


Abstract
(±)-Fenfluramine is an amphetamine analog that was once widely prescribed as an appetite suppressant. Although (±)-fenfluramine is no longer clinically available, the mechanisms underlying its anorectic properties are still of interest. Upon peripheral administration, stereoisomers of (±)-fenfluramine are N-deethylated to form the metabolites, (+)- and (-)-norfenfluramine. It is well accepted that isomers of (±)-fenfluramine and (±)-norfenfluramine interact with 5-hydroxytryptamine (serotonin, 5-HT) transporters to release 5-HT from neurons. However, the effects of these drugs on other monoamine transporters are not well characterized. In this study, we examined the interaction of stereoisomers of (±)-fenfluramine and (±)-norfenfluramine with transporters for 5-HT, norepinephrine (NE), and dopamine (DA). Results from in vitro assays confirmed these drugs are potent substrates for 5-HT transporters: (+)-fenfluramine, (-)-fenfluramine, (+)-norfenfluramine, and (-)-norfenfluramine released [3H]5-HT from synaptosomes with EC50 values of 52, 147, 59, and 287 nM, respectively. Importantly, (+)-fenfluramine and (+)-norfenfluramine released [3H]NE with EC50 values of 302 and 73 nM. Results from in vivo microdialysis experiments showed that intravenous injection of (+)-norfenfluramine elevates extracellular levels of 5-HT, NE, and DA in rat frontal cortex. The effects of (+)-norfenfluramine on NE and DA were antagonized by pretreatment with the NE uptake blocker nisoxetine. In summary, administration of fenfluramines can increase synaptic levels of 5-HT, NE, and DA in the cortex, and (+)-norfenfluramine likely contributes to these effects. Release of NE and DA evoked by (+)-norfenfluramine is at least partly mediated via NE transporters. Our results emphasize the potential involvement of noradrenergic mechanisms in the actions of fenfluramines.
The search for effective appetite suppressants is underscored by the continuing problem of obesity and its associated medical complications. (±)-Fenfluramine (Pondimin) and its more potent stereoisomer, (+)-fenfluramine (dexfenfluramine, Redux), are anorectic agents that were widely prescribed until their removal from the market due to adverse effects (Connolly and McGoon, 1999). Although fenfluramines are no longer clinically available, the mechanisms responsible for their powerful anorectic actions are of great interest. Pharmacokinetic investigations have shown that stereoisomers of (±)-fenfluramine are N-deethylated by liver enzymes to yield the metabolites (+)- and (-)-norfenfluramine (Caccia et al., 1985). In humans and animals receiving systemic (±)-fenfluramine, circulating concentrations of (+)- and (-)-norfenfluramine are similar to or greater than concentrations of fenfluramine itself (Marchant et al., 1992). Moreover, stereoisomers of (±)-fenfluramine and (±)-norfenfluramine cross the blood-brain barrier to accumulate in the central nervous system. Thus, peripheral administration of (±)-fenfluramine gives rise to four pharmacological agents with potential neurobiological activity.
Historical evidence shows that stereoisomers of (±)-fenfluramine stimulate 5-hydroxytryptamine (serotonin, 5-HT) transmission (Pinder et al., 1975). Specifically, these agents increase synaptic and extrasynaptic levels of 5-HT in nervous tissue by a mechanism involving 5-HT transporter proteins (SERTs) (Rothman and Baumann, 2002). Drugs that interact with SERTs, or other membrane-bound transporter proteins, can be divided into two classes: uptake inhibitors and substrate-type releasers. Uptake inhibitors bind to transporter proteins but are not transported. These drugs increase extracellular concentrations of neurotransmitters by interfering with neurotransmitter recapture from the synaptic cleft. Substrates, in contrast, are transported into the nerve terminal where they promote the release of intracellular neurotransmitters by a process of carrier-mediated exchange (Rudnick and Clark, 1993). Most findings indicate that stereoisomers of (±)-fenfluramine and (±)-norfenfluramine increase extracellular 5-HT by acting as substrates for SERTs (for review, see Garattini et al., 1986). Recent in vivo microdialysis studies confirm that 5-HT release evoked by (±)-fenfluramine or (+)-fenfluramine is antagonized by pretreatment with the SERT inhibitor fluoxetine (Prozac) (Gundlah et al., 1997; Tao et al., 2002).
In our laboratory, we have developed a high-throughput in vitro method that can be used to discriminate between drugs that act as transporter uptake inhibitors versus substrate-type releasers. Using this method, it is possible to profile the mechanism of action of test drugs at norepinephrine (NE) transporters (NETs), dopamine (DA) transporters (DATs), and SERTs using nearly identical assay conditions (Rothman et al., 2001). With few exceptions (Pettersson, 1995; Cozzi et al., 1998), investigations examining the neuropharmacology of fenfluramines and norfenfluramines have focused on the 5-HT effects of these drugs (for review, see Garattini et al., 1986). For this reason, we studied the interaction of (±)-fenfluramine, (±)-norfenfluramine, and their stereoisomers, with SERTs, NETs, and DATs using in vitro assay methods and in vivo microdialysis methods. Here, we report that (+)-fenfluramine and (+)-norfenfluramine are potent substrates for NETs, with (+)-norfenfluramine being about 4-fold more potent than the parent compound. Both drugs are capable of elevating extracellular levels of NE, along with 5-HT, at doses that produce anorexia. Our findings suggest the possibility that central noradrenergic mechanisms are involved in at least some in vivo effects produced by systemic administration of (±)-fenfluramine.
Materials and Methods
Animals. Male Sprague-Dawley rats (Charles River Laboratories, Inc., Wilmington, MA) weighing 300 to 400 g were used as subjects in these experiments. Rats were housed in standard conditions (lights on from 7:00 AM to 7:00 PM) with food and water freely available. Animals were maintained in facilities fully accredited by the American Association of the Accreditation of Laboratory Animal Care, and experiments were performed in accordance with the Institutional Care and Use Committee of the National Institute on Drug Abuse, Intramural Research Program.
Drugs and Reagents. The National Institute of Mental Health Chemical Synthesis and Drug Supply Program provided the following compounds: (±)-norfenfluramine, (+)-norfenfluramine, and (-)-norfenfluramine. (±)-Fenfluramine and (+)-fenfluramine were obtained from the National Institute on Drug Abuse Drug Supply Program (Rockville, MD). (-)-Fenfluramine and 1-(2-diphenylmethoxyethyl)-4-(3-phenylpropyl)piperazine (GBR12935) were purchased from Sigma/RBI (Natick, MA). 3β-(4-Iodophenyl)tropane-2β-pyrrolidine carboxamide tartrate (RTI-229) was provided by Dr. F. Ivy Carroll (Research Triangle Institute, Research Triangle Park, NC). [3H]DA (specific activity, 27.5 Ci/mmol), [3H]NE (specific activity, 55 Ci/mmol), and [3H]5-HT (specific activity, 27.5 Ci/mmol) were purchased from PerkinElmer Life Sciences (Boston, MA). The sources of other reagents are published (Baumann et al., 2001; Rothman et al., 2001).
[3H]DA, [3H]5-HT, and [3H]NE Uptake Assays. The effects of test agents on [3H]DA, [3H]5-HT, and [3H]NE uptake were evaluated using published methods (Rothman et al., 2001). Briefly, synaptosomes were prepared from rat caudate for [3H]DA reuptake, or from whole brain minus caudate and cerebellum for [3H]5-HT and [3H]NE reuptake. Fresh tissue was homogenized in ice-cold 10% sucrose using a Potter-Elvehjem homogenizer. Homogenates were centrifuged at 1000g for 10 min at 4°C and supernatants were retained on ice (synaptosomal preparation). Polystyrene test tubes (12 × 75 mm) received 50 μl of Krebs-phosphate buffer (final pH 7.4) consisting of 0.5 mM Na2SO4, 0.5 mM KH2PO4, 126 mM NaCl, 2.4 mM KCl, 0.83 mM CaCl2, 0.8 mM MgCl2, and 11.1 mM glucose at pH 7.4, with 1 mg/ml ascorbic acid, 1 mg/ml bovine serum albumin (BSA), and 50 μM pargyline added (uptake buffer). Subsequently, 750 μl of [3H]DA (5 nM), [3H]5-HT (2 nM), or [3H]NE (5 nM) diluted in uptake buffer without BSA, and 100 μl of test agent in uptake buffer, were added to the tubes. Nonspecific uptake was defined using 10 μM tyramine for [3H]DA and [3H]NE assays, or 100 μM tyramine for [3H]5-HT assays.
The uptake assay was initiated by adding 100 μl of the synaptosomal preparation to the tubes. Inhibition curves were generated by incubating [3H]ligand with test agent (1 nM–100 μM final tube concentration) diluted in uptake buffer. [3H]5-HT uptake was conducted in the presence of 100 nM nomifensine and 100 nM GBR12935 to prevent uptake of [3H]5-HT into NE or DA nerve terminals. [3H]NE uptake was conducted in the presence of 5 nM RTI-229 to prevent uptake of [3H]NE into DA nerve terminals. Incubations were carried out at 25°C for a periods of 10, 15, and 30 min for [3H]NE, [3H]DA, and [3H]5-HT, respectively. The incubations were terminated by adding 4 ml of wash buffer containing 10 mM Tris-HCl (pH 7.4) in 0.9% NaCl at 25°C, followed by rapid filtration over GF/B filters (Whatman, Maidstone, UK) and two additional wash cycles. The tritium retained on the filters was counted in a beta counter (Taurus; Titertek, Huntsville, AL) at 40% efficiency after an overnight extraction into Cytoscint cocktail (ICN Biomedicals, Inc., Costa Mesa, CA).
[3H]DA, [3H]NE, and [3H]5-HT Release Assays. Rat caudate (for [3H]DA release) or whole brain minus cerebellum and caudate (for[3H]NE and [3H]5-HT release) was homogenized in ice-cold 10% sucrose containing 1 μM reserpine. Nomifensine (100 nM) and GBR12935 (100 nM) were also added to the sucrose solution for [3H]5-HT release experiments to block any potential [3H]5-HT uptake into NE and DA nerve terminals. After 12 strokes with a Potter-Elvehjem homogenizer, homogenates were centrifuged at 1000g for 10 min at 0 to 4°C, and the supernatants were retained on ice (synaptosomal preparation). Each rat brain (approximately 1200 mg) produced enough tissue for 250 test tubes for the [3H]DA and [3H]5-HT release assays, and for 125 test tubes for the [3H]NE release assay.
Synaptosomal preparations were incubated to steady state with 5 nM [3H]DA (30 min), 7 nM [3H]NE (60 min), or 5 nM [3H]5-HT (60 min) in uptake buffer without BSA, plus 1 μM reserpine, in a polypropylene beaker with stirring at 25°C. Nomifensine (100 nM) and GBR12935 (100 nM) were added to the buffer for [3H]5-HT release experiments, whereas RTI-229 (5 nM) was added to the buffer for [3H]NE release experiments. After incubation to steady state, 850 μl of synaptosomes preloaded with [3H]neurotransmitter was added to 12-× 75-mm polystyrene test tubes that contained 150 μl of test drug in uptake buffer. After 5 min ([3H]DA and [3H]5-HT) or 30 min ([3H]NE), the release reaction was terminated by dilution with 4 ml of wash buffer (10 mM Tris-HCl, pH 7.4, containing 0.9% NaCl at 25°C) followed by rapid vacuum filtration over GF/B filters (Whatman) using a harvester (Brandel, Inc., Gaithersburg, MD). The filters were rinsed twice with 4 ml of wash buffer using the harvester (Brandel, Inc.), and the retained tritium was counted by a Taurus liquid scintillation counter at 40% efficiency after an overnight extraction in 3 ml of Cytoscint (ICN Biomedicals, Inc.).
Surgery. For the microdialysis studies, rats received sodium pentobarbital (60 mg/kg i.p.) for surgical anesthesia. Indwelling jugular catheters made of Silastic Medical Grade tubing (Dow Corning, Midland, MI) were implanted to allow for i.v. drug administration. Indwellling intracerebral guide cannulae made of plastic (CMA 12; CMA/Microdialysis, Acton, MA) were implanted into the frontal cortex according to the following coordinates (medialateral, -2.5 mm; anterioposterior, +3.0 mm from bregma; dorsoventral, -0.8 mm from dura). The guide cannulae were secured to the skull using stainless steel screws and dental acrylic. Animals were housed individually and allowed 7 to 10 days for recovery.
In Vivo Microdialysis. Microdialysis sampling was carried out as described previously with minor modifications (Baumann et al., 2001). On the evening before an experiment, rats were moved to the testing room and lightly anesthetized with methohexitol (5 mg/kg i.v.). While anesthetized, a tether collar was placed on each rat. A microdialysis probe with a 3-× 0.5-mm exchange surface (CMA/12; CMA/Microdialysis) was lowered into the guide cannula and an extension tube was attached to the jugular catheter. Each rat was placed into its own plastic container and connected to the tethering system that allowed motor activity within the container. The microdialysis inflow and outflow tubing, as well as the catheter extension tubing, was connected to a fluid swivel (Instech Laboratories, Inc., Plymouth Meeting, PA). Artificial Ringer's solution containing 150.0 mM Na+, 3.0 mM K+, 1.4 mM Ca2+, 0.8 mM Mg2+, 1.0 mM P, and 155 mM Cl- (Harvard Apparatus, Inc., Holliston, MA) was pumped through the probe overnight at 0.5 μl/min. On the next morning, the flow rate was increased to 1.1 μl/min and dialysate samples were collected at 20-min intervals. Samples were split so that 10 μl was assayed for DA and 5-HT, and 10 μl were assayed for NE by high pressure-liquid chromatography with electrochemical detection as described below. When three stable baseline samples were obtained, drug treatments were administered.
Analysis of Monoamines. Aliquots of the dialysate (10 μl) were injected onto a microbore high pressure-liquid chromatography column (5 μm, C18, 100 × 1 mm; UniJet; Bioanalytical Systems, Inc., West Lafayette, IN) that was coupled to an amperometric detector (model LC-4C; BAS Bioanalytical Systems, Inc., West Lafayette, IN). A glassy carbon electrode was set at a potential of +650 mV relative to Ag/AgCl reference. Mobile phase for DA and 5-HT determinations consisted of 150 mM monochloroacetic acid, 145 mM NaOH, 1.5 mM sodium octanesulfonic acid, and 215 μM disodium EDTA, with 1 ml of triethylamine, 6% MeOH, and 6% CH3CN/l of water (final pH 5.3). Mobile phase for NE determinations consisted of 61 μM disodium EDTA, 62 mM lithium acetate, 4.2 mM heptanesulfonic acid, and 7% MeOH/l of water (final pH 4.8). Mobile phase was pumped (model 260D; ISCO, Lincoln, NE) at a rate of 60 μl/min. Chromatographic data were acquired on-line and exported to a Millennium software system (Waters, Milford, MA) for peak amplification, integration, and analysis. Standards of NE, DA, and 5-HT were run daily before dialysate samples, and standard curves were linear over a wide range of concentrations (0.1–100 pg). A monoamine standard mix containing NE, DA, and 5-HT, and their respective acid metabolites, was injected before and after the experiment to ensure validity of the constituent retention times. Peak heights of unknowns were compared with peak heights of standards and the lower limit of assay sensitivity (3 times baseline noise) was100 fg/5-μl sample.
Data Analysis and Statistics. For the in vitro experiments, IC50 and EC50 values were determined using the nonlinear least-squares curve fitting program MLAB-PC (Civilized Software, Bethesda, MD) as described previously (Rothman et al., 1993). Ki values were calculated from uptake assay results according to the following formula: Ki = IC50/(1 + L/Km), where L is the concentration of the radiolabeled drug ([3H]DA, [3H]NE, or [3H]5-HT) (Cheng and Prusoff, 1973). For the in vivo microdialysis experiments, raw neurotransmitter data were converted to a percentage of baseline values. Neurotransmitter baseline was determined from three dialysate samples collected immediately before drug or vehicle treatments, and each animal served as its own control. In vivo data were evaluated by analysis of variance (ANOVA): one-factor ANOVA (dose) for the dose-response experiments and two-factor ANOVA (pretreatment × acute treatment) for the nisoxetine pretreatment experiments. When significant F values were obtained, Duncan's post hoc test was used to determined statistical significance between group means (P < 0.05).
Results
As reported in Table 1, (±)-fenfluramine and its stereoisomers were essentially inactive in the DA release assay. The most potent action of (±)-fenfluramine and its stereoisomers was to evoke [3H]5-HT release. (+)-Fenfluramine released [3H]5-HT with an EC50 value of 51.7 nM, whereas (-)-fenfluramine released [3H]5-HT with an EC50 value of 147 nM, a 2.8-fold difference in potency. In the [3H]NE release assay, (+)-fenfluramine displayed appreciable activity (EC50 = 302 nM) that was about 6-fold weaker than its activity in the [3H]5-HT release assay. (-)-Fenfluramine was very weak in NE release assay. Thus, (-)-fenfluramine seems more selective than (+)-fenfluramine as a [3H]5-HT releaser, but (-)-fenfluramine is somewhat less potent in this regard.
EC50 values of test drugs for release of DA, NE, and 5-HT
Release assays were conducted as described under Materials and Methods. Values are mean ± S.D. for n = 3 experiments.
(±)-Norfenfluramine and its stereoisomers were more potent than fenfluramines at evoking [3H]DA release. However, norfenfluramines were much more potent at releasing [3H]5-HT compared with [3H]DA. (+)-Norfenfluramine released [3H]5-HT with an EC50 value of 59.3 nM, whereas (-)-norfenfluramine released [3H]5-HT with an EC50 value of 287 nM. (±)-Norfenfluramine and its stereoisomers were much more potent at releasing [3H]NE than fenfluramines. For example, (+)-norfenfluramine released [3H]NE with an EC50 value of 72.7 nM, compared with (+)-fenfluramine, which released [3H]NE with an EC50 value of 302 nM (see above). It is important to note that norfenfluramines released [3H]NE and [3H]5-HT with roughly equivalent potency. Additionally, the potency of (+)-norfenfluramine to evoke [3H]NE release was similar to the potency of phentermine, a known NE-releasing agent.
Results obtained in the uptake inhibition assays paralleled the results of the release assays, and these findings are summarized in Table 2. In general, as observed with other transporter substrates (Rothman et al., 2001), the substrates tested here were more potent in the release assays than in the uptake inhibition assays.
Ki values of test drugs for uptake inhibition
Uptake inhibition assays were conducted as described under Materials and Methods. Values are mean ± SD for n = 3 experiments.
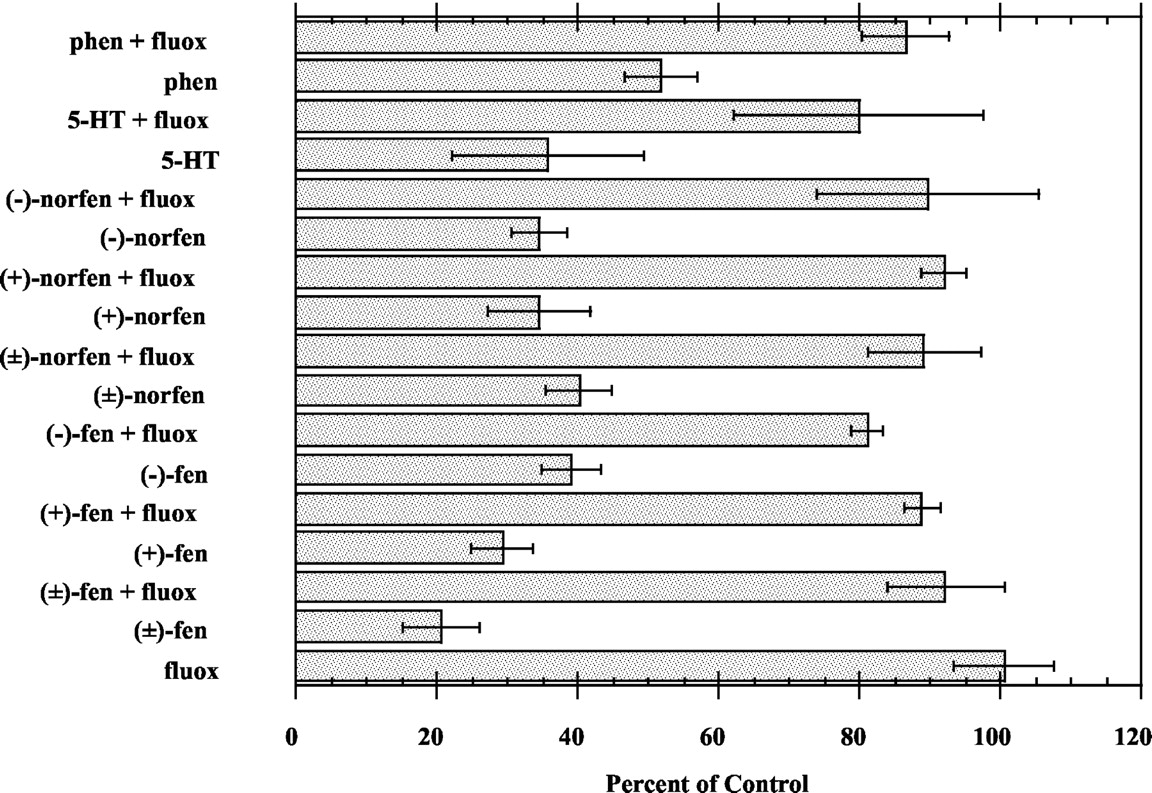
Substrate reversal experiments were performed to determine the role of SERT and NET in mediating the neurotransmitter releasing activity of (±)-fenfluramine, (±)-norfenfluramine, and their stereoisomers. Our previous findings have shown that uptake inhibitors reliably block the releasing activity of transporter substrates; therefore, the reversal of substrate-induced3H transmitter efflux by uptake inhibitors (i.e., substrate reversal) is used as a defining criterion to classify drugs as true substrate type-releasing agents. As reported in Fig. 1, the 5-HT uptake inhibitor fluoxetine antagonized the ability of (±)-fenfluramine, (±)-norfenfluramine, and their stereoisomers to release [3H]5-HT. Fluoxetine also reversed the ability of 5-HT and phentermine to release [3H]5-HT. Similarly, the NE uptake inhibitor desipramine reversed the ability of test drugs, NE, and DA to release [3H]NE (Fig. 2).

Fluoxetine reversal of the 5-HT-releasing activity of test drugs. Drug concentrations were as follows: fluoxetine (100 nM), (±)-fenfluramine (100 nM), (-)-fenfluramine (300 nM), (+)-fenfluramine (100 nM), (±)-norfenfluramine (200 nM), (-)-norfenfluramine (600 nM), (+)-norfenfluramine (100 nM), 5-HT (100 nM), and phentermine (3000 nM). Each column represents the mean ± S.E.M. for n = 3 experiments.

Desipramine (DMI) reversal of the NE-releasing activity of test drugs. Drug concentrations were as follows: DMI (166 nM), (±)-fenfluramine (1000 nM), (+)-fenfluramine (600 nM), (±)-norfenfluramine (200 nM), (-)-norfenfluramine (1000 nM), (+)-norfenfluramine (100 nM), NE (300 nM), DA (60 nM), and phentermine (50 nM). Each column represents the mean ± S.E.M. for n = 3 experiments.
The in vivo microdialysis data depicted in Fig. 3 demonstrate that (+)-fenfluramine produces dose-related increases in extracellular NE (F[8,45] = 8.47, P < 0.001), DA (F[8,63] = 5.92, P < 0.001), and 5-HT (F[8,63] = 10.83, P < 0.0001) in rat frontal cortex. The stimulatory effect of (+)-fenfluramine on dialysate 5-HT was the predominant action of this drug, with 5-HT levels reaching about 1000% above baseline (10-fold increase) at the 1-mg/kg dose and about 2200% of baseline (22-fold increase) at the 3-mg/kg dose. The (+)-fenfluramine-induced rise in extracellular NE was significant at both the 1- and 3-mg/kg doses of drug (P < 0.05), but this effect was 5-fold lower in magnitude compared with 5-HT effects. (+)-Fenfluramine was weak as a releaser of DA, and the drug was only effective at the high dose.

Dose-response effects of (+)-fenfluramine on extracellular levels of NE, DA, and 5-HT in rat frontal cortex. Rats undergoing in vivo microdialysis sampling received i.v. injections of 1 and 3 mg/kg (+)-fenfluramine at time 0 and 60 min, respectively. Dialysate samples were collected at 20-min intervals and assayed for NE, DA, and 5-HT, as described under Materials and Methods. Data are mean ± S.E.M. expressed as percentage of baseline. *, P < 0.05 with respect to preinjection control.
Like (+)-fenfluramine, (+)-norfenfluramine produced dose-related elevations in extracellular NE (F[8,45] = 12.17, P < 0.0001), DA (F[8,54] = 7.89, P < 0.001), and 5-HT (F[8,54] = 20.05, P < 0.0001) in rat cortex. As shown in Fig. 4, the rise in extracellular 5-HT evoked by (+)-norfenfluramine was similar in magnitude to that observed with (+)-fenfluramine. (+)-Norfenfluramine increased dialysate NE levels to a greater extent than that observed with (+)-fenfluramine (P < 0.05; Duncan's t test). In contrast to (+)-fenfluramine, (+)-norfenfluramine produced increases in extracellular DA that were similar in magnitude to its effect on extracellular NE.

Dose-response effects of (+)-norfenfluramine on extracellular levels of NE, DA, and 5-HT in rat frontal cortex. Rats undergoing in vivo microdialysis sampling received i.v. injections of 1 and 3 mg/kg (+)-norfenfluramine at time 0 and 60 min, respectively. Dialysate samples were collected at 20-min intervals and assayed for NE, DA, and 5-HT, as described under Materials and Methods. Data are mean ± S.E.M. expressed as percentage of baseline. *, P < 0.05 with respect to preinjection control.
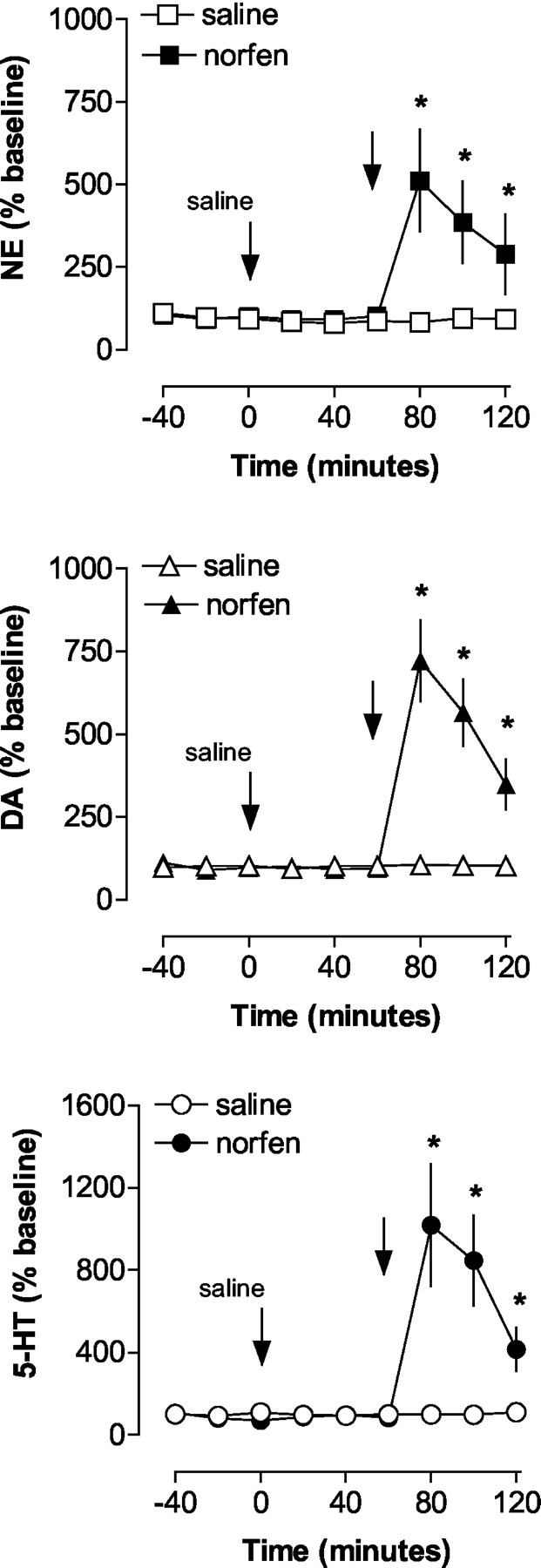
To determine the possible role of NET in mediating the actions of (+)-norfenfluramine, we tested the ability of the selective NET inhibitor nisoxetine (Tejani-Butt, 1992) to alter neurotransmitter release evoked by (+)-norfenfluramine. The data in Fig. 5 show the expected increase in extracellular NE, DA, and 5-HT in frontal cortex produced by (+)-norfenfluramine (2 mg/kg) in saline-pretreated rats. As shown in Fig. 6, pretreatment with nisoxetine alone (1 mg/kg i.v.) produced modest, albeit significant, elevations in dialysate NE (F[1,21] = 5.92, P < 0.02) and DA (F[1,24] = 5.07, P < 0.03), but had no effect on 5-HT (F[1,24] = 0.76, P < 0.76). Compared with the saline pretreatment condition, nisoxetine did not significantly alter (+)-norfenfluramine-induced increases in extracellular NE, DA, or 5-HT (nisoxetine by norfenfluramine interaction; Figs. 5 and 6). However, interpretation of the nisoxetine pretreatment effects was complicated by the effect of nisoxetine alone on extracellular NE and DE.
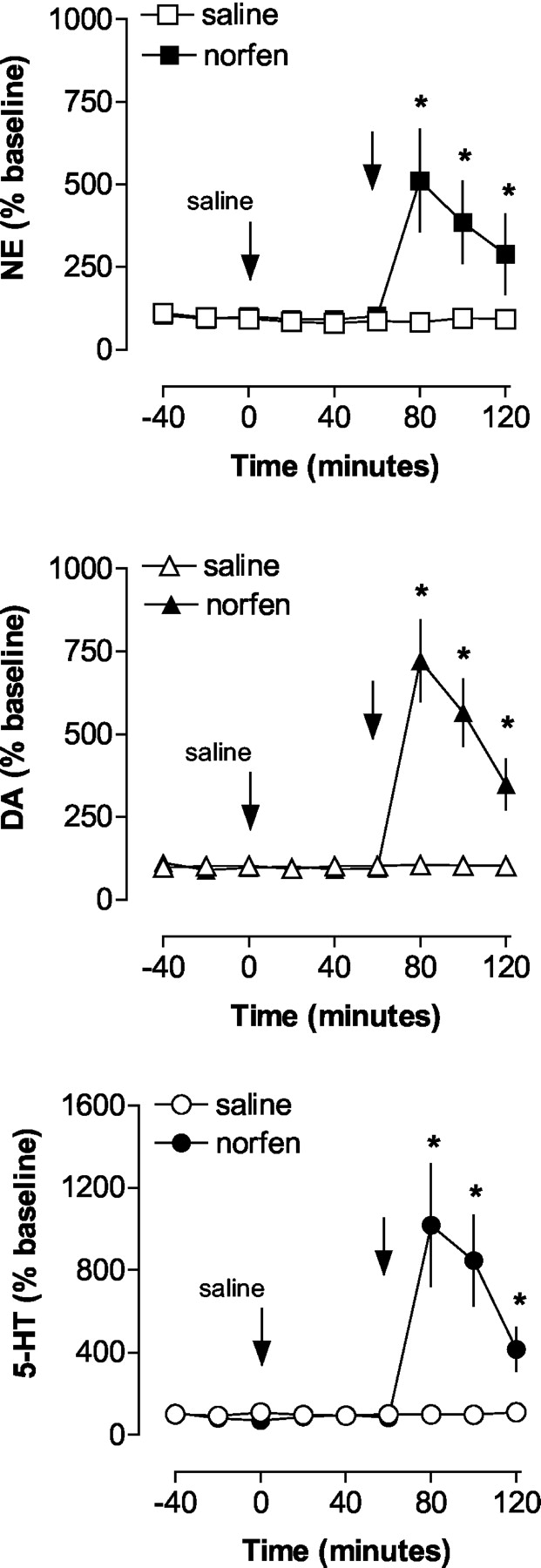
In vivo monoamine release evoked by acute (+)-norfenfluramine in frontal cortex of saline-pretreated rats. Rats undergoing microdialysis received saline pretreatment (1 ml/kg i.v.) at time 0, followed by either saline or 2 mg/kg (+)-norfenfluramine 60 min later. Dialysate samples were collected at 20-min intervals and assayed for NE, DA, and 5-HT, as described under Materials and Methods. Data are mean ± S.E.M. expressed as percentage of baseline. *, P < 0.05 with respect to saline/saline control group.

In vivo monoamine release evoked by (+)-norfenfluramine in frontal cortex of nisoxetine-pretreated rats. Rats undergoing microdialysis received nisoxetine pretreatment (1 mg/kg i.v.) at time 0, followed by either saline or 2 mg/kg (+)-norfenfluramine 60 min later. Dialysate samples were collected at 20-min intervals and assayed for NE, DA, and 5-HT, as described under Materials and Methods. Data are mean ± S.E.M. expressed as percentage of baseline. *, P < 0.05 with respect to nisoxetine/saline control group.
The data in Fig. 7 depict the effects of nisoxetine pretreatment on transmitter release evoked by (+)-norfenfluramine, when peak effects of (+)-norfenfluramine are calculated a percentage of preexisting baseline. The dialysis data from Figs. 5 and 6 were used to calculate means as follows: transmitter level at 80 min (peak effect of acute drug or saline) divided by transmitter level at 60 min (immediately before acute challenge) times 100. Stated more simply, the data in Fig. 7 are expressed to factor out the effect of nisoxetine alone. When the data are normalized in this manner, it is apparent that nisoxetine significantly blunts the maximal effect of (+)-norfenfluramine on NE and DA release (P < 0.05; Duncan's t test), but not 5-HT release.

Effect of nisoxetine pretreatment on peak monoamine release evoked by (+)-norfenfluramine. Rats were pretreated with saline or nisoxetine, followed by acute saline or (+)-norfenfluramine as described in Figs. 5 and 6. Data are mean ± S.E.M. expressed as the percentage of change in transmitter level produced by (+)-norfenfluramine (percentage of change from preexisitng baseline). *, P < 0.05 with respect to corresponding saline-pretreated group.
Discussion
The main goal of the present study was to assess the activity of stereoisomers of (±)-fenfluramine and (±)-norfenfluramine at monoamine transporters. Our in vitro results confirm that these drugs stimulate 5-HT release by a mechanism involving SERT proteins (Garattini, 1995; Rothman and Baumann, 2002). (+)-Fenfluramine and (+)-norfenfluramine displayed similar potency as releasers of [3H]5-HT in brain tissue, and (+)-isomers of both drugs were more potent than (-)-isomers. Perhaps the most striking finding reported here is that (+)-fenfluramine and (+)-norfenfluramine are also substrates for NETs. (+)-Norfenfluramine was 4-fold more potent than (+)-fenfluramine in this regard, and (+)-norfenfluramine was essentially equipotent as a releaser of NE and 5-HT. The in vivo microdialysis data revealed that (+)-isomers of fenfluramine and norfenfluramine produce dose-related elevations in extracellular 5-HT, NE, and DA in rat frontal cortex. It is noteworthy that the magnitude of drug-induced 5-HT release was always greater than the corresponding NE and DA release at a given dose. Consistent with the in vitro results, (+)-norfenfluramine was more potent than (+)-fenfluramine in its ability to elevate extracellular NE and DA in vivo. Collectively, the data indicate that systemic administration of anorectic doses of (±)-fenfluramine or (+)-fenfluramine can stimulate NE and DA release, in addition to 5-HT release, in certain brain regions.
The fact that (+)-isomers of fenfluramine and norfenfluramine are able to elevate extracellular DA in vivo is surprising based on the low potency of these drugs as DA releasers in vitro (Table 1). One possibility is that NET sites are involved in the DA-releasing action of these drugs. It is well established, for example, that DA has comparable affinity as a substrate for NETs or DATs and that DA is translocated from the extracellular medium into cells via either transporter (Rothman et al., 2001). Indeed, NETs contribute significantly to the neuronal uptake of DA in rat cortex, a region where the density of NET sites is greater than that of DAT sites (Carboni et al., 1990; Tanda et al., 1997; Linner et al., 2001). Our microdialysis results show that blockade of NE uptake with nisoxetine increases extracellular DA along with NE, suggesting NE nerve terminals in rat frontal cortex might contain both DA and NE. Consistent with this notion, (+)-norfenfluramine released DA and NE to a similar extent. The ability of (+)-norfenfluramine to evoke in vivo release of NE and DA was partially antagonized by nisoxetine, indicating that NETs play at least some role in the catecholamine-releasing activity of (+)-norfenfluramine (Fig. 7).
Chronic administration of (±)-fenfluramine or (+)-fenfluramine to humans leads to steady-state plasma levels of (±)-norfenfluramine and (+)-norfenfluramine that are about one-half the plasma levels of parent drugs (Caccia et al., 1985). In nonhuman primates, plasma levels of (+)-norfenfluramine exceed those of the parent drug by more than 10-fold due to rapid elimination of (+)-fenfluramine in these species. In rats, circulating levels of (+)-norfenfluramine can surpass those of (+)-fenfluramine for hours after peripheral administration of (+)-fenfluramine (De Souza et al., 1991). Importantly, (+)-fenfluramine and (+)-norfenfluramine accumulate in rat brain tissue, with brain-to-plasma ratios reaching 40-to-1 (De Souza et al., 1991). These pharmacokinetic factors indicate that administration of (±)-fenfluramine or (+)-fenfluramine will produce enough (+)-norfenfluramine to evoke significant release of NE throughout the neuraxis. Our in vivo microdialysis data provide direct evidence that (+)-norfenfluramine releases NE in the rat cortex at the same doses that release 5-HT (Fig. 4).
Clinical pharmacokinetic studies indicate that anorectic doses of (+)-fenfluramine produce brain concentrations of (+)-norfenfluramine high enough to elicit NE release. For example, Christensen et al. (1999) used19F magnetic resonance spectroscopy to measure steady-state levels of (+)-fenfluramine and (+)-norfenfluramine in plasma and brain tissue of human subjects. These investigators reported that daily oral administration of 30 mg of (+)-fenfluramine produced a combined concentration of (+)-fenfluramine and (+)-norfenfluramine of about 4 μM in the brain. Given that plasma (+)-norfenfluramine concentrations are approximately one-half those of (+)-fenfluramine (Caccia et al., 1985), brain levels of (+)-norfenfluramine are likely to be in the range of 1 μM, a concentration that is sufficient to release NE.
The collective findings suggest that NE mechanisms might be involved in mediating pharmacological actions of fenfluramines. As noted under Introduction, (±)-fenfluramine and (+)-fenfluramine are effective appetite suppressants, and anorectic effects of the drugs are presumably related to stimulation of 5-HT transmission in the central nervous system (Pinder et al., 1975; Garattini et al., 1986). However, emerging evidence indicates that 5-HT release per se is not required for fenfluramine-induced anorexia (Curzon et al., 1997). For example, certain pharmacological manipulations that completely block fenfluramine-evoked 5-HT release do not alter hypophagic effects of the drug (Gibson et al., 1993; Raiteri et al., 1995). Such observations have fueled speculation that (±)-fenfluramine and (+)-fenfluramine produce their anorectic effects via direct activation of 5-HT2C receptor sites. Table 3 summarizes the binding affinities of stereoisomers of (±)-fenfluramine and (±)-norfenfluramine at various 5-HT2 receptor subtypes: (+)-isomers of fenfluramine and norfenfluramine display Ki values of 6245 and 324 nM at 5-HT2C receptors (Rothman et al., 2000). In the present study, (+)-fenfluramine and (+)-norfenfluramine released [3H]NE with EC50 values of 320 and 72 nM (Table 1), thereby demonstrating NE release would be occurring at lower doses than those producing direct 5-HT2C receptor agonism. Because 5-HT2C receptor antagonists can only partially reduce the hypophagic effects of (+)-fenfluramine and (+)-norfenfluramine, other non-5-HT2C mechanisms are implicated (Vickers et al., 2001). Thus, NE might be involved in the anorectic actions of fenfluramines, and further investigation of this proposal is warranted.
Ki values of test drugs for 5-HT2 receptor subtypes
Values are mean ± S.D. for n = 3 experiments. Data taken from Rothman et al. (2000).
Probably the strongest evidence for a physiologically relevant noradrenergic effect of fenfluramine comes from studies examining renin secretion. Activation of the sympathetic nervous system increases the secretion of renin from the kidneys (Cody, 1997). Conversely, suppression of sympathetic nervous activity via stimulation of central α2-adrenergic receptors decreases renin secretion (Oates et al., 1978; Schoeppe and Brecht, 1980). In rats, administration of (±)-fenfluramine produces an initial stimulation of renin secretion that is mediated by 5-HT release, followed by a long-lasting inhibition of renin secretion that is mediated by central NE nerves (Van de Kar et al., 1994). Administration of the NE uptake blocker desipramine completely abolishes the delayed suppression of renin release produced by (±)-fenfluramine (Van de Kar et al., 1994). Based on the present findings, it seems likely that (±)-fenfluramine administration leads to the formation of (+)-norfenfluramine, which then acts as a substrate for NETs in the brain. The ensuing elevations in extracellular NE stimulate α2-receptors to decrease sympathetic outflow and renin secretion.
Clinical studies provide further support for the importance of NE mechanisms in mediating fenfluramine-induced inhibition of renin secretion. In humans, (+)-fenfluramine decreases sympathetic nervous system activity (Hirsch et al., 2000), plasma NE (Andersson et al., 1991; Kolanowski et al., 1992; Flechtner-Mors et al., 1998), blood pressure (Andersson et al., 1991; Kolanowski et al., 1992; Flechtner-Mors et al., 1998), and plasma renin (Andersson et al., 1991). Interestingly, administration of the 5-HT precursor 5-hydroxytryptophan causes cardiovascular effects similar to (+)-fenfluramine but does not decrease plasma renin (Maestri et al., 1988). Thus, two agents that produce comparable increases in synaptic 5-HT differ with respect to their effects on plasma renin. One reason for this discrepancy might be related to NE release associated with the formation of (+)-norfenfluramine after systemic (+)-fenfluramine. It is noteworthy that amphetamine releases NE from neurons and increases blood pressure (Weiner, 1989). The fact that (±)-fenfluramine, which releases NE via its metabolite (±)-norfenfluramine, does not increase blood pressure is probably because rapid and simultaneous elevations of synaptic 5-HT produce an overall reduction in blood pressure (Zimmermann and Ganong, 1980; Ramirez et al., 1982; Itskovitz et al., 1989).
Viewed collectively, the present results emphasize the complex pharmacology of (±)-fenfluramine. From a pharmacokinetic perspective, administration of (±)-fenfluramine generates four bioactive agents: (+)-fenfluramine, (-)-fenfluramine, (+)-norfenfluramine, and (-)-norfenfluramine. These compounds interact to varying degrees with SERTs, NETs, and multiple 5-HT2 receptor subtypes. We have previously proposed that substrate activity at SERTs is one factor responsible for the increased incidence of primary pulmonary hypertension in patients who have taken fenfluramines (Rothman et al., 1999). As discussed above, activation of 5-HT2C receptors is thought to mediate the anorectic effects of fenfluramines, and (+)-norfenfluramine is probably involved in this effect due to its potent affinity for 5-HT2C receptors (Garattini, 1995; Curzon et al., 1997; Vickers et al., 2001). Accumulating evidence shows that activation of 5-HT2B receptors underlies cardiac valve disease associated with (±)-fenfluramine and (+)-fenfluramine (Fitzgerald et al., 2000; Rothman et al., 2000) and also might contribute to the development of pulmonary hypertension (Launay et al., 2002); in this case, (+)-norfenfluramine is a likely culprit based on its potent activity at 5-HT2B sites. Further research is warranted to assess the precise role of NE in mediating the diverse pharmacological actions of fenfluramines.
Acknowledgments
We acknowledge the expert technical assistance of Mario Ayestas.
Footnotes
-
This work was generously supported by the Intramural Research Program of the National Institute on Drug Abuse (National Institutes of Health, Bethesda, MD) and the National Institute of Mental Health Psychoactive Drug Screening Program and the National Institute of Mental Health's Chemical Synthesis and Drug Supply Program.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
DOI: 10.1124/jpet.103.049684.
-
ABBREVIATIONS: 5-HT, 5-hydroxytryptamine, serotonin; SERT, serotonin transporter; NE, norepinephrine; NET, norepinephrine transporter; DA, dopamine; DAT, dopamine transporter; BSA, bovine serum album; ANOVA, analysis of variance; GBR12935, 1-(2-diphenylmethoxyethyl)-4-(3-phenylpropyl)piperazine.
- Received January 28, 2003.
- Accepted March 11, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}