive to the selective α7 nAChR antagonist methyllycaconitine (MLA) and enhanced GABAergic synaptic activity when applied to hippocampal slices. Amphetamine-induced sensory gating deficit, determined by auditory-evoked potentials in hippocampal CA3 region, was restored by systemic administration of PNU-282987 in chloral hydrate-anesthetized rats. Auditory gating of rat reticular thalamic neurons was also disrupted by amphetamine; however, PNU-282987 normalized gating deficit only in a subset of tested neurons (6 of 11). Furthermore, PNU-282987 improved the inherent hippocampal gating deficit occurring in a subpopulation of anesthetized rats, and enhanced amphetamine-induced hippocampal θ oscillation. We propose that the α7 nAChR agonist PNU-282987, via modulating/enhancing hippocampal GABAergic neurotransmission, improves auditory gating and enhances hippocampal oscillatory activity. These results provide further support for the concept that drugs that selectively activate α7 nAChRs may offer a novel, potential pharmacotherapy in treatment of schizophrenia.
It is recognized that development of schizophrenia is genetically influenced, and a subset of genes are predisposing to the illness. Among a number of genetic linkage sites, the homomeric, α7 nicotinic acetylcholine receptor (α7 nAChR) subunit gene CHRNA7 has been implicated in schizophrenia (Stassen et al., 2000; Gault et al., 2003). Thus, a genetic linkage of the 15q13-15 region of chromosome 15 containing CHRNA7 has been established to impaired auditory gating, a presumed indicator of dysfunctional sensory processing in schizophrenia (Freedman et al., 1997; Leonard et al., 2002). Deficiency in auditory (P50) gating has been regarded as a manifestation of an impaired sensory filtering mechanism leading to inefficient sensory processing and disturbed perception in schizophrenic patients (Light and Braff, 1999; Freedman et al., 2003; Thoma et al., 2003). Based on the previous clinical observation that nicotine transiently improves auditory gating in schizophrenics (Adler et al., 1993) and the association between α7 nAChRs and auditory gating in preclinical models (Stevens et al., 1998), it has been proposed that activation of α7 nAChRs would improve sensory processing and thus provide benefit for positive and/or negative symptoms, or impaired cognitive function in schizophrenic patients (Stevens et al., 1998; Hajos et al., 2003b; Bodnar et al., 2004; Martin et al., 2004).
We have recently described PNU-282987 [N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] as a potent and selective α7 nAChR agonist (Bodnar et al., 2004). This compound showed high affinity for the rat α7 nAChR (Ki = 26 nM) and activity at the α7-5-HT3 chimera (EC50 = 128 nM) and showed a negligible block of α1β1γδ and α3β4 nAChRs (≥60 μM). In addition, PNU-282987 was found to be inactive at all tested monoamine, muscarine, glutamate, and GABA receptors at 1 μM concentration, except 5-HT3 receptors (Ki = 930 nM; Bodnar et al., 2004). In the present study, we further evaluated its in vitro pharmacological characteristics and its action on auditory gating processes. PNU-282987 was compared with reference α7 nAChR agonists for functional activity using cultured rat hippocampal neurons and for the ability to modulate GABAergic synaptic activity in isolated rat hippocampal slices. To evaluate the in vivo activity of PNU-282987, auditory gating experiments were carried out in anesthetized rats. Physiological gating in the hippocampal CA3 region or reticular thalamic nucleus (nRT) was disrupted by systemic administration of amphetamine (Stevens et al., 1996; Krause et al., 2003), and the efficacy of PNU-282987 to reverse the amphetamine-induced gating deficit was determined. The efficacy of the partial α7 nAChR agonist GTS-21 [DMXBA; 3-[(2,4-dimethoxy)benzylidene]-anabaseine dihydrochloride] (Briggs et al., 1997) was also evaluated in our hippocampal gating model since GTS-21 has been shown previously to improve the inherent auditory gating deficits in DBA mice (Stevens et al., 1998) or in isolation-reared rats (O'Neill et al., 2003).
It is known that enhanced catecholamine neurotransmission in the hippocampal formation leads to synchronized activity, i.e., θ oscillation in the hippocampus (Berridge and Foote, 1991; Hajos et al., 2003a), and pronounced hippocampal θ activity has been demonstrated after systemic administration of amphetamine (Krause et al., 2003). Since hippocampal θ activity is thought to be associated with synaptic plasticity and hippocampal-dependent cognitive processes (Buzsaki 2002; Seager et al., 2002), and cognitive-enhancing compounds have been shown to augment evoked θ activity (Kinney et al., 1999), possible modulations of amphetamine-induced hippocampal θ activity by PNU-282987 were also analyzed. Interestingly, a subset of rats used in the present study (approximately 5%) showed consistent impairment in hippocampal auditory gating at control measurements. These animals were not treated with amphetamine; instead, the ability of PNU-282987 and GTS-21 was tested to normalize their inherent gating deficit.
Materials and Methods
Cell Isolation and Culture Conditions. Sprague-Dawley rats (postnatal day 3) were killed by decapitation, and their brains were removed and placed in ice-cold Hibernate-A medium. Hippocampal regions were gently removed, cut into small pieces and placed in Hibernate-A medium with 1 mg/ml papain for 60 min at 35°C. After digestion, the tissues were washed several times in Hibernate-A media and transferred to a 50-ml conical tube containing 6 ml of Hibernate-A medium with B27 supplement (2%). Neurons were dissociated by gentle trituration through a series of three 9-inch Pasteur pipettes with decreasing tip diameters. Cells were purified over a Nycoprep gradient according to the methods of Brewer (1997). Cells were plated onto poly-d-lysine/laminin-coated coverslips at a density of 300 to 700 cells/mm2, allowed to adhere for 1 h at room temperature, and then transferred to 24-well tissue culture plates containing warmed culture medium composed of Neurobasal-A medium, B27 supplement (2%), l-glutamine (0.5 mM), 100 U/ml penicillin, 100 mg/ml streptomycin, and 0.25 mg/ml Fungizone. Cells were maintained in a humidified incubator at 37°C and 6% CO2 for 1 to 2 weeks. The medium was changed after 24 h and then approximately every 3 days thereafter.
Brain Slice Isolation. Sprague-Dawley rats ranging from postnatal day 16 to 21 were anesthetized with halothane, decapitated, and the brains were removed and blocked. The region containing the hippocampus was sectioned into 350-μm slices (Microslicer, DSK model 1500E; Dosaka, Tokyo, Japan) under ice-cold slicing buffer composed of 130 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 3 mM KCl, 0.5 mM CaCl2, 10 mM MgCl2, 10 mM glucose, 0.4 mM ascorbic acid, and 0.2 mM lidocaine continuously bubbled with a mixture of O2, CO2 (95:5). Slices were warmed slowly to room temperature in the same bath solution as described above but with 1 mM Ca2+ and no lidocaine; the slices were allowed to recover for at least 1 h before recording.
Patch-Clamp Electrophysiology. Whole cell currents were recorded using an Axopatch 200B amplifier (Axon Instruments, Union City, CA). Analog signals were filtered at one-fifth the sampling frequency, digitized, stored, and measured using pCLAMP software (Axon Instruments). Patch pipettes were pulled from borosilicate capillary glass using a Flaming/Brown micropipette puller (P97; Sutter Instrument Company, Novato, CA) and filled with an internal pipette solution composed of 126 mM CsCH3SO3, 10 mM CsCl, 4 mM NaCl, 1 mM MgCl2, 0.5 mM CaCl2, 5 mM EGTA, 10 mM HEPES, 3 mM ATP-Mg, 0.3 mM GTP-Na, and 4 mM phosphocreatin, pH 7.2. QX314 (4 mM) was included in the pipette solution for experiments measuring synaptic activity in brain slices. The resistances of the patch pipettes when filled with internal solution ranging between 3 and 6 MΩ. All experiments were conducted at room temperature. Cultured cells were continuously superfused with an external bath solution containing 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4. Bicuculline (10 μM), CNQX (5 μM), and tetrodotoxin (0.5 μM) were included in the bath solution to diminish spontaneous synaptic activity. Compounds were delivered via a multibarrel fast perfusion exchange system (Warner Instrument, Hamden, CT). For experiments with brain slices, tissue was transferred to a recording chamber superfused with a recording buffer composed of 130 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 3 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 0.4 mM ascorbic acid, 0.01 mM AP-5, and 0.005 mM CNQX, saturated with O2/CO2 (95:5). The recording chamber was mounted on the stage of a Zeiss Axioscope microscope with infrared-differential interference contrast optics and water immersion objectives. Slices were continuously superfused with the recording buffer at 3 to 4 ml per minute. Either PNU-282987 or DMSO was applied by bath application; solution exchange was achieved in <2 min. All data are reported as mean ± S.E.M. Statistical analysis was performed with a two-tailed Student's t test for populations of unequal variance.
Animals and Surgical Procedures. Experiments were performed on male Sprague-Dawley rats (weighing 250-300 g) in chloral hydrate anesthesia (400 mg/kg i.p.), under an approved animal use protocol and were in compliance with the Animal Welfare Act Regulations (9 CFR parts 1, 2, and 3) and with the Guide for the Care and Use of Laboratory Animals, National Institutes of Health guidelines. The femoral artery and vein were cannulated for monitoring arterial blood pressure and administration of test compounds or additional doses of anesthetic, respectively. The anesthetized rat was placed in a Kopf stereotaxic frame, and unilateral craniotomy was performed above the regions of the reticular thalamus or CA3 region of the hippocampus. Body temperature of the rat was maintained at 37°C by means of an isothermal (37°C) heating pad (Braintree Scientific, Braintree, MA). After conclusion of experiments, animals were euthanized with an i.v. bolus of chloral hydrate; brains were removed, blocked, and frozen for histological verification of electrode placement.
Hippocampal EEG Recordings. Field potentials (electroen-cephalogram, EEG) were recorded from the CA3 region of the left hippocampus, 3.8 mm ventral, 3.5 mm posterior, and 3.0 mm lateral from bregma (Paxinos and Watson 1986), using a monopolar, stainless steel macroelectrode (Rhodes Medical Instruments, Woodland Hills, CA). Data were digitized and stored using the Spike2 software package (Cambridge Electronic Design, Cambridge, UK). Rhythmic synchronized (θ) and large-amplitude irregular hippocampal activities were distinguished in the EEG; quantitative EEG analysis was performed by means of Fast Fourier transformation (Hajos et al., 2003a). Power spectrum density of EEG was calculated between 0 and 12 Hz and determined in periods of 10 min before or after drug treatment. θ peak was defined as the highest power between 3 and 6 Hz. Auditory-evoked potentials were determined by measuring the potential difference between the positive and the negative deflection 20 and 40 ms after stimulation (P20 and N40), respectively. For quantification, 50 sweeps were averaged, and the amplitude was determined and the ratio of the response after the second stimulus (test, T) and the first stimulus (conditioning, C) was calculated. This T/C ratio is used as a measure of sensory (auditory) gating. Statistical significance was determined by means of two-tailed paired Student's t test.
Single Unit Recordings from Reticular Thalamic Nucleus. Glass microelectrodes filled with 2 mol/l NaCl and 2% pontamine sky blue (impedance 4-10 MΩ) were lowered 5.2 to 5.6 mm into the left nRT (3.0 mm posterior and 3.6 mm lateral with respect to bregma), using a hydraulic microdrive (Kopf Instruments, Tujunga, CA). To identify neurons in the auditory sector of the reticular thalamus, continuous auditory stimulation was presented during electrode descent. Spontaneously active nRT neurons were recorded extracellularly, and only those neurons that responded with activation to auditory stimuli were selected for our studies (Krause et al., 2003). Extracellular signals were amplified, low-pass filtered, and action potentials discriminated on-line (Neurolog System, Hertfordshire, UK). At the end of each experiment, dye was deposited iontophoretically from the recording electrode, and location of the electrode tip was verified by microscopic inspection of slide-mounted and cresyl violet-stained sections. Data were digitized, stored, and analyzed using the Spike3 software package. Firing rates and interspike time interval histograms were determined at baseline and after drug administration. Raster displays and peristimulus time histograms (PSTH) were constructed from the evoked responses to auditory stimulation on-line. The number of events (i.e., extracellularly recorded action potentials) before auditory stimulation and after the conditioning and test stimuli were determined using PSTHs. The number of events after the test stimulus divided by the number of events after the conditioning stimulus was called the T/C ratio. Statistical significance was determined by means of two-tailed paired Student's t test.
Auditory Stimulation. Auditory stimulation consisted of two consecutive tone bursts 10-ms duration at a frequency of 5 kHz. The sound pressure level was 95 dB between the ear bars as determined with a sound level meter (RadioShack, Fort Worth, TX). Tones were delivered through hollow ear bars. Recording hippocampal auditory gating, delay between the first “conditioning” stimulus and second “test” stimulus was 0.5 s. Due to the long-lasting activation of nRT neurons to auditory stimulus, gating of nRT neurons was tested by paired tones with 1-s interval between conditioning and test stimuli. The time interval between tone-pairs was 10 s for both hippocampal and nRT recordings.
Experimental Design and Drug Treatment. Baseline auditory gating was determined by an average of 50 sequential evoked potentials (hippocampal CA3 region) or PSTH (nRT neurons) in response to conditioning and test stimuli. Amphetamine (d-amphetamine sulfate, 1 mg/kg i.v.) was administered to disrupt sensory gating. Recordings of evoke potentials or PSTHs commenced 5 min after amphetamine administration, and four blocks of 25 evoked potentials were computed. Disruption in auditory gating was affirmed if the mean of the last 50 evoked potentials showed gating deficit equal or exceeding a 0.2 increase in T/C ratio. Auditory gating measurements started 5 min after i.v. administration of the drug or vehicle. Levels of auditory gating (T/C rations) have been determined from means of 50 subsequent evoked potentials at time intervals between 5 and 15 min, as well as between 15 and 30 min after drug or vehicle administration. In addition, auditory gating was calculated from all 100 evoked potentials after drug or vehicle treatment.
Materials. Cell culture reagents were purchased from Invitrogen (Carlsbad, CA). (-)-Nicotine tartrate salt, papain, bicuculline methiodide, CNQX, d-amphetamine sulfate, and terodotoxin with citrate buffer were purchased from Sigma-Aldrich (St. Louis, MO). Methyllycaconitine (MLA) was purchased from Sigma/RBI (Natick, MA). PNU 282987 and GTS-21 were obtained from the Department of Medicinal Chemistry (Pfizer, Inc., Kalamazoo, MI). For auditory gating experiments, compounds were dissolved in phosphate-buffered saline (PBS) based upon their salt weights and the concentrations were adjusted so that injection volumes equaled 1 ml/kg b.wt. Control animals received PBS.
Results
Activation of α7 nAChRs on Cultured Rat Hippocampal Neurons by PNU-282987 and Reference Agonists
Examples of whole-cell currents evoked by the α7 nAChR agonists nicotine, GTS-21 and PNU-282987 are shown in Fig. 1A. Agonists were applied for 1 s once every 30 s at a series of concentrations. Because hippocampal neurons express varying levels of functional α7 nAChRs, the nonselective agonist (-)-nicotine (100 μM) was applied to each cell to normalize the data for comparisons between cells. In addition, because multiple nicotinic receptor subtypes are expressed by these neurons (Alkondon and Albuquerque, 1993), nicotine-evoked currents were recorded in the absence and presence of the selective α7 nAChR antagonist MLA. To minimize the influence of other receptor subtypes, cells were included in this study only if the current evoked by nicotine was inhibited >90% by 10 nM MLA. As illustrated in Fig. 1B, some cells did express a small but measurable amount of nicotine-evoked current that was resistant to MLA, reflecting the fraction of current mediated by non-α7 nAChRs (traces shown in Fig. 1B were recorded from the same cell as those shown in the third row in Fig. 1A; peak nicotine-evoked currents were -289 and -19 pA in the absence and presence of MLA, respectively). In contrast, the current evoked by PNU-282987 was completely inhibited by 10 nM MLA in every cell tested, even those that had a MLA-resistant component to the nicotine response (Fig. 1B). These results suggest that PNU-282987 activated only MLA-sensitive or α7-containing nAChRs on the cell soma and/or proximal dendrites. The concentration-response of the three compounds are shown in Fig. 1C normalized to the peak current evoked by 100 μM nicotine.

Agonist-activation of nAChRs on cultured rat hippocampal neurons. A, whole-cell currents evoked by 1-s applications of nicotine (100 μM) and three concentrations of either nicotine (top row), GTS-21 (middle row), and PNU-282987 (bottom row). Sequential agonist challenges were separated by a 30-s washout period. Traces shown on any row were all recorded from the same cell. B, example of currents evoked by nicotine (100 μM) and PNU-282987 (30 μM) in the presence of 10 nM MLA. Both traces were recorded from the same cell as the traces shown in the third row of A. C, concentration-response relationships for nicotine, GTS-21, and PNU-282987. Data points represent the peak current evoked by the indicated concentration of the test compound normalized to the peak current evoked by 100 μM nicotine from the same cell.
PNU-282987 Elevates Spontaneous GABAergic Synaptic Activity in Hippocampal Slices
Previous work has demonstrated that within the rat hippocampus, nAChRs are predominantly expressed on GABAergic interneurons and that activation of those receptors modulates GABAergic synaptic activity (Alkondon et al., 1997; Jones and Yakel, 1997; Ji and Dani, 2000; Köfalvi et al., 2000). We therefore evaluated the ability of PNU-282987 to modulate GABAergic synaptic activity in acutely isolated rat hippocampal slices. Spontaneous GABAergic synaptic events were recorded from CA1 pyramidal neurons for 3 to 10 min under baseline conditions and then for an additional 10 min in the presence of either vehicle (0.1% DMSO) or PNU-282987. Bath application of 30 and 300 nM PNU-282987 more than doubled frequency of synaptic activity in about one-half the cells tested (3 of 6 cells and 5 of 11 cells for 30 and 300 nM, respectively), but the average change in frequency was significantly different from the vehicle control only for cells treated with 300 nM PNU-282987 (p = 0.002; Fig. 2). No clear effect was observed with 1 μM PNU-282987, possibly reflecting the desensitization of α7 nAChRs during the 10-min treatment.

PNU-282987 produces a long-lasting enhancement of GABAergic synaptic activity in hippocampal slices. A, example of synaptic events recorded under baseline conditions (left) and in the presence of 300 nM PNU-282987 (right). B, summary of change in frequency of spontaneous synaptic activity relative to baseline for 0.1% DMSO (vehicle) and PNU-282987 (30, 300, and 1000 nM). The mean change in synaptic activity was evaluated by comparing the activity measured during a 3- to 10-min baseline period to the activity measured during 10-min treatment with the vehicle or PNU-282987 from the same cell. The mean change in frequency was as follows: -8 ± 9% (n = 10) for 0.1% DMSO, 143 ± 65% (n = 6) for 30 nM PNU-282987, 103 ± 28% (n = 11) for 300 nM PNU-282987, and -13 ± 30% (n = 6) for 1000 nM PNU-282987.
Effects of nAChR Agonists on Auditory Gating in Anesthetized Rats
Auditory Gating in the Hippocampus. Hippocampal field potential recordings revealed evoked responses to auditory stimulation in anesthetized rats. Auditory gating, expressed as the ratio between evoked potentials to paired C and T stimuli was determined at baseline by an average of 50 subsequently evoked potentials. Systemic administration of amphetamine (1 mg/kg i.v.) disrupted auditory gating in the majority of the treated rats, as indicated by a significant increase of the T/C ratio (Figs. 3 and 4, A and B). The increase in T/C ratio was due both to an increase in amplitude in response to the test stimuli and a decrease in amplitude in response to the conditioning stimuli (Figs. 3 and 4C). Because the absolute level of disruption induced by amphetamine was somewhat variable, only rats showing ≥0.2 increase in the T/C ratio (approximately 70% of tested animals) were used for subsequent evaluation of α7 nAChR agonists or vehicle. In addition, rats with appropriate auditory gating displayed average amplitudes of conditioning-evoked potential over 100 μV, providing excellent signal/noise ratio for evaluating parallel changes in amplitudes of evoked potentials to conditioning and test stimuli induced by drug treatments.

Typical hippocampal auditory evoked potentials (summation of 50 subsequent evoked potentials) in response to conditioning and test stimuli (intertone interval 0.5 s) in control conditions, after administration of amphetamine (AMP; 1.0 mg/kg i.v.), and after a subsequent administration of PBS (A) or PNU-282987 (1 mg/kg i.v.) (B).

Hippocampal auditory gating expressed as a ratio between evoke potential amplitudes (n = 50) to test and conditioning stimuli (T/C ratio). Administration of amphetamine (1.0 mg/kg i.v.) disrupted auditory gating indicated by an increase in T/C ration. A, after a subsequent administration of vehicle (PBS; 1 ml/kg i.v., n = 6) auditory gating remained disrupted. B, administration of PNU-282987 (1 mg/kg i.v.) restored auditory gating (n = 6; p < 0.01). C, amplitudes of hippocampal evoked potentials: amphetamine-induced decrease in the amplitude of the conditioning response and an increase in the amplitude of the test response were reversed by PNU-282987.
Administration of vehicle (PBS 1 ml/kg i.v.; n = 6) did not alter amphetamine-induced gating deficit as determined from the average of 50 evoked potentials measured between 5 to 15 min after vehicle application (Fig. 4A). Disrupted auditory gating prevailed for at least 30 min after amphetamine administration, as indicated by a significant increase in T/C ratio calculated over this time period from 100 evoked potentials (T/C; 0.59 ± 0.11; p < 0.02 versus control). In contrast, administration of PNU-282987 (1 mg/kg i.v.; n = 6) significantly restored auditory gating (Fig. 4B; determined from the average of 50 evoked potentials) by reversing the action of amphetamine on the amplitude of evoked potentials, particularly on test stimuli (Fig. 4C). Significant drug action was also established when the degree of auditory gating was calculated from 100 evoked potentials (T/C; 0.37 ± 0.07; p < 0.03 versus amphetamine). The partial α7 nAChR agonist GTS-21 also reversed amphetamine-induced gating deficit: T/C values were 0.14 ± 0.04 at baseline, 0.48 ± 0.03 after amphetamine (1 mg/kg i.v.; p < 0.005), and 0.09 ± 0.05 after subsequent administration of GTS-21 (1 mg/kg i.v.; p < 0.005 versus amphetamine; n = 4).
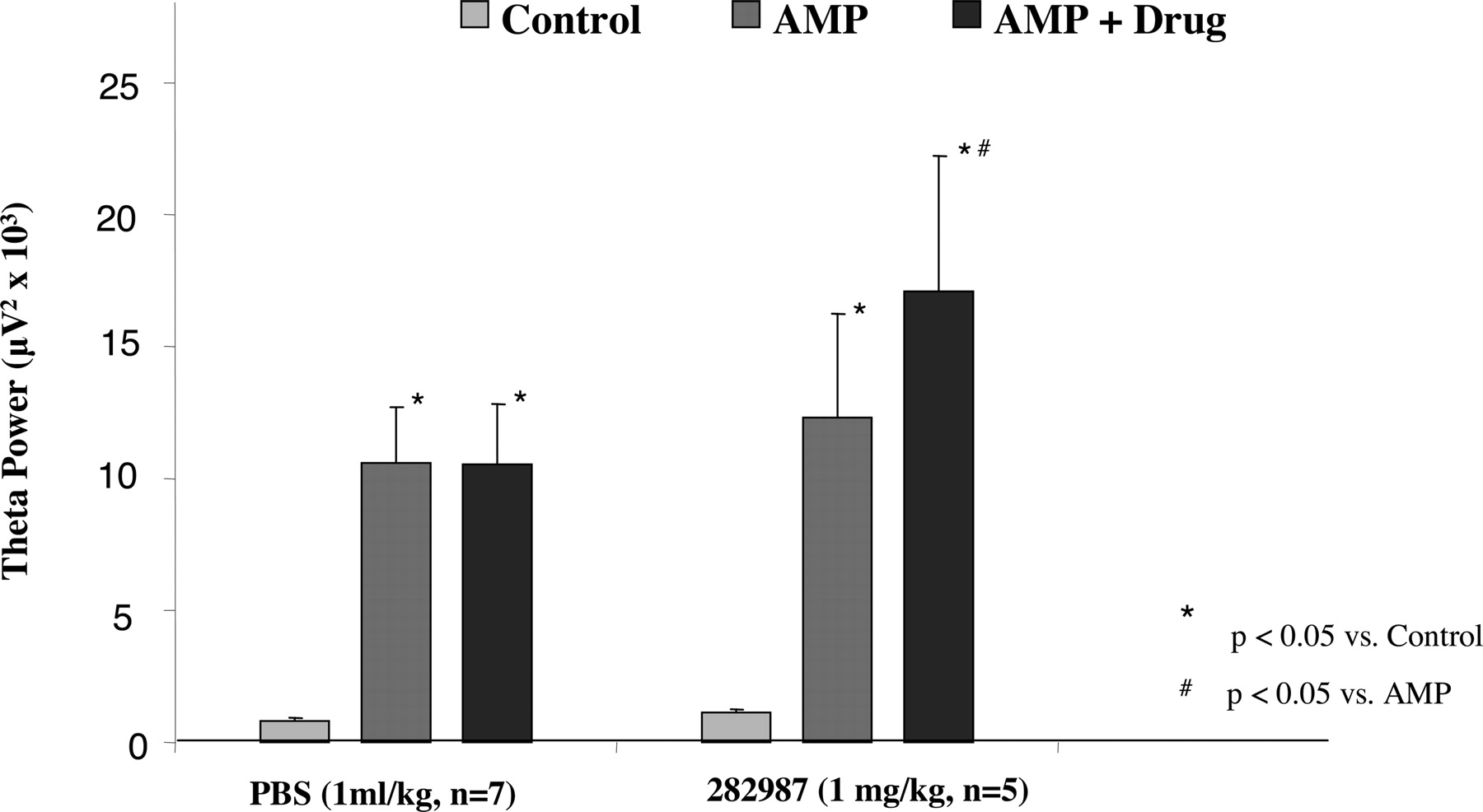
In agreement with our previous observations (Krause et al., 2003), administration of amphetamine resulted in synchronization of hippocampal EEG (Fig. 5). Quantitative EEG analysis showed a significant increase in EEG power, resulting in a peak frequency of 4.4 ± 0.1 Hz (baseline value 1.6 ± 0.2 Hz; p < 0.0001; n = 12), thereby indicating an increased synchrony in the θ frequency range (Figs. 5 and 6). Interestingly, amphetamine elicited pronounced hippocampal θ activity irrespective of its effect on auditory gating, indicating different mechanisms involved in these two pharmacological responses. Subsequent administration of vehicle (PBS, 1 ml/kg i.v.) or PNU-282987 (1 mg/k i.v.) did not alter peak frequency of hippocampal EEG (data not shown); however, PNU-282987 significantly enhanced θ power (Fig. 6).
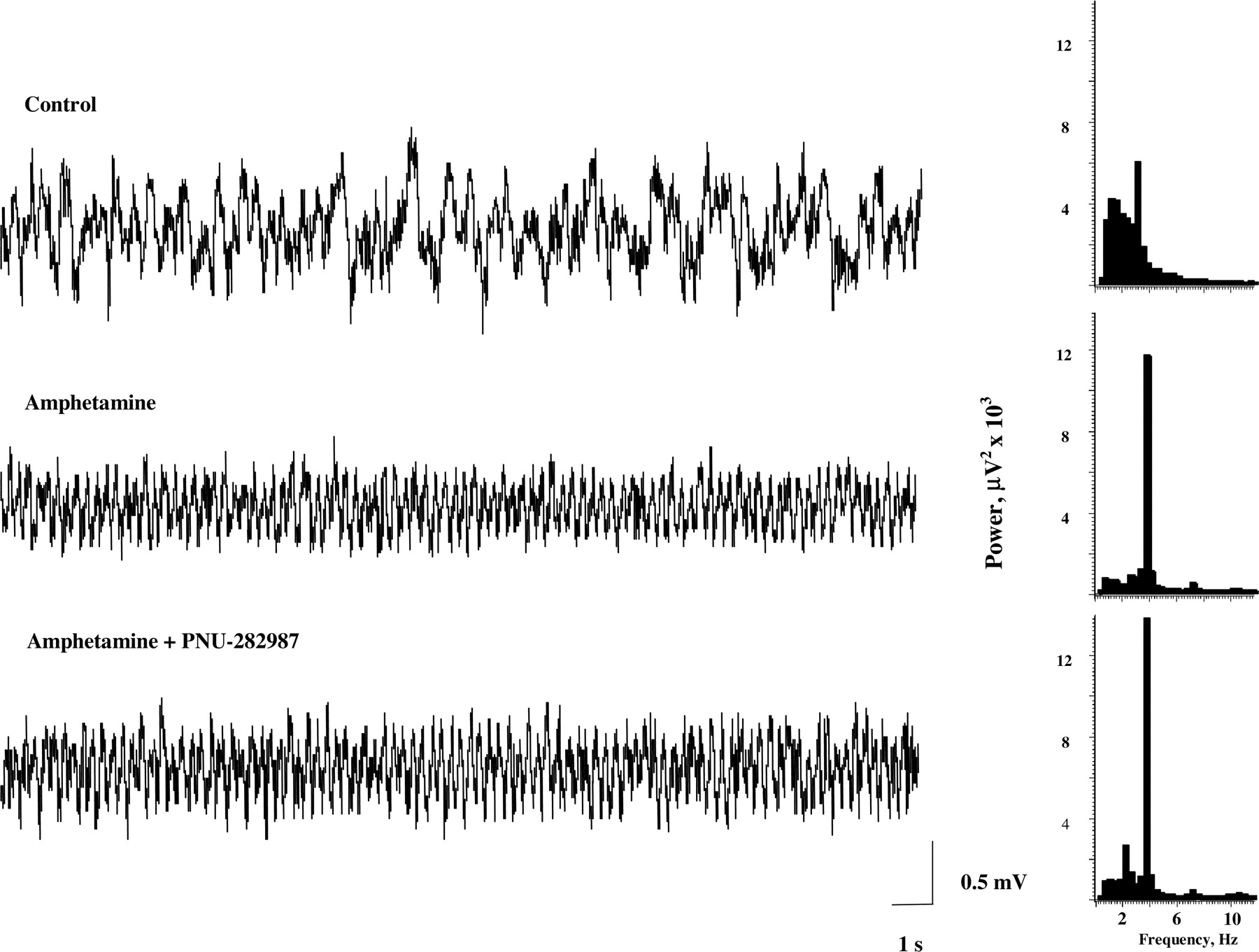
Effects of amphetamine and PNU-282987 on rhythmic activity in the hippocampal EEG. Hippocampal EEG (left) and power spectra (right) under control conditions, after administration of amphetamine (1.0 mg/kg i.v.), and after subsequent administration of PNU-282987 (1 mg/kg i.v.). Amphetamine induced a slow rhythmic activity in the hippocampal EEG in the θ frequency range, indicated by an increase in power between 3 and 6 Hz. The power of the rhythmic θ activity was enhanced after administration of PNU-282987.

Summary graph showing changes in EEG power at peak θ frequency after amphetamine and a subsequent administration of either vehicle (PBS; 1 ml/kg i.v.; n = 7) or PNU-282987 (1 mg/kg i.v.; n = 5).
Although most of chloral hydrate anesthetized rats showed normal auditory gating (characterized by a T/C ration lower than 0.2), approximately 5% of rats displayed persistent auditory gating deficits (monitored by blocks of subsequent averages of 25 evoked potentials) with a T/C ratio ≥0.5 under baseline conditions. Administration of the α7 nAChR partial agonist GTS 21 (1 mg/kg i.v.; n = 4) or the α7 nAChR agonist PNU-282987 (1 mg/kg i.v.; n = 4) significantly improved auditory gating in these rats (Fig. 7).

Effects of the α7 nAChR partial agonist GTS-21 and full agonist PNU-282987 on auditory gating in rats showing inherent auditory gating deficit. Both compounds improved gating as indicated by a significant reduction in T/C ratio.
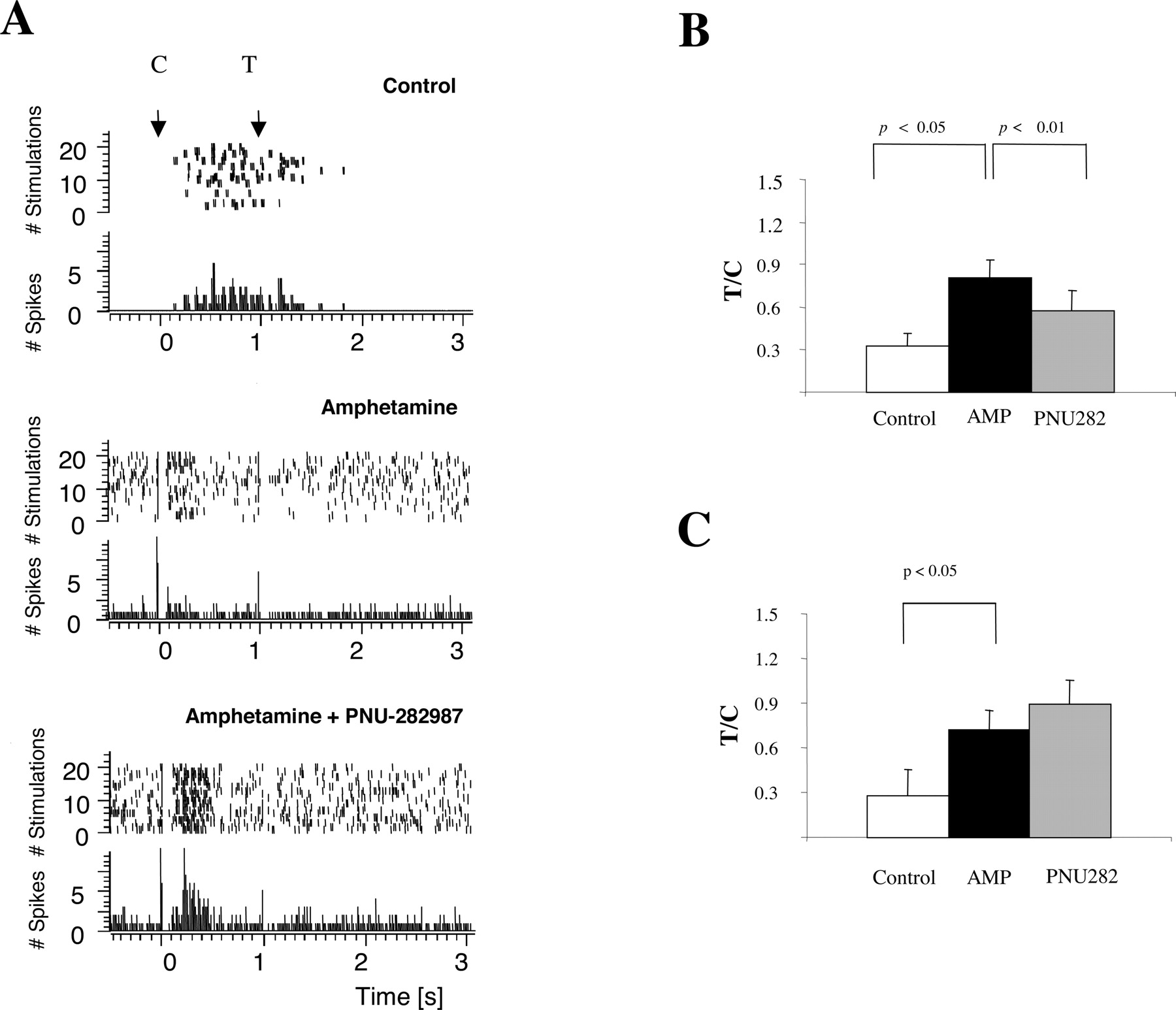
Auditory Gating in the Thalamic Reticular Nucleus. Reticular thalamic neurons responded to auditory stimuli with a typical discharge of bursts of action potentials (n = 11 neurons from 11 rats; Fig. 8). Auditory evoked activity of reticular thalamic neurons showed oscillations at 7 to 12 Hz, with each burst comprising approximately six action potentials, as it has been described previously (Krause et al., 2003). The number of evoked potentials was determined in 800-ms poststimulus interval after conditioning and test stimuli, and the ratio between the number of spikes after test stimuli and the number of spikes after the conditioning stimuli represented auditory gating (Krause et al., 2003). Administration of amphetamine (1 mg/kg i.v.; n = 11) disrupted auditory gating in each tested neuron (Fig. 8). Subsequent administration of the α7 nAChR agonist PNU-282987 (1 mg/kg i.v.) restored auditory gating in about one-half of reticular thalamic neurons (n = 6 of 11; Fig. 8, B and C). As has been reported previously (Krause et al., 2003), amphetamine changed the firing pattern of reticular thalamic neurons from burst firing to single-spike firing mode (Fig. 8A). Administration of PNU-282987 did not reverse amphetamine-induced firing pattern change in reticular thalamic neurons (n = 11).

A, typical recordings from a single unit in the reticular nucleus of the thalamus showing auditory gating. Control, distribution of spikes over a period of 24 stimulations (top) and post stimulus time histogram (bottom, bin size 2 ms) after conditioning (I) and test pulse (T) of a single unit recorded in the reticular nucleus of the thalamus. Amphetamine, administration of amphetamine (1 mg/kg i.v.) reduces the number of spikes after the conditioning stimulus and increases the number of spikes after the test stimulus. At the same time burst firing is abolished and the unit fires in a phasic manner. Amphetamine + PNU-282987, subsequent administration of the α7 nAChR agonist PNU-282987 (1.0 mg/kg i.v.) restores amphetamine-induced gating deficit. Note that amphetamine-induced tonic-firing mode is still prevailing. B, summary graphs for auditory gating in single units in the reticular thalamic nucleus when PNU-282987 restored amphetamine-induced gating deficit (n = 6 animals). Gating is expressed as a ratio of number of spikes after test pulse and conditioning pulse. Averaged T/C ratios after administration of amphetamine (AMP; 1.0 mg/kg i.v.) were significantly higher from control condition; PNU-282987 (PNU282; 1.0 mg/kg i.v.) significantly reversed this effect. C, summary graphs for auditory gating in single units in the reticular thalamic nucleus when PNU-282987 failed to restore amphetamine-induced gating deficit (n = 5 animals). Gating is expressed as a ratio of number of spikes after test pulse and conditioning pulse. Averaged T/C ratios after administration of AMP (1.0 mg/kg i.v.) or amphetamine + PNU-282987 (1 mg/kg i.v.) were significantly different from control condition.
Discussion
Previous studies have shown that within the rat hippocampus α7 nAChRs are expressed predominantly on GABAergic interneurons where they function to modulate inhibitory synaptic transmission (Alkondon et al., 1997; Jones and Yakel, 1997; Ji and Dani, 2000; Köfalvi et al., 2000). Impaired function of these interneurons, due in part to decreased expression of α7 nAChRs, has been proposed to contribute to the neuropathology of schizophrenia (Freedman et al., 2000). Thus, activation of α7 nAChRs by selective agonists could provide an effective therapy for treating the cognitive deficits of schizophrenia (Levin and Rezvani, 2002). We recently reported that PNU-282987 is a potent and selective agonist of human and rat α7 nAChRs (Bodnar et al., 2004). When applied to cultured rat hippocampal neurons, PNU-282987 evoked MLA-sensitive currents that were readily detectable when briefly applied at concentrations ≥300 nM or approximately 30-fold lower than that required for either nicotine or GTS-21 (Fig. 1). It should be noted, however, that although these results provide good evidence that PNU-282987 selectively activated α7-containing nAChRs on the cell body and/or proximal dendrites, they do no exclude the possibility that PNU-282987 activated MLA-resistant currents in the axon terminals. The effects of prolonged application of PNU-282987 on GABAergic synaptic transmission was evaluated in acutely isolated rat hippocampal slices. In agreement with the reported role of the α7 nAChR in the hippocampus, bath application of 30 and 300 nM PNU-282987 increased the frequency of synaptic events by >2-fold in about one-half the cells tested although the average effect was significant only for the 300 nM group, and no clear effect was observed at the highest tested concentration of 1 μM. These results suggest that 300 nM PNU-282987 activated a sufficient number of receptors to produce a measurable change in synaptic activity and that a balance was achieved between receptor activation and receptor desensitization that allowed for a relatively long-lasting response. The high cell to cell variability observed with 30 and 300 nM PNU-282987 likely reflects both inhibitory and disinhibitory actions produced by excitation of multiple hippocampal interneurons within the circuit influencing the recorded pyramidal cell (Ji and Dani, 2000).
To analyze in vivo activity of α7 nAChR agonists, auditory gating experiments were carried out in anesthetized rats. Physiological auditory gating was disrupted by amphetamine since impaired hippocampal gating is well demonstrated after systemic administration of amphetamine (Stevens et al., 1996; Krause et al., 2003). Impairment of gating was apparent as a result of a significant decrease in amplitude of evoked potentials to conditioning stimuli, and a significant increase in amplitude of evoked potentials to test stimuli, leading to an increased T/C ratio. Since dopamine D2 receptor antagonists reverse the amphetamine-induced gating deficit, it has been proposed that enhanced dopamine neurotransmission results in disrupted gating (Stevens et al., 1996; Krause et al., 2003). Furthermore, enhanced catecholamine neurotransmission by amphetamine (Light et al., 1999) or cocaine (Adler et al., 2001) leads to impaired gating in humans. Interestingly, it has been demonstrated that amphetamine- or cocaine-induced gating deficit can be reversed not only with D2 antagonists, but with nicotine or nicotinic agonists as well (Stevens et al., 1995, 1999; Adler et al., 2001), presumably interacting with inhibitory neuronal circuitry involved in gating, i.e., GABAergic interneurons in the hippocampus (Stevens et al., 1999; Freedman et al., 2000). Subsequent experiments indicated a key role for the α7 nAChR in nicotine-induced improvement in auditory gating and in gating mechanisms in general. It was shown that α7 nAChR stimulation normalizes chronic cocaine-induced loss of hippocampal sensory inhibition in C3H mice (Stevens et al., 1999). Furthermore, inherently impaired auditory gating in DBA/2 mice was normalized by GTS-21 (1-10 mg/kg s.c.; Stevens et al., 1998). Our current findings provide further evidence that α7 nAChR agonists can normalize abnormal auditory sensory gating, since we confirmed the efficacy of the partial agonist GTS-21 and demonstrated that the structurally distinct, highly selective, and potent PNU-282987 reversed amphetamine-induced gating deficit. Interestingly, a subset of rats in our experiments showed a gating deficit at baseline measurements. Although it is unclear what mechanisms contributed to this pathological gating, it was normalized by both GTS-21 and PNU-282987. Recently, it has been reported that social isolation (O'Neill et al., 2003) or early maternal deprivation (Ellenbroek et al., 2004) can also impair sensory gating in adult rats, suggesting that early life events such as stress could contribute to gating abnormality. Similar to our current finings, GTS-21 can normalize auditory gating deficits in isolation-reared rats (O'Neill et al., 2003).
Systemic administration of amphetamine disrupted auditory gating in nRT neurons as we reported previously (Krause et al., 2003). Although PNU-282987 reversed hippocampal gating deficit in all amphetamine-treated rats, it reversed gating deficit only in one-half of the tested nRT neurons. The reason for the heterogeneous response of the nRT neurons is presently unknown, but it could reflect heterogeneity in expression of α7 nAChRs by nRT neurons, or a disparity in the synaptic input/circuit connectivity of nRT neurons. Although within the human thalamus the highest α-bungarotoxin binding, reflecting α7 nAChR expression has been localized in nRT (Spurden et al., 1997), recent publication on rat brain nAChRs indicates a predominant presence of heteromeric nAChRs (labeled with epibatidine) in the thalamus, including nRT (Tribollet et al., 2004). In addition, PNU-282987 did not modify amphetamine-induced changes in firing pattern characteristics, in contrast to the D2 antagonist haloperidol (Krause et al., 2003).
In line with our previous findings, amphetamine not only disrupted auditory gating in anesthetized rats but also induced a slow rhythmic activity in the hippocampal EEG, with a significant increase in θ power and frequency (Krause et al., 2003). Subsequent administration of the selective α7 nAChR agonist PNU-282987 further enhanced hippocampal rhythmic activity as revealed by a significant increase in θ power. In contrast, administration of vehicle (or the D2 antagonist haloperidol; Krause et al., 2003) did not heighten θ activity, although haloperidol normalized amphetamine-induced gating deficit. The fact that PNU-282987 further synchronized hippocampal activity and significantly augmented θ power could be a contributing mechanism to procognitive actions of α7 nAChR agonists described recently both in animal models (Van Kampen et al., 2004; Young et al., 2004) and humans (Kitagawa et al., 2003).
In conclusion, the highly selective and potent α7 nAChR agonist PNU-282987 enhances GABAergic synaptic activity in the hippocampus in vitro, and reverses amphetamine-induced auditory gating deficit in anesthetized rats. In addition, PNU-282987 improves the inherent gating deficit observed in a subset of rats and enhances amphetamine-induced hippocampal θ activity. These results support the concept that α7 nAChR agonists represent a novel, potential pharmacotherapy in treatment of schizophrenia.
Footnotes
-
↵1 Current address: Baylor College of Medicine, Division of Neuroscience, Houston, TX.
-
↵2 Current address: MPI-CardIon Laboratories, Kalamazoo, MI.
-
doi:10.1124/jpet.104.076968.
-
ABBREVIATIONS: nAChR, α7 nicotinic acetylcholine receptor; nRT, reticular thalamic nucleus; PNU-282987, N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride; GTS-21, 3-[(2,4-dimethoxy)benzylidene]-anabaseine dihydrochloride (DMXBA); CNQX, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid/kainate 6-cyano-7-nitroquinoxaline-2,3-dione; DMSO, dimethyl sulfoxide; T, test; C, conditioning; PSTH, peristimulus time histograms; MLA, methyllycaconitine; PBS, phosphate-buffered saline.
- Received August 31, 2004.
- Accepted October 18, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}