


Abstract
Roflumilast, a potent and selective phosphodiesterase 4 (PDE4) inhibitor, has been demonstrated to be an effective anti-inflammatory agent in airway inflammatory diseases. In the present study, we investigated the mechanism of anti-inflammatory effects of roflumilast in murine macrophage cell line RAW264.7 cells. Roflumilast inhibited NO, tumor necrosis factor (TNF)-α, and interleukin (IL)-1β production via suppression of their gene expressions in lipopolysaccharide (LPS)-stimulated macrophages. To elucidate the mechanism by which roflumilast inhibits the production of inflammatory mediators, we examined the effect of roflumilast on the activation of nuclear factor-κB (NF-κB) in these cells. Roflumilast inhibited the DNA binding activity of NF-κB by preventing inhibitor κBα phosphorylation and degradation. The phosphorylation of mitogen-activated protein (MAP) kinases, including stress-activated protein kinase/c-Jun NH2-terminal kinase (JNK) and p38 MAP kinase, was also markedly inhibited by roflumilast. Similar to the effects of roflumilast, treatment of either SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)imidazole] or SP600125 [anthra(1,9-cd)pyrazol-6(2H)-one 1,9-pyrazoloanthrone], specific inhibitors of p38 MAP kinase and JNK, respectively, suppressed NO, TNF-α, and IL-1β production. Consistent with in vitro results, administration of roflumilast recovered the survival rate of LPS-treated mice, with concurrent suppression of plasma levels of nitrite/nitrate, TNF-α, and IL-1β. These results suggest that the inhibitory activity of roflumilast on the production of inflammatory mediators seems to be mediated via inhibition of NF-κB, p38 MAP kinase, and JNK activation in macrophages.
Roflumilast, the most potent and advanced PDE4 inhibitor so far, has been demonstrated to be an effective anti-inflammatory agent in many inflammatory diseases, including asthma (Bundschuh et al., 2001). For example, roflumilast significantly inhibited airway inflammation and remodeling in antigen-sensitized mice (Kumar et al., 2003), collagen-induced arthritis in mice (Barsig et al., 2001), and inflammatory bowel disease in mice (Banner and Trevethick, 2004). In in vitro studies, roflumilast has been shown to inhibit neutrophil elastase, myeloperoxidase, and matrix metalloproteinase-9 release from TNF-α-stimulated human neutrophils (Jones et al., 2005), to inhibit MUC5AC gene expression in epidermal growth factor-stimulated airway epithelial cells (Mata et al., 2005), and to suppress lipopolysaccharide (LPS)-induced TNF-α secretion in macrophages (Hatzelmann and Schudt, 2001). Although roflumilast exhibits several beneficial effects, its downstream mechanism after PDE4 inhibition in inflammatory cells remains unclear.
Upon inflammatory stimulation, macrophages produce NO and proinflammatory cytokines such as IL-1β and TNF-α (Zembowicz and Vane, 1992; Tracey and Cerami, 1994). Overproduction of these mediators as well as their mRNA expressions has been found in macrophages of many inflammatory diseases, including rheumatoid arthritis, atherosclerosis, and hepatitis (Tilg et al., 1992; Isomaki and Punnonen, 1997; Libby et al., 2002). A transcription factor, nuclear factor-κB (NF-κB), is known as an important regulator of iNOS, TNF-α, and IL-1β expression in LPS-activated inflammatory cells, such as macrophages and microglias (Xie et al., 1994; Colasanti et al., 1995). NF-κB is present in the cytoplasm as a heterodimer consisting of p50, p65, and IκBα subunits. In response to an activation signal, IκBα is phosphorylated and degraded, thus exposing the nuclear localization signals on the p50-p65 heterodimer. The p65 is then phosphorylated, leading to nuclear translocation and binding to a specific sequence in DNA, which in turn results in gene transcription (Pahl, 1999).
On the other hand, phosphorylation of MAP kinases (Ajizian et al., 1999) is also known to be a key component of production of NO and proinflammatory cytokines in activated macrophages. Three MAP kinase families have been identified in mammalian cells, such as extracellular signal-regulated kinase 1/2 (ERK1/2), SAPK/JNK, and p38 (Murga et al., 1999). LPS activates ERK, SAP/JNK, and p38 MAP kinase, which mediate LPS-induced inflammatory gene expression (Bhat et al., 1998; Ajizian et al., 1999). Therefore, suppression of NF-κB and MAP kinase activation has been used as a biomarker for the screening of anti-inflammatory activity. It was previously reported that elevation of intracellular cAMP by rolipram controlled cytokine secretion by T cells via inhibition of NF-κB activation (Jimenez et al., 2001). In addition, rolipram inhibited the interferon-γ-stimulated phosphorylation of p38 MAP kinase in U937 monocytic cells (MacKenzie and Houslay, 2000).
In this study, we tested whether roflumilast suppresses LPS-stimulated proinflammatory mediators NO, TNF-α, and IL-1β in macrophages and whether NF-κB and MAP kinases are involved in the actions of roflumilast. The results indicate that roflumilast suppresses NO, TNF-α, and IL-1β production by inhibiting the activation of NF-κB, SAPK/JNK, and p38. The suppressive effect of roflumilast was further confirmed by in vivo study in which roflumilast inhibited plasma levels of NO, TNF-α, and IL-1β triggered by LPS, with increased survival rate of septic shock.
Materials and Methods
Chemicals. RPMI 1640 medium, fetal bovine serum, and other tissue culture reagents were purchased from Invitrogen (Carlsbad, CA). Roflumilast was synthesized by Korea Research Institute of Chemical Technology (Taejon, Korea) and dissolved in dimethyl sulfoxide and freshly diluted in culture media for all in vitro experiments. Specific antibodies against iNOS, p-SAPK/JNK, SAPK/JNK, p-p38, and p38 were the products of Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and p-IκBα and IκBα were purchased from Cell Signaling Technology Inc. (Beverly, MA). Bay11-7082, SB203580, and SP600125 were obtained from Calbiochem (San Diego, CA). Cytotox 96 nonradioactive assay kit was obtained from Promega (Madison, WI). All other reagents were the products of Sigma-Aldrich (St. Louis, MO) unless indicated otherwise.
Animals. Male C57BL6/J (∼8- to 9-week-old) mice, weighing ∼20 to 25 g, were produced and maintained in specific pathogen-free conditions at our animal breeding facility. All animal studies were performed in accordance with National Institutes of Health guidelines (National Academy Press, 1996).
Peritoneal Macrophage Isolation and Cell Culture. The murine macrophage cell line RAW264.7 (American Type Culture Collection, Manassas, VA) cells were cultured in complete RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin G (100 U/ml), streptomycin (100 μg/ml), and l-glutamine (2 mM). C57BL6/J mice were intraperitoneally injected with 1.5 ml of 4% thioglycollate broth. After 4 days, macrophages were collected from the peritoneal cavity. Primary macrophages were cultured in six-well plates (4 × 105 cells/well) at 37°C in a humidified atmosphere containing 5% CO2 and 95% air.
Cell Cytotoxicity Assay. Cellular toxicity was determined by lactate dehydrogenase release in the medium. In brief, cells (4 × 105 cells/ml) were incubated with roflumilast at concentrations of 0.1 to 30 μM. After 18-h incubation, lactate dehydrogenase activity in the media was measured by using the assay kit (Promega). Optical density at 490 nm was measured and quantified.
cAMP Assay. RAW264.7 cells (1 × 106 cells/well) were incubated for 20 min in 200 μl of media alone or with roflumilast in the indicated concentrations. Ten microliters of isoprenaline (10 μM) was added and incubated for 20 min. After washing the media, 200 μl of lysis buffer was added as provided by Biotrak cAMP enzyme immunoassay kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK). The enzyme immunoassay kit was used according to the manufacturer's instruction.
Measurement of Nitrite, TNF-α, and IL-1β. Accumulation of nitrite in the medium was measured by a colorimetric assay method based on the Griess reaction. In brief, samples were reacted with 1% sulfanilamide, 0.1% naphthyl ethylenediamine dihydrochloride, and 2.5% phosphoric acid at room temperature for 10 min, and nitrite concentration was determined by absorbance at 540 nm in comparison with sodium nitrite as a standard. Plasma levels of nitrite plus nitrate were determined using a nitrate assay kit (Promega). The levels of TNF-α and IL-1β were measured in cell culture media and plasma using BD Biosciences (San Jose, CA) and R&D Systems (Minneapolis, MN) ELISA kits, respectively.
RNA Extraction and Reverse Transcription-Polymerase Chain Reaction. Total RNA was isolated from the cells using Easy-Blue reagent as suggested by the manufacturer (Intron, Seoul, Korea). Three micrograms of RNA was used for reverse transcriptionpolymerase chain reaction (RT-PCR) according to the manufacturer's instructions (Promega). Polymerase chain reaction amplification was initiated with 2 min of denaturation at 94°C followed by 30 cycles at 94°C for 20 s, 60°C for 30 s, and 72°C for 30 s (Mastercycler gradient; Eppendorf AG, Hamburg, Germany). Primer sequences for analysis of iNOS, IL-1β, and β-actin mRNA were as follows: iNOS, 5′-CAGAAGCAGAATGTGACCATC-3′ (sense) and 5′-CTTCTGGTCGATGTCATGAGC-3′ (antisense); IL-1β, 5′-TGCAGAGTTCCCCAACTGGTACATC-3′ (sense) and 5′-GTGCTGCCTAATGTCCCCTTGAATC-3′ (antisense); and β-actin, 5′-CCTTCTACAATGAGC-3′ (sense) and 5′-ACGTCACACTTCATG-3′ (antisense). After amplification, the samples were separated on a 1.5% agarose gel containing 0.5 μg/ml ethidium bromide and photographed. Digital photographs were assessed with image analysis software, and mRNA expression was evaluated as the ratio to β-actin.
Western Blot Analysis. Protein samples of whole lysate (30 μg) were mixed with an equal volume of 2× SDS sample buffer, boiled for 5 min, and then separated by 10% SDS-polyacrylamide gels. After electrophoresis, proteins were transferred to nylon membrane. The membranes were blocked in 5% dry milk for 1 h, rinsed, and incubated with specific antibodies against iNOS, SAPK/JNK, p-SAPK/JNK, p38, p-p38, IκBα, and p-IκBα in Tris-buffered saline containing Tween 20 (0.1%) (TBS-T) overnight at 4°C. Primary antibody was removed by washing the membranes three times in TBS-T and incubated for 1 h with horseradish peroxidase-conjugated secondary antibody (1:1000). After three times of washing in TBS-T, bands were visualized by enhanced chemiluminescence and exposed to X-ray film.
Electrophoretic Mobility Shift Assay. Nuclear extracts were prepared as described previously (Jeon et al., 2000). The protein content of the nuclear extracts was determined using a Bio-Rad protein assay kit according to the manufacturer's instructions. The oligonucleotide sequence for NF-κB/Rel was 5′-GATCTCAGAGGGGACTTTCCGAGAGA-3′. Double-stranded oligonucleotides were end-labeled with [γ-32P]ATP. Nuclear extracts (5 μg) were incubated with 2 μg of poly(dI-dC) and a 32P-labeled DNA probe, and DNA binding activity was analyzed using a 4.8% polyacrylamide gel. After electrophoresis, the gel was dried and subjected to autoradiography. The specificity of binding was examined by competition with an unlabeled oligonucleotide.
Induction of Septic Shock. Endotoxin shock was induced by administration of a single i.p. dose of LPS/GalN (20 mg/700 mg/kg) in saline (Lehmann et al., 1987). To examine whether roflumilast has protective effects on endotoxin shock, mice were divided into four groups and treated with vehicle (0.5% carboxymethyl cellulose; n = 10), LPS/GalN (n = 10), or LPS/GalN plus roflumilast (2 and 10 mg/kg; n = 10, respectively). Roflumilast was administered intraperitoneally 1 h before LPS administration. Six hours later, blood sample was collected by orbital puncture, and plasma was prepared by centrifugation at 12,000g for 20 min at 4°C for measuring NOx, TNF-α, and IL-1β levels. Survival of mice was monitored for 24 h.
Statistical Analysis. Data are represented as means ± S.D. of more than three separate experiments performed in triplicates. The significance of difference from the respective control for each experimental test condition was assessed by using Student's t test for each paired experiments except in vivo experiment of LPS-induced septic shock. The statistical significance of survival rate in in vivo experiment was determined by the Fisher's exact test. A p value <0.05 was regarded as a significant difference.
Results
Effect of Roflumilast on cAMP Accumulation. We examined the effect of roflumilast on isoprenaline-induced cAMP production in RAW264.7 cells. As expected, treatment with roflumilast for 20 min augmented intracellular cAMP levels in a concentration-dependent manner, with an EC50 value of 0.9 nM and effects being saturated at 1 μM (Fig. 1).
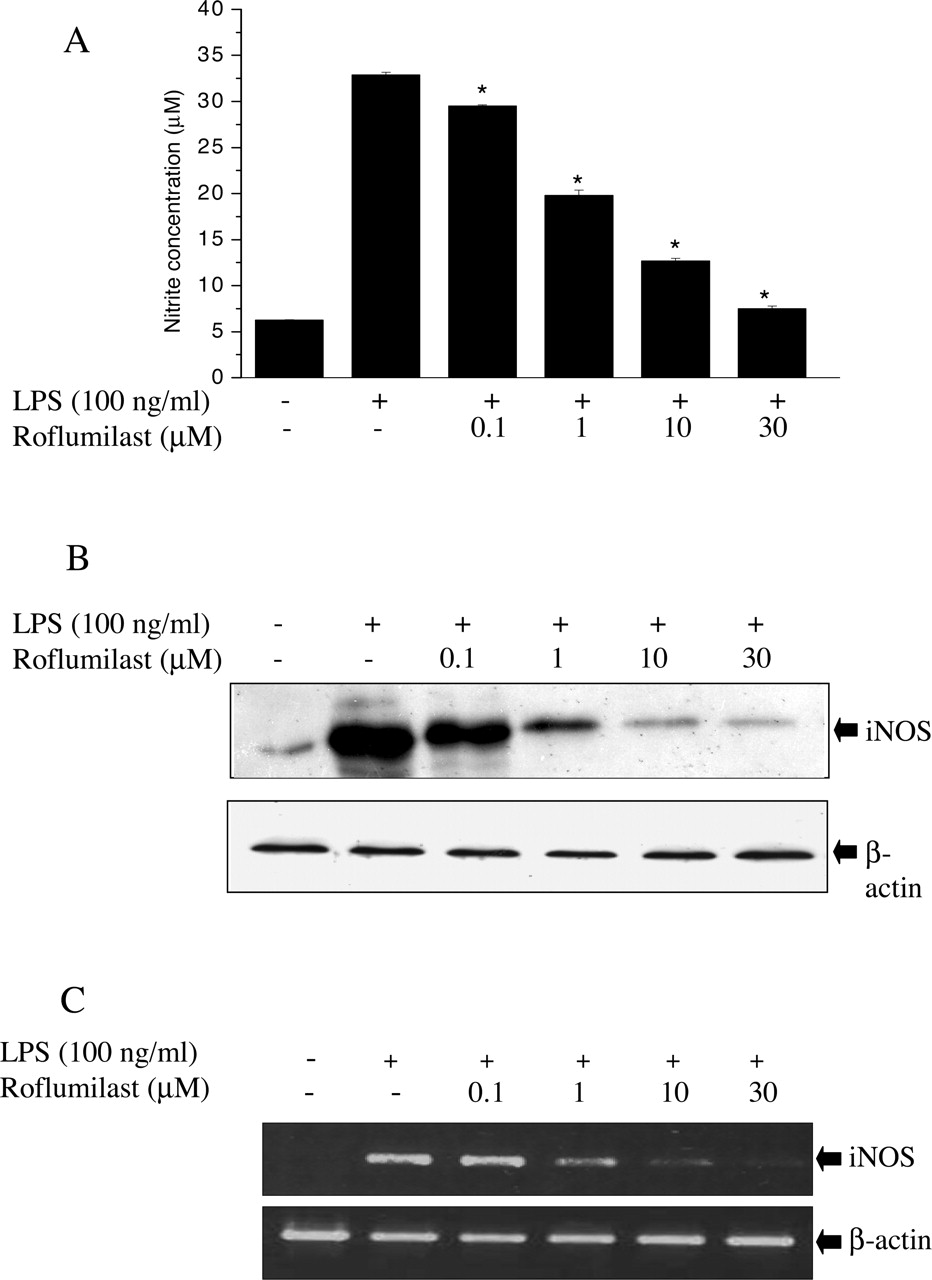
Inhibition of NO Production and iNOS Gene Expression by Roflumilast. Unstimulated macrophages produced a background level of NO (∼6 μM) in the culture medium. When RAW264.7 cells were stimulated with LPS (100 ng/ml) for 18 h, the levels of nitrite, a stable oxidized product of NO, were markedly increased in the culture medium to 30 to 35 μM. To investigate whether roflumilast is capable of inhibiting LPS-induced NO production, RAW264.7 cells were pretreated for 1 h with various concentrations of roflumilast followed by 100 ng/ml LPS treatment for 18 h. Roflumilast inhibited LPS-induced NO production and iNOS protein levels in a concentration-dependent manner with almost complete inhibition achieved at 30 μM (Fig. 2, A and B). To examine whether the suppression of iNOS protein by roflumilast was due to reduced iNOS mRNA expression, RT-PCR analyses from LPS-stimulated cells were conducted. The results showed that roflumilast concentration dependently reduced iNOS mRNA levels (Fig. 2C). No cytotoxic effect of roflumilast was observed in this experimental condition as measured by lactate dehydrogenase (data not shown).
Inhibition of TNF-α and IL-1β by Roflumilast. We further examined whether roflumilast inhibits LPS-induced TNF-α and IL-1β by ELISA. RAW264.7 cells were stimulated with LPS for 18 h in the absence or presence of roflumilast pretreatment. TNF-α and IL-1β levels were increased in LPS-stimulated RAW264.7 cells, and these elevations were significantly decreased in a concentration-dependent manner by roflumilast treatment (Fig. 3, A and B). In addition, increased expression of IL-1β mRNA by LPS was suppressed by roflumilast (Fig. 3C). Complete suppression of either TNF-α or IL-1β by roflumilast was achieved at 30 μM.

Effects of roflumilast on intracellular cAMP levels. RAW264.7 cells (1 × 106 cell) were incubated for 20 min with media alone and indicated concentrations of roflumilast. Lysates of the cells were then assayed for cAMP using a radioimmunoassay. Data shown are means ± S.D. *, p < 0.05 versus isoprenaline-induced group. Each experiment was carried out three times in triplicates.
Inhibition of NO, TNF-α, and IL-1β by Roflumilast in Primary Peritoneal Macrophages. Next, we examined the effects of roflumilast on NO, TNF-α, and IL-1β production in primary cultured murine peritoneal macrophages. As seen with RAW264.7 cells (Figs. 2 and 3), roflumilast (30 μM) inhibited NO, TNF-α, and IL-1β production in primary peritoneal macrophages stimulated with LPS (Fig. 4). No cytotoxic effect of roflumilast was observed in this experimental condition as measured by lactate dehydrogenase (data not shown).
Effects of Roflumilast on LPS-Induced NF-κB Activation and IκBα Phosphorylation and Degradation. Previous reports have demonstrated that the transcription factor NF-κB is involved in the LPS-induced activation of iNOS or other inflammatory cytokines (Heiss et al., 2001). To determine whether NF-κB is an important target for the action of roflumilast in RAW264.7 cells, we performed an electrophoretic mobility shift assay (EMSA). As shown in Fig. 5A, the induction of specific NF-κB DNA binding activity by LPS was decreased by roflumilast pretreatment in a concentration-dependent manner. These results suggest that roflumilast inhibits LPS-induced iNOS, TNF-α, and IL-1β expression in RAW264.7cells, possibly by attenuation of NF-κB activity. To gain further insight into the mechanism of roflumilast-mediated regulation of NF-κB, we examined the effects of roflumilast on IκBα phosphorylation and degradation. LPS treatment of RAW264.7 cells resulted in the IκBα phosphorylation and degradation, and exposure of cells to higher concentrations than 10 μM roflumilast for 1 h before LPS stimulation significantly inhibited IκBα phosphorylation and degradation (Fig. 5, B and C).

Effects of roflumilast on NO production and iNOS expression in LPS-stimulated RAW264.7 cells. RAW264.7 cells were preincubated with indicated concentrations of roflumilast (0.1–30 μM) for 1 h followed by the incubation with LPS (100 ng/ml). A, nitrite levels were measured in the cultured media by Griess reaction after incubation with LPS for 18 h. B, levels of iNOS protein were measured in cultured cells by Western blot using antibody for murine iNOS. C, levels of iNOS mRNA were measured in cultured cells by RT-PCR after incubation with LPS for 10 h. Data shown are means ± S.D. *, p < 0.05 versus LPS alone. Each experiment was carried out three times in triplicates.
Effects of Roflumilast on the LPS-Induced Activation of SAPK/JNK and p38 MAP Kinase. LPS is known to activate ERK, SAPK/JNK, and p38 MAP kinase pathways in macrophages (DeFranco et al., 1995; Guha and Mackman, 2001). The possibility was raised that roflumilast inhibited production of LPS-induced inflammatory mediators by down-regulating MAP kinases. Therefore, we examined the effect of roflumilast on LPS-induced activation of MAP kinases. As shown in Fig. 6, LPS treatment induced a strong increase in the phosphorylated JNK and p38 levels. Treatment with roflumilast significantly suppressed LPS-induced activation of JNK and p38 MAP kinase concentration dependently (Fig. 6). To more closely assess the relative role of MAP kinases in LPS-induced inflammatory mediator production, we checked the effects of selective MAP kinase inhibitors on the LPS-induced NO, TNF-α, and IL-β production. SP600125 (a JNK inhibitor), SB203580 (a p38 MAP kinase inhibitor), and Bay11-7082 (an NF-κB inhibitor) profoundly inhibited LPS-mediated NO, TNF-α, and IL-1β production. However, treatment with PD98059 (mitogen-activated kinase kinase inhibitor) had little effect (Fig. 7, A–C). These results suggest that roflumilast-mediated inactivation of JNK and p38 MAP kinase may be, at least in part, responsible for the suppression of NO and proinflammatory cytokines in RAW264.7 cells.

Effects of roflumilast on production of TNF-α and IL-1β in LPS-stimulated RAW 264.7 cells. RAW264.7 cells were preincubated with indicated concentrations of roflumilast (0.1–30 μM) for 1 h followed by the incubation with LPS (100 ng/ml). TNF-α (A) and IL-1β (B) concentrations were measured in the cultured media by using commercial ELISA kits after 18-h incubation with LPS. C, levels of IL-1β mRNA were measured in cultured cells by RT-PCR after 10-h incubation with LPS. Data shown are means ± S.D. *, p < 0.05 versus LPS alone. Each experiment was carried out three times in triplicates.
Effects of Roflumilast on LPS-Induced Lethal Shock in Mice. The LPS-induced murine endotoxic shock model was used to assess the protective effect of roflumilast in vivo. As shown in Fig. 8, the administration of LPS/GalN to the mice resulted in a survival rate of 0% within 12 h. However, pretreatment (1 h before LPS) of roflumilast produced a dose-dependent protection against endotoxin shock. Administration of roflumilast (10 mg/kg i.p.) significantly increased the survival rate up to 60% (6/10 mice survived). Even at the lower dose (2 mg/kg i.p.), roflumilast increased the survival rate to 30%. These results clearly show that roflumilast is effective in protecting LPS/GalN-induced lethal shock in mice. Concurrent with its protective effects in vivo, roflumilast treatment markedly decreased the elevated plasma levels of NOx, TNF-α, and IL-1β (Table 1).
Effects of roflumilast on the plasma levels of NOx, TNF-α, and IL-1β in the LPS-injected mice
Different groups of mice (n = 10) were treated with vehicle (n = 10), LPS/GalN (20 mg/700 mg/kg) (n = 10), LPS/GalN (20 mg/700 mg/kg), and roflumilast (2 or 10 mg/kg, 1 h before LPS treatment) (n = 10). Six hours later, blood samples were prepared from mice. The concentrations of plasma NOx, TNF-α, and IL-1β were determined as described under Materials and Methods. Treat group values are in milligrams per kilogram. Data shown are means ± S.D.

Effects of roflumilast on NO, TNF-α, and IL-1β production in primary peritoneal macrophages. Peritoneal macrophages isolated from mouse peritoneal cavity were precultured overnight and incubated with roflumilast (30 μM) for 1 h followed by the incubation with LPS (100 ng/ml) for 18 h. A, nitrite levels were measured by Griess reaction. TNF-α (B) and IL-1β (C) concentrations were measured by using a commercial ELISA kit. Data shown are means ± S.D. *, p < 0.05 versus LPS alone. Each experiment was carried out three times in triplicates.
Discussion
PDE4 inhibitors are considered as promising anti-inflammatory drugs based on their abilities to suppress the activation of many inflammatory cells (Teixeira et al., 1997; Souness et al., 2000). However, the detailed mechanism of PDE4 inhibitors involved in the anti-inflammatory activities is largely unknown. Recently, it has been proposed that PDE4 inhibitors interfere with cellular signaling leading to activation of transcription factor, consequently blocking leukocyte activation and cytokine synthesis (Wang et al., 1997; Matsumori et al., 2000; Jimenez et al., 2001; Haddad et al., 2002). Roflumilast is a novel potent PDE4-selective inhibitor being effective in airway inflammation (Bundschuh et al., 2001; Timmer et al., 2002), and its potential usefulness in human asthma has been demonstrated in clinical studies (Bayes et al., 2002). In the present study, we examined the mechanism of anti-inflammatory action of roflumilast and found that roflumilast inhibits the production of several inflammatory mediators such as NO, TNF-α, and IL-1β in RAW264.7 cells via inhibition of NF-κB activation and phosphorylation of SAPK/JNK and p38 MAP kinase.

Inhibition of NF-κB activation, IκBα phosphorylation, and degradation by roflumilast in LPS-stimulated RAW264.7 cells. RAW264.7 cells were pretreated with indicated concentrations of roflumilast for 1 h before the addition of 100 ng/ml LPS, and the cells were further incubated for 1 h. A, after isolation of nuclear extracts, EMSA was carried out. The binding specificity was determined using unlabeled wild-type probe (100-fold in excess) to compete with the labeled oligonucleotide. B, lysates were subjected to immunoblot analysis using IκBα and p-IκBα-specific antibodies. Each experiment was carried out three times. C, densitometric analysis of IκBα and p-IκBα bands obtained from B; the results are expressed as ratio of p-IκBα and IκBα.

Effects of roflumilast on the LPS-induced activation of SAPK/JNK and p38 MAP kinase in RAW264.7 cells. Cells were pretreated with indicated concentrations of roflumilast for 1 h, incubated with LPS (100 ng/ml) for 1 h, and then assayed for activation of SAPK/JNK and p38 MAP kinase using Western blot analysis. Each experiment was carried out three times.

Effects of MAP kinase inhibitors on NO, TNF-α, and IL-1β production. RAW264.7 cells were pretreated with roflumilast (30 μM), SB203580 (10 μM), PD98059 (30 μM), SP600125 (10 μM), and Bay11-7082 (10 μM) for 1 h followed by the incubation with LPS (100 ng/ml) for 18 h. A, nitrite levels were measured by Griess reaction. TNF-α (B) and IL-1β (C) concentrations were measured by using a commercial ELISA kit. Data shown are means ± S.D. *, p < 0.05 versus LPS alone. Each experiment was carried out three times in triplicates.

Effects of roflumilast on survival of LPS/GalN-treated mice. Different groups of mice (n = 10) were intraperitoneally treated with vehicle (▪), LPS/GalN (20 mg/700 mg/kg) (•), LPS/GalN (20 mg/700 mg/kg) plus 2 mg/kg roflumilast (▴), or LPS/GalN (20 mg/700 mg/kg) plus 10 mg/kg roflumilast (▾). Survival rate of mice was monitored for 24 h, and the statistical significance was determined by the Fisher's exact test (*, p < 0.05 versus LPS alone).
Increased production of NO, TNF-α, and IL-1β plays a critical role in the process of macrophage activation and is associated with acute and chronic inflammations. It was previously reported that PDE4 inhibitors rolipram and pentoxifylline inhibited NO production in islet cells (Beshay and Prud'homme, 2001). In addition, rolipram was shown to inhibit the production of TNF-α on acute and chronic models of inflammation, producing anti-inflammatory activities (Beshay et al., 2001). In the present study, we have demonstrated that roflumilast inhibits NO, TNF-α, and IL-1β production in macrophages as well as in vivo stimulated by LPS treatment.
The control of iNOS, IL-1β, and TNF-α transcription is mediated by the activation of NF-κB, which is an important proximal mechanism for the overproduction of the inflammatory mediators in macrophages in response to LPS (Shapira et al., 1996). The involvement of NF-κB in the suppressive action of roflumilast on inflammatory mediators was supported in the present study by several results, including EMSA, inhibition of IκBα phosphorylation, and effects of NF-κB inhibitor Bay11-7082. On the other hand, the relationship between cAMP elevation and NF-κB inhibition seems to be controversial. It was reported that elevated cAMP inhibited NF-κB activation in transformed T cells (Chen and Rothenberg, 1994; Haraguchi et al., 1995) and that PDE4 inhibition by rolipram regulated transcription of particular cytokines through inhibition of NF-κB activation in T cells (Jimenez et al., 2001). On the contrary, inhibition of PDE1, PDE3, and PDE4 isozymes in alveolar epithelial cells exhibited no effect on LPS-mediated translocation of NF-κB subunits (Haddad et al., 2002). Based on the results of the present study, roflumilast caused the increased intracellular cAMP in RAW264.7 cells, accompanied by NF-κB inhibition, consistent with previous reports in T cells (Chen and Rothenberg, 1994; Haraguchi et al., 1995). The mechanism of action of roflumilast on NF-κB inhibition seems to be an inhibition of IκBα protein phosphorylation and degradation, possibly via modulation mediated by cAMP-dependent protein kinases. However, the possibility that the anti-inflammatory action of roflumilast is mediated by the mechanism independent of cAMP elevation cannot be excluded at present.
Among various intracellular signaling pathways, MAP kinase pathway has been known to be important in the regulation of cytokine gene expression showing that SAPK/JNK and p38 MAP kinase are involved in the iNOS gene expression (Chan and Riches, 1998; Jeon et al., 2000). Recently, MacKenzie and Houslay (2000) reported that elevated cAMP by rolipram inhibited the interferon-γ-stimulated phosphorylation of p38 MAP kinase in U937 cells. Consistent with the results, our results also demonstrated that roflumilast inhibited SAPK/JNK and p38 MAPK pathways and that the JNK inhibitor SP600125 and p38 MAPK inhibitor SB203580 suppressed NO, TNF-α, and IL-1β production induced by LPS stimulation. These results suggest that besides NF-κB inhibition, roflumilast may inhibit proinflammatory gene expression via an additional mechanism of JNK and p38 MAPK inactivation in RAW264.7 cells. To our knowledge, the current data are the first demonstration of inhibitory effects of roflumilast on the activity of NF-κB and MAP kinases. However, the possible link between the effects on NF-κB and MAP kinases activity and their relative contributions to cytokine regulation needs to be further elucidated.
Septic shock is frequently induced in experimental conditions by LPS, a major constituent of Gram negative bacterial outer membrane, with sensitization using GalN. LPS is known to stimulate production or release of inflammatory cytokines from monocytes and macrophages, thus triggering the development of septic shock. In the present study, roflumilast attenuated the lethality induced by LPS/GalN in vivo, concurrent with the suppression of plasma levels of NOx, TNF-α, and IL-1β. These effects seem to be linked, at least in part, with the actions of roflumilast on NF-κB, JNK, and p38 kinases in macrophages based on the in vitro results using RAW264.7 macrophages and primary peritoneal macrophages. Furthermore, selective inhibitors of p38 kinase (SB203580) and NF-κB (PC-SPES, an eight-herb mixture) have been shown to rescue the mice from endotoxin-induced shock (Badger et al., 1996; Ikezoe et al., 2003), supporting the involvement of p38 kinase and NF-κB in in vivo endotoxin-induced septic shock.
In summary, we have found that the well known PDE4 inhibitor roflumilast controls LPS-induced NO, TNF-α, and IL-1β production through inhibition of NF-κB activation and SAPK/JNK and p38 MAPK pathways in RAW264.7 macrophages. In addition, our results indicate that roflumilast exerted potent in vivo anti-inflammatory effects and increased the survival rate of mice in sepsis model. The current data would provide a new insight into the mechanisms responsible for the anti-inflammatory and pharmacological activities of roflumilast.
Footnotes
-
This research was supported by a grant from the Center for Biological Modulators of the 21st Century Frontier Research & Development Program, The Ministry of Science and Technology, Taejon, Korea.
-
doi:10.1124/jpet.105.092056.
-
ABBREVIATIONS: PDE4, phosphodiesterase 4; TNF, tumor necrosis factor; IL, interleukin; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; iNOS, inducible nitric-oxide synthase; ERK, extracellular signal-regulated kinase; SAPK, stress-activated protein kinase; JNK, c-Jun NH2-terminal kinase; Bay11-7082, (E)-3[(4-methylphenyl)sulfonyl]-2-propenentrile; SB203580, 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)imidazole; SP600125, anthra(1,9-cd)pyrazol-6(2H)-one 1,9-pyrazoloanthrone; NOx, nitrate and nitrite; ELISA, enzyme-linked immunosorbent assay; RT-PCR, reverse transcription-polymerase chain reaction; TBS-T, Tris-buffered saline-Tween 20; GaIN, d(+)galactosamine; EMSA, electrophoretic mobility shift assay; PD98059, 2′-amino-3′-methoxyflavone.
- Received July 4, 2005.
- Accepted August 25, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}