


Abstract
The catecholaldehyde hypothesis posits that 3,4-dihydroxyphenylacetaldehyde (DOPAL), an obligate intermediary metabolite of dopamine, is an autotoxin that challenges neuronal homeostasis in catecholaminergic neurons. DOPAL toxicity may involve protein modifications, such as oligomerization of α-synuclein (AS). Potential interactions between DOPAL and other proteins related to catecholaminergic neurodegeneration, however, have not been systemically explored. This study examined DOPAL-induced protein-quinone adduct formation (“quinonization”) and protein oligomerization, ubiquitination, and aggregation in cultured MO3.13 human oligodendrocytes and PC12 rat pheochromocytoma cells and in test tube experiments. Using near-infrared fluorescence spectroscopy, we detected spontaneous DOPAL oxidation to DOPAL-quinone, DOPAL-induced quinonization of intracellular proteins in both cell lines, and DOPAL-induced quinonization of several proteins related to catecholaminergic neurodegeneration, including AS, the type 2 vesicular monoamine transporter, glucocerebrosidase, ubiquitin, and l-aromatic-amino-acid decarboxylase (LAAAD). DOPAL also oligomerized AS, ubiquitin, and LAAAD; inactivated LAAAD (IC50 54 μM); evoked substantial intracellular protein ubiquitination; and aggregated intracellular AS. Remarkably, N-acetylcysteine, which decreases DOPAL-quinone formation, attenuated or prevented all of these protein modifications and functional changes. The results fit with the proposal that treatments based on decreasing the formation and oxidation of DOPAL may slow or prevent catecholaminergic neurodegeneration.
Introduction
Diseases involving progressive catecholaminergic neurodegeneration, such as Parkinson’s disease (PD) and multiple system atrophy (MSA), are associated with buildup of 3,4-dihydroxyphenylacetaldehyde (DOPAL), a toxic obligate intermediary metabolite of dopamine (DA), in affected brain regions (Goldstein et al., 2011, 2013, 2015b). According to the “catecholaldehyde hypothesis,” DOPAL causes or contributes to neuronal malfunctions and death in these diseases (Burke et al., 2003; Goldstein et al., 2014a). In cells and animals, DOPAL exerts substantial cytotoxicity (Mattammal et al., 1995; Burke et al., 2003; Panneton et al., 2010). The catecholaldehyde is far more toxic than are several other DA metabolites and DA itself (Kristal et al., 2001; Burke et al., 2003).
A potential mechanism of DOPAL toxicity is via protein modifications. For instance, DOPAL oligomerizes the protein α-synuclein (AS) (Burke et al., 2008), a major component of Lewy bodies in PD (Spillantini et al., 1997) and of glial cytoplasmic inclusions (GCIs) in MSA (Wakabayashi et al., 1998), and oligomerized AS may be pathogenic (Winner et al., 2011). DOPAL-induced AS oligomers interfere with vesicular storage of DA (Plotegher et al., 2017), and blockade of vesicular uptake shifts the fate of cytoplasmic DA toward DOPAL formation (Goldstein et al., 2012)—a potentially lethal vicious cycle. Interactions between DOPAL and other proteins involved in catecholaminergic neurodegeneration, however, have not been explored systemically.
DOPAL-induced protein modifications may occur as a result of oxidation of DOPAL to DOPAL-quinone (Follmer et al., 2015). In the present study, we applied a recently published assay method to detect and quantify quinones by near-infrared fluorescence (nIRF) spectroscopy (Mazzulli et al., 2016). We used this method to measure DOPAL-quinone and DOPAL-derived protein-quinone adducts (“quinonization”). In cultured cells, we examined whether DOPAL quinonizes intracellular proteins, and in test tubes, we examined whether DOPAL quinonizes proteins involved in the pathogenesis of synucleinopathies, such as l-aromatic-amino-acid decarboxylase (LAAAD) (Goldstein et al., 2017b), the type 2 vesicular monoamine transporter (VMAT2) (Pifl et al., 2014), glucocerebrosidase (GBA) (Goker-Alpan et al., 2010), ubiquitin (UBQ) (Cartier et al., 2012), AS, and the A53T mutant form of AS [which causes the PARK1 form of familial PD (Polymeropoulos et al., 1997)].
Of the 140 amino acids in the AS molecule, a relatively high percentage (11%) are lysines, and DOPAL-derived protein adduct formation seems to occur via interactions with the lysine residues (Plotegher et al., 2017; Werner-Allen et al., 2017). UBQ, a key mediator of proteasomal function, also contains several lysine residues (9%). We therefore explored whether DOPAL quinonizes and oligomerizes UBQ.
Since ubiquitination is a key cellular process for disposing of misfolded proteins in proteasomes, we used DOPAL-induced ubiquitination of intracellular proteins as an indirect measure of protein misfolding.
DOPAL aggregates intracellular AS in SHSY-5Y neuroblastoma cells (Burke et al., 2008). Whether DOPAL aggregates AS in human oligodendrocytes has been unknown. This could be relevant to the pathogenesis of MSA, because in MSA, AS deposition occurs in the cytoplasm of oligodendrocytes (Dickson et al., 1999b). We assessed this possibility by incubating cultured human oligodendrocyte MO3.13 cells with AS and DOPAL.
LAAAD catalyzes the formation of DA from 3,4-dihydroxyphenylalanine (DOPA), and putamen LAAAD activity is decreased in PD and MSA (Goldstein et al., 2017b). The mechanisms of this decrease are unknown. We therefore also examined whether DOPAL quinonizes, oligomerizes, and inhibits the activity of LAAAD.
To explore whether protein modifications exerted by DOPAL depend on its oxidation, we assessed effects of the antioxidant N-acetylcysteine (NAC). A variety of antioxidants interfere with DOPAL-induced oligomerization of AS (Follmer et al., 2015; Anderson et al., 2016). We chose NAC for the present experiments for several reasons. NAC is a Food and Drug Administration–approved drug (to treat acetaminophen overdose) and is already undergoing testing in many experimental therapeutics trials (Deepmala et al., 2015), including to treat PD (Monti et al., 2016), so that movement from preclinical to clinical studies could proceed relatively quickly. In rat pheochromocytoma PC12 cells, monoamine oxidase (MAO) inhibitors increase the spontaneous oxidation of endogenous DA (Goldstein et al., 2016), and NAC attenuates this effect of MAO inhibition without interfering with the decrease in endogenous DOPAL production (Goldstein et al., 2017a). NAC decreases endogenous DOPAL levels in PC12 cells (Goldstein et al., 2017a) and prevents DA-evoked formation of intracellular quinoprotein adducts (Banerjee et al., 2014). Finally, NAC is known to inhibit DOPAL-induced oligomerization of AS (Anderson et al., 2016).
The MO3.13 human oligodendrocyte cell line was used because of the potential relevance to the pathogenesis of MSA. GCIs containing AS—especially in oligodendrocytes—are a histopathologic hallmark of this disease (Dickson et al., 1999a). Given that oligodendrocytes normally do not express AS, where does the AS come from that characterizes the GCIs in this disease? Based on our previous report that DOPAL oligomerizes AS in human glial cells (Jinsmaa et al., 2016), we hypothesized that GCIs are derived from both DOPAL and monomeric AS being transmitted (Longhena et al., 2017), resulting in AS aggregation during coincubation of MO3.13 cells with DOPAL and AS.
The rat pheochromocytoma PC12 cell line was used, because this cell line produces DOPA, DA, DOPAL, 3,4-dihydroxyphenylacetic acid (DOPAC), and 3,4-dihydroxyphenylethanol endogenously, enabling a comprehensive analysis of the synthesis and fate of endogenous DA (Goldstein et al., 2012). Moreover, endogenous DOPAL contributes to rotenone-induced cytotoxicity (Lamensdorf et al., 2000) and DA-induced apoptosis (Goldstein et al., 2012) in this cell line.
Therefore, the aims of this study were to evaluate comprehensively DOPAL-induced protein modifications in MO3.13 and PC12 cells and in test tube experiments and to determine whether NAC mitigates or prevents these effects.
Materials and Methods
Reagents and Chemicals.
Human recombinant AS was purchased from Calbiochem (La Jolla, CA). DOPAL was from Santa Cruz Biotech (Dallas, TX). Mutant A53T-AS, NAC, UBQ, dithiothreitol, glycine, saponin, and the aldehyde dehydrogenase inhibitor benomyl were from Sigma-Aldrich (St. Louis, MO). The aldehyde reductase inhibitor AL-1576 (spiro-(2,7-difluoro-9H-fluoren 9,4′-imidazoline) 2′,5′-dione was a gift from Alcon Laboratories (Fort Worth, TX). Tolcapone was from Orion Pharma (Espoo, Finland). GBA was a gift from Dr. Ellen Sidransky (National Heart, Lung, and Blood Institute/National Institutes of Health, Bethesda, MD). Cell culture media were from Invitrogen (Camarillo, CA). Phosphate-buffered saline was from KD Medical (Columbia, MD).
Benomyl, AL-1576, and tolcapone were dissolved in dimethylsulfoxide (American Bio, Natick, MA) and stored at −20°C. NAC was freshly prepared in ultrapure water (KD Medical) before use.
MO3.13 and PC12 Cell Cultures.
Human oligodendrocytes (MO3.13) were from Cellutions Biosystems Inc. (Burlington, ON, Canada). The MO3.13 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum (FCS). For experiments, MO3.13 cells (1.5 × 105 or 2 × 105 cells/well) were plated in 12- or six-well plates and incubated for 24 hours with 10 μM tolcapone in the medium to block catechol-O-methyltransferase prior to the acute experimental treatments. On the day of the acute experiments, the medium was replaced with 2.5% FCS in Dulbecco’s modified Eagle’s medium containing 10 µM tolcapone, 10 µM benomyl, and 1 µM AL-1576; the cells were further exposed to the compounds as described in the following sections.
Suspended PC12 cells were from the American Type Culture Collection (PC12 cells catalog no. CRL-1721; Manassas, VA). The PC12 cells were cultured in flasks containing F-12K medium, 15% horse serum, and 2.5% FCS. As noted earlier, prior to each acute experiment, the cells in flasks (∼1.6 × 107 cells) were incubated for 24 hours with 10 µΜ tolcapone added to the medium. On the day of the acute experiments, cells were harvested and counted, and 5 × 105 cells/well were resuspended in F-12K medium containing 2.5% FCS with 10 µM tolcapone, 10 µM benomyl, and 1 µM AL-1576; the cells were then exposed to additional compounds as described in the following sections.
We reported previously that inhibition of aldehyde dehydrogenase (ALDH) and aldehyde/aldose reductase (AR) increases cell and medium concentrations of DOPAL in PC12 cells (Goldstein et al., 2012), and that there is efficient conversion of DOPAL to DOPAC via ALDH in human glial cells (Jinsmaa et al., 2016). Therefore, the current experiments involving addition of DOPAL to culture medium were done in the setting of ALDH and AR inhibition.
Quinonization by nIRF.
We applied the nIRF method of Mazzulli et al. (2016) to detect and quantify quinones in test tube and cell experiments.
First, we examined the formation of DOPAL-quinone in wells with incubation medium and no cells. Different concentrations of DOPAL (3, 10, 30, and 100 μM) or, for comparison, DA (100 μM) were added to the medium in 12-well plates, and the cells were incubated at 37°C. The nIRF signals were read at 0, 1, 2, and 24 hours with an excitation wavelength of 685 nm using an Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE). There was one experiment, with one replicate for each observation point. To assess whether NAC affects the nIRF responses, 1 mM NAC was added to the wells after 1 hour of DOPAL incubation. There was one experiment, with two replicates at each observation point.
We then examined DOPAL-induced quinones in wells plated with human oligodendrocyte MO3.13 cells (1.5 × 105 cells/well). After 24 hours of incubation with medium containing tolcapone as described in the previous sections about MO3.13 and PC12 cell cultures, the cells were treated with different concentrations of DOPAL (3, 10, 30, and 100 μM) or, for comparison, DA (100 μM) with or without NAC (10, 30, and 100 μM). The nIRF signals in the wells were read at 0, 0.5, 1, 2, 24, and 48 hours of incubation. There were two experiments, with three replicates at each observation point.
For detection of intracellular DOPAL-induced quinones, MO3.13 cells (2 × 105 cells/well) were exposed to DOPAL for 5 or 24 hours. The cells were then lysed with radioimmunoprecipitation assay buffer (Millipore, Temecula, CA) containing one tablet per 10 ml of Complete Mini protease inhibitors (Roche Diagnostics, Indianapolis, IN). Ten microliters of each cell lysate were added to the plastic well. The plates were then warmed to dryness in an incubator. To obtain detectable signals by nIRF, the same amounts of cell lysate were applied to the same dried spot two more times. There was one experiment, with three replicates at each observation point.
Experiments on concentration- and time-related formation of intracellular DOPAL-quinonized proteins were done using MO3.13 (2 × 105 cells/well) and suspended PC12 cells (5 × 105 cells/well). Cell protein lysates (15 μl) obtained as described earlier were electrophoresed on NuPAGE 4%–12% of Bis-Tris gels (Life Technologies, Cartsbad, CA), and the protein gels were read by nIRF spectroscopy. In some experiments, the gels were also stained for proteins with AcquaStain (Bulldog Bio, Portsmouth, NH). There was one experiment, with three replicates at each observation point. NAC (30, 100, and 300 µM) was added at the same time as DOPAL in two experiments, with three replicates at each observation point.
Test tube experiments were done to explore DOPAL-induced quinonization of AS and other proteins thought to be related to PD pathogenesis. The tested proteins were AS, the A53T mutant AS, UBQ, VMAT2, GBA, and LAAAD. Each protein [1 μM solution, except UBQ (50 μM solution)] was incubated with different concentrations of DOPAL (10, 30, and 100 μM) or, for comparison, DA (10, 30, and 100 μM), or in some cases only 100 μM DOPAL (considering the cost of the proteins), with or without NAC (1–100 μM) for 1 hour at 37°C. The protein gels were then read for nIRF signals. There was one experiment, with three replicates at each observation point.
Whether AS quinonization depends on available lysine residues has been unknown. Therefore, we studied the effects of acetylation of the lysine residues using the method of Rees et al. (2007). In these experiments, 1 mg/ml of AS was incubated with 5 or 10 mM citraconic anhydride (total volume 100 µl; Mullica Hill, Acros Organics, NJ) at room temperature for 24 hours. The samples were then electrophoresed, and the protein gels were read by nIRF spectroscopy or were treated further for western blotting. There was one experiment, with three replicates at each observation point.
DOPAL-UBQ Interactions.
We examined whether DOPAL quinonizes or oligomerizes UBQ and compared the results for UBQ with those for AS. Western blotting was done to detect UBQ and AS oligomers in test tubes and ubiquitinated proteins in cell lysates. The proteins (5 µl) were heated in NuPAGE LDS (Lithium dodecyl sulfate) sample buffer with 50 mM dithiothreitol for 5 minutes at 70°C. The reaction mixtures were then electrophoresed on NuPAGE 4%–12% Bis-Tris gels and transferred to nitrocellulose membranes using an iBlot Dry Blotting system (Novex Life Technologies, Thermo Fisher Scientific, Waltham, MA). AS detection was done with mouse anti-human AS antibody (1:200; Invitrogen) and the secondary antibody goat anti-mouse IRDye 800CW (1:10,000; Abcam, Cambridge, MA). UBQ detection was performed with rabbit anti-ubiquitin antibody (1:1000; Abcam) and the secondary antibody goat anti-rabbit IRDye 680CW (1:10,000; Abcam). The detector was an Odyssey Infrared Imaging System (LI-COR Biosciences).
DOPAL-induced protein modifications would be expected to augment protein misfolding, targeting the proteins for the UBQ-proteasome system. We tested whether DOPAL increases ubiquitination of intracellular proteins. PC12 cells (1 × 106 cells/well) were exposed to different concentrations of DOPAL (10, 30, and 100 µM) for 5 hours. The cells were then lysed with radioimmunoprecipitation assay buffer containing Complete Mini protease inhibitors. The proteins (15 µl) were subjected to electrophoresis and western blotting using UBQ antibody as described earlier. The loading control β-actin was detected using mouse anti-β-actin antibody (1:700; Abcam) and goat anti-mouse IRDye 800CW secondary antibody (1:10,000; Abcam). Relative intensities of each sample were calculated by dividing the integrated intensity by the intensity of β-actin and expressed as a fraction of control or of DOPAL alone. There were two experiments, with three replicates at each observation point.
DOPAL-LAAAD Interactions.
DOPAL-induced quinonization and oligomerization of LAAAD (PROSpec Protein Specialists, East Brunswick, NJ) were assessed as described earlier for other proteins of interest. For LAAAD oligomer detection, we used goat anti-rabbit LAAAD antibody (1:500; Abcam) and goat anti-rabbit IRDye 680CW secondary antibody (1:10,000; Abcam). There were two experiments, with three replicates at each observation point.
We tested the effects of DOPAL on LAAAD activity by the ability of the enzyme to convert DOPA to DA. The enzymatic reaction of LAAAD (RD Systems Biotechne, Minneapolis, MN) was done in 50 mM phosphate buffer (pH 7.5) with 1 mM DOPA, 1 mM pyridoxal phosphate (Sigma-Aldrich), and LAAAD (0.5 μg/ml) incubated at 37°C for 1 hour. To study inhibition of LAAAD by DOPAL, different concentrations of DOPAL (0, 10, 30, 100, 300, and 1000 μM) were added to the reaction mixtures at 0 minutes. After 1 hour of incubation, the reactions were stopped by adding 90 μl of a 20:80 solution of 40 mM H2PO4:200 mM acetic acid to 10 μl of the reaction mixtures. DA concentrations in the reaction mixtures were analyzed by liquid chromatography with series electrochemical detection (Goldstein et al., 2012).
To determine the effects of NAC on DOPAL-induced inhibition of LAAAD, different concentrations of NAC (0, 10, 30, 100, 300, and 1000 μM) were added to reaction mixtures containing 300 μM DOPAL (corresponding to 90% LAAAD inhibition). There was one experiment, with four replicates at each observation point.
DOPAL-Induced Cytoplasmic AS Aggregation by Immunofluorescence Microscopy.
We used immunofluorescence microscopy to detect DOPAL-induced intracellular AS aggregation in MO3.13 cells. For these experiments, MO3.13 cells (2 × 105 cells/slide) were exposed to 3 µl of AS (70 µM) for 2–3 hours to allow AS to enter the cells and then were treated for 4–5 hours with DOPAL (100 µM) in the presence or absence of NAC (10 and 100 µM). We did not examine how AS monomer added to the medium gets into the cells. After treatment, the cells were fixed with ice-cold methanol for 20 minutes. Permeabilization and blocking were done with 200 µl of 0.1% Tween 20 (Affymetrix USB Products, Cleveland, OH) in phosphate-buffered saline and 200 µl of a solution containing 0.1% goat serum (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), 1.5 mg/ml glycine, and 10 mg/ml saponin for 1 hour. For AS detection, the solutions were incubated with mouse anti-human AS (1:500; Invitrogen) overnight at 4°C and then with the secondary antibody Alexa Fluor 488 goat anti-mouse (1:500; Molecular Probes, Thermo Fisher Scientific) for 1 hour at room temperature. Cells were also stained for 4,6-diamidino-2-phenylindole (1:2000; Abcam) to visualize cell nuclei. Images were obtained using a Zeiss 880 confocal microscope (Zeiss, Jena, Germany). There were three experiments, with one set of observations per experiment.
To measure intracellular DOPAL concentrations attained during incubation of cells with 100 µM DOPAL, we measured intracellular concentrations of DOPAL and other catechols after incubation of MO3.13 cells (1.5 × 105 cells/well) with DOPAL for up to 5 hours. Cells were collected at 0, 0.5, 1, 3, and 5 hours in 400 μl of a 40 mM H2PO4:200 mM acetic acid solution and frozen at −80°C until assayed for catechol contents. For catechol assays, 200μl aliquots of 400 μl of disrupted cells were used (Goldstein et al., 2012). Catechol concentrations in cell lysates were expressed in units of pmoles per well. There was one experiment, with four replicates at each observation point.
Data Analysis and Statistics.
Values were expressed as the mean ± S.E.M. Statistical analyses were done by one-way factorial analyses of variance with Dunnett’s post-hoc test to compare experiment with control (or DOPAL alone) mean values (GraphPad Software, La Jolla, CA). Statistical significance was defined by P < 0.05.
Results
DOPAL Quinonization in Medium without and with Cells.
DOPAL standard added to a cellular incubation medium resulted in concentration- and time-dependent spontaneous formation of a quinone (Fig. 1A). In this regard, DOPAL was far more potent than DA. Addition of NAC after 1 hour attenuated the quinone signal evoked by DOPAL (Fig. 1B). Simultaneous addition of DOPAL and tyrosinase to the incubation medium (to oxidize DOPAL enzymatically) resulted in essentially immediate quinone formation (data not shown).

DOPAL-quinone formation and NAC effect in wells. (A) DOPAL or DA. (B) Effects of NAC added into wells after 1 hour of DOPAL incubation. DOPAL spontaneously formed DOPAL-quinone. NAC partly reversed the quinonization of DOPAL.
When MO3.13 cells in wells were incubated with DOPAL, the medium showed concentration- and time-dependent increases in quinones (Fig. 2A), with DOPAL again being more potent than DA. NAC concentration-dependently attenuated the appearance of quinones in the medium (Fig. 2B). Similar results were obtained by analyzing cell lysates (Fig. 2, C and D).

DOPAL-induced quinone formation in MO3.13 cells. MO3.13 cells were exposed to DOPAL or DA without (A and C) and with NAC (B and D). Quinone detection was performed in the medium (A and B) and cell lysates (C and D). Above the graphs are black-and-white photographs of the quinone signals. Mean (± S.E.M.) values for nIFR signal intensities relative to control. *P < 0.05; **P < 0.01; ****P < 0.0001 versus control or DOPAL. Incubation of MO3.13 cells with DOPAL produced concentration- and time-dependent quinones in both the medium and cell lysates. NAC prevented the quinonization of DOPAL.
DOPAL-Induced Quinonization of Intracellular Proteins and NAC Effects.
When MO3.13 or PC12 cells were incubated with 100 μM DOPAL and then lysed, and the lysates were electrophoresed in 4%–12% Bis-Tris gels over different exposure times, there was time-dependent DOPAL-induced quinonization of numerous relatively low-molecular-weight proteins (Fig. 3A shows results from an experiment in MO3.13 cells), bands at about 50–60 kDa, and a discrete band at about 62 kDa (Fig. 3, A and D). In both cell lines, NAC concentration-dependently attenuated formation of the 62-kDa band (Fig. 3, C and E).

DOPAL-quinonized proteins in MO3.13 (A–C) and PC12 (D and E) cells, with NAC effects. (A) nIRF signal intensity as a function of time of incubation of MO3.13 with 100 µM DOPAL. There are numerous relatively low-molecular-weight DOPAL-quinonized proteins and high-intensity bands at about 62 kDa. MW, molecular weight. (B) Mean (± S.E.M.) values for nIFR signal intensities at about 62 kDa. (C) Effects of various concentrations of NAC on nIRF signal at 62 kDa at 24 hours of incubation of MO3.13 cells with 100 µM DOPAL. *P < 0.05 versus DOPAL alone. (D) nIRF signal at 62 kDa in PC12 cells exposed to various DOPAL concentrations for 24 hours. (E) Effects of various concentrations of NAC on nIRF signal at 62 kDa after 24 hours of incubation of PC12 cells with 100 µM DOPAL. ***P < 0.001 versus DOPAL alone. DOPAL incubation resulted in formation of intracellular quinonized proteins, especially at about 62 kDa.
DOPAL-AS Interactions and NAC Effects.
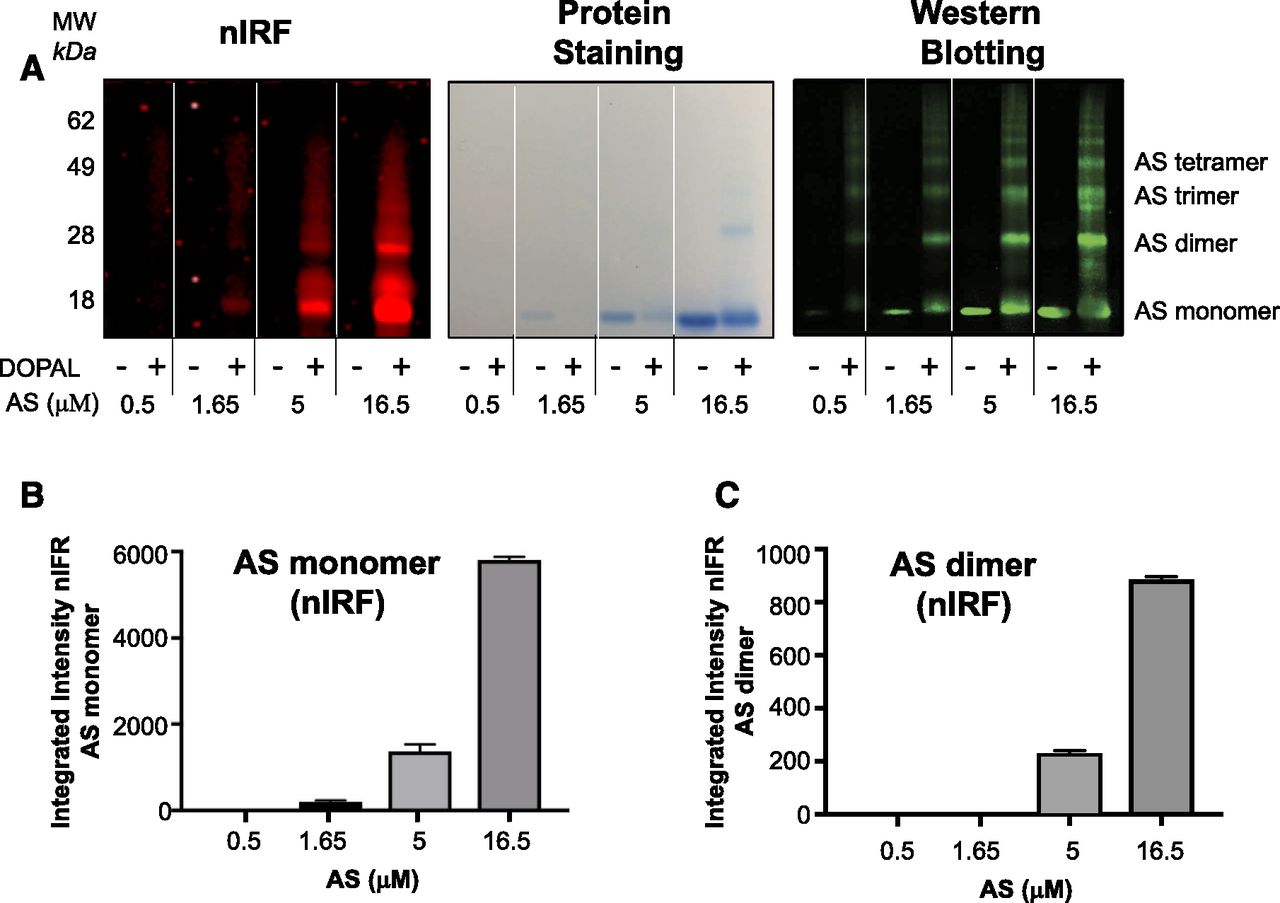
Test tube experiments showed that DOPAL concentration-dependently quinonized and oligomerized AS (Fig. 4A). DOPAL-induced quinonization of AS monomer (Fig. 4B) and dimer (Fig. 4C) was noted. DOPAL was far more potent than DA in quinonizing and oligomerizing AS (Fig. 5A). NAC concentration-dependently attenuated both the quinonization and oligomerization of AS evoked by DOPAL (Fig. 5, B and C).
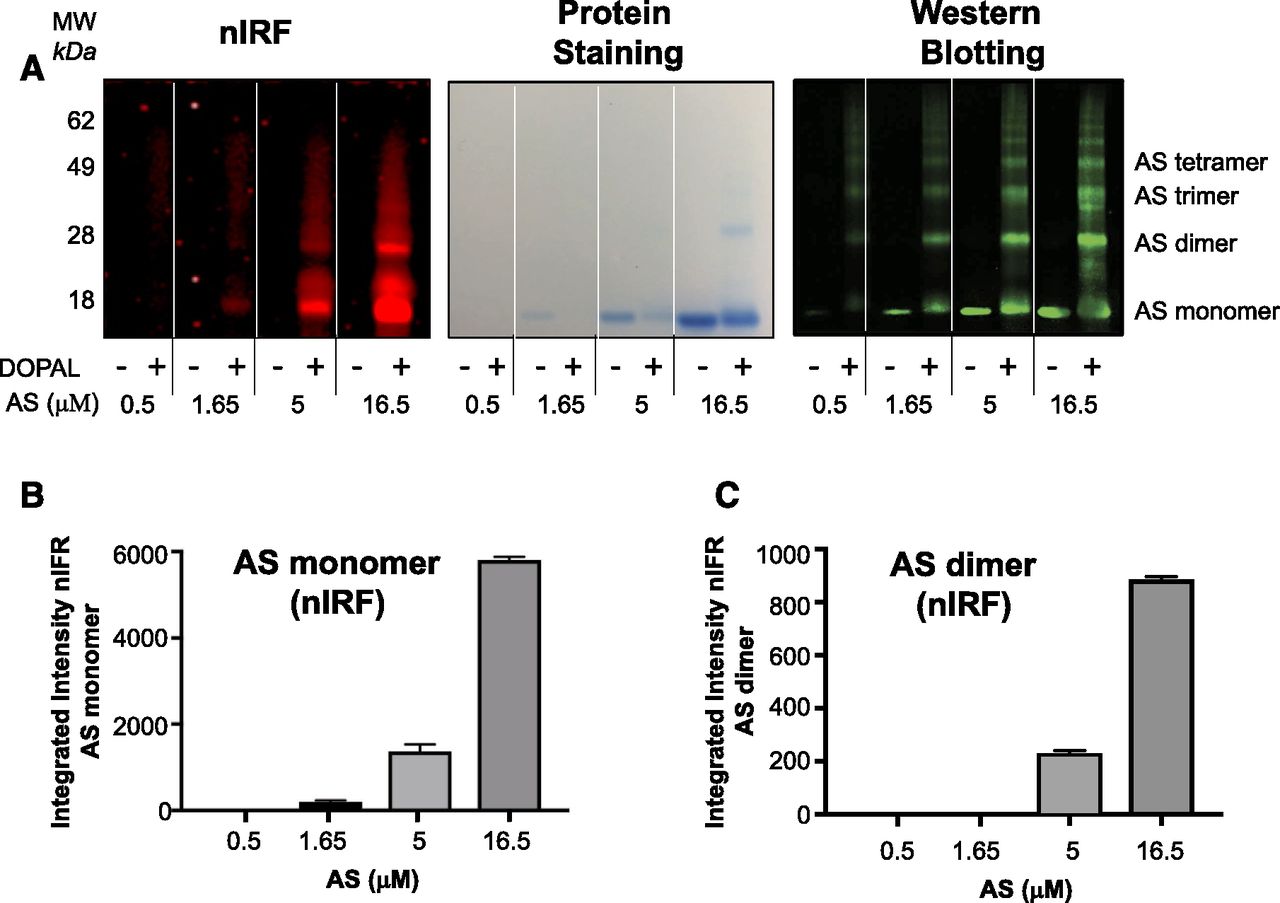
DOPAL-induced quinonization and oligomerization of AS. Reaction mixtures containing AS and DOPAL (100 μM) were incubated at 37°C for 1 hour. (A) nIRF, protein staining, and western blotting of the gel. MW, molecular weight. (B) Mean (± S.E.M.) values nIRF signal intensities of AS monomer. (C) Mean (± S.E.M.) values nIRF signal intensities of AS dimer. DOPAL exposure resulted in quinonization of AS monomer and dimers.

Comparison of AS quinonization by DOPAL versus DA, with NAC effects. Reaction mixtures containing AS (1.65 μM) and DOPAL (100 μM) or DA (100 μM) were incubated without (A) or with NAC (B and C) at 37°C for 1 hour. For nIRF signals, 15 µl of protein gels were read, and for western blotting, 5 µl of protein gels were read. Western blots of AS oligomers (green) and corresponding nIRF signals (red). (C) Mean (± S.E.M.) values for nIRF signals from AS monomer with 100 µM DOPAL alone or DOPAL with NAC. DOPAL quinonized and oligomerized AS, with both effects attenuated by NAC. MW, molecular weight.
DOPAL-Induced Quinonization of Other Synucleinopathy-Related Proteins and NAC Effects.
DOPAL also quinonized UBQ, A53T mutant AS, GBA, LAAAD, and VMAT2 (Fig. 6, A, C, E, G, and H). For all of these proteins, the monomers were quinonized, although for some, quinonized dimers were also detected (Fig. 6, A and G). NAC concentration-dependently attenuated all of these effects of DOPAL (Fig. 6, B, D, and F–H).

DOPAL-quinonized proteins in test tubes, with NAC effects: UBQ (50 µM) (A); UBQ and NAC (B); A53T-AS (1 µM) (C); A53T-AS and NAC (D); GBA (1 µM) (E); GBA and NAC (F); LAAAD (1 µM) (G) NAC; (H) VMAT2 (1 µM) and NAC. DOPAL quinonized all of the tested proteins. NAC attenuated the 100 µM DOPAL-induced quinonization of the proteins. MW, molecular weight.
Dependence of Quinonization and Oligomerization of AS on Lysine Residues.
To assess whether DOPAL-induced quinonization and oligomerization of AS depend on lysine residues, AS was treated with citraconic anhydride. Citraconic anhydride blocked both AS quinonization and oligomerization evoked by DOPAL (Fig. 7).

Involvement of lysine residues in DOPAL-induced AS quinonization and oligomerization. Amino groups of lysine residues of AS (1.65 µM) were acetylated with 5 and 10 mM citraconic anhydride overnight at room temperature. After electrophoresis on 4%–12% Bis-Tris gels, quinonized AS was detected by nIRF (red) and oligomerized AS by western blotting (green). DOPAL-induced quinonization and oligomerization of AS were blocked by citraconic anhydride at both the 5 and 10 mM concentrations. MW, molecular weight.
DOPAL-UBQ Interactions and NAC Effects.
We carried out additional experiments on the effects of DOPAL and other catechols on oligomerization of UBQ. DOPAL oligomerized UBQ, although to a lesser extent than DOPAL oligomerized AS (Fig. 8, A and B). DOPAL was more potent than DA, 3,4-dihydroxyphenylethanol, and DOPAC in these effects (Fig. 8, C and D). NAC significantly reduced DOPAL-induced oligomerization of UBQ (Fig. 8E).

DOPAL-induced UBQ oligomerization, with NAC effects. (A) Western blotting of AS oligomers (green) and UBQ oligomers (red) after AS (1.65 µM) or UBQ (50 μM) was incubated with DOPAL (30 μM) at 37°C for 1 hour. (B) Mean (± S.E.M.) values for AS or UBQ dimers relative to no DOPAL group. (C) UBQ oligomers produced by incubation of UBQ (50 µM) with different catechols (100 µM each). (D) Mean (± S.E.M.) values for UBQ dimers relative to control. *P < 0.05 versus control. (E) Effects of NAC on UBQ dimers. Mean (± S.E.M.) values for UBQ dimers relative to DOPAL alone. *P < 0.05; ***P < 0.001 versus DOPAL alone. DOPAL oligomerized AS and UBQ. The effect of DOPAL was greater than those of other catechols. NAC attenuated the DOPAL-induced oligomerization of UBQ. DOPET, 3,4-dihydroxyphenylethanol; MW, molecular weight.
Incubation of PC12 cells with DOPAL resulted in marked concentration-dependent protein ubiquitination (Fig. 9, A and B), and NAC prevented this effect (Fig. 9, C and D). DOPAL was more potent than DA (Fig. 9, E and F). Incubation of the cells with DA and the MAO inhibitor selegiline (to block DOPAL formation) prevented DA-induced protein ubiquitination (Fig. 9, G and H).

DOPAL-induced protein ubiquitination in PC12 cells, with NAC effects. (A) Western blotting of DOPAL-induced ubiquitinated proteins in PC12 cells incubated at 37°C for 5 hours. (B) Mean (± S.E.M.) values for DOPAL-induced ubiquitinated proteins expressed as fractions of control. *P < 0.05; **P < 0.01 versus control. (C) Western blotting of ubiquitinated proteins after incubation with DOPAL (30 µM), without or with NAC. (D) Mean (± S.E.M.) values for DOPAL-induced ubiquitinated proteins expressed as fractions of control, without or with NAC. ***P < 0.001; ****P < 0.0001 versus DOPAL alone. (E) DOPAL- or DA-induced ubiquitinated proteins. (F) Mean (± S.E.M.) values for DOPAL- or DA-induced ubiquitinated proteins expressed as fractions of control. *P < 0.05; **P < 0.01 versus control. (G) Ubiquitinated proteins after incubation of PC12 with DA (100 µM), without or with selegiline. (H) Mean (± S.E.M.) values for DA-induced ubiquitinated proteins expressed as fractions of control without or with selegiline. *P < 0.05 versus DA alone. DOPAL evoked ubiquitination of intracellular proteins. NAC attenuated DOPAL-induced ubiquitination. DA at 100 µM also ubiquitinated intracellular proteins, and selegiline attenuated this effect. MW, molecular weight.
DOPAL-Induced Oligomerization and Inhibition of LAAAD and NAC Effects.
DOPAL oligomerized LAAAD (Fig. 10A) and decreased LAAAD activity (Fig. 10C). NAC attenuated the quinonization (Fig. 6G) and oligomerization (Fig. 10, A and B) of LAAAD and attenuated the inhibitory effect of DOPAL on the activity of the enzyme (Fig. 10D).

DOPAL-induced oligomerization and inhibition of LAAAD, with NAC effects. (A) Western blotting of LAAAD oligomers after incubation with 100 µM DOPAL at 37°C for 1 hour, without or with NAC. (B) Mean (± S.E.M.) values for LAAAD trimers relative to DOPAL alone. *P < 0.05 versus DOPAL alone. (C) Percentage of LAAAD activity was inhibited by DOPAL after incubation at 37°C for 1 hour. (D) Percentage of control LAAAD activity as a function of NAC concentration. DOPAL oligomerized LAAAD and decreased LAAAD activity. NAC attenuated both effects. MW, molecular weight.
DOPAL-Induced Aggregation of AS and NAC Effects.
Incubation of MO3.13 cells with AS and then DOPAL resulted in intracytoplasmic foci of immunoreactive AS (Fig. 11, A–C). NAC prevented this effect (Fig. 11, D and E). As indicated in Fig. 11F, intracellular DOPAL concentrations were far below the concentration in the incubation medium, and DOPAC levels increased progressively over time.

DOPAL-induced AS aggregation in MO3.13 cells, with NAC effects (A–E) and intracellular catechols after DOPAL treatment (F). (A) MO3.13 cells incubated at 37°C with vehicle for 2–3 hours. (B) MO3.13 cells incubated with 1.65 µM AS for 2–3 hours. (C) MO3.13 cells incubated with 1.65 µM AS and then with DOPAL (100 µM) for an additional 4–5 hours. (D) MO3.13 cells incubated with AS and DOPAL, with NAC (10 µM) added at the same time as DOPAL. (E) MO3.13 cells incubated with AS and DOPAL, with NAC (100 µM) added at the same time as DOPAL. (F) Mean (± S.E.M.) cellular concentrations of catechols as a function of time after adding DOPAL (100 µM) to MO3.13 cells. DOPAL augmented intracellular AS aggregation. NAC attenuated AS augmentation. Intracellular DOPAL concentrations were far below 100 µM and decreased over time as DOPAC increased. DOPET, 3,4-dihydroxyphenylethanol.
Discussion
According to the catecholaldehyde hypothesis, DOPAL acts as an endogenous neurotoxin (“autotoxin”) in diseases characterized by catecholaminergic neurodegeneration (Mattammal et al., 1995; Burke et al., 2003; Panneton et al., 2010; Goldstein et al., 2014a). Here we report several DOPAL-induced protein modifications that, taken together, could threaten neuronal homeostasis. We also report that NAC is remarkably effective in mitigating the protein modifications induced by DOPAL.
In contrast with other catechols (e.g., DOPA, DA, DOPAC), where oxidizing agents are required to produce a detectable nIRF signal (Mazzulli et al., 2016), this was not the case for DOPAL, which we found oxidizes spontaneously to DOPAL-quinone.
The present experiments demonstrate that DOPAL incubation results in quinone adduct formation with many proteins. We call this “quinonization.” Specifically, we found that DOPAL quinonizes AS at physiologically relevant AS concentrations (Wills et al., 2010) as well as other proteins likely related to the pathogenesis of synucleinopathies, including GBA, VMAT2, LAAAD, and UBQ. DOPAL also quinonized AS dimers and trimers and UBQ dimers at physiologically relevant concentrations of the proteins.
Several studies have reported that DOPAL potently oligomerizes AS (Burke et al., 2008; Follmer et al., 2015; Plotegher et al., 2017), especially in the setting of divalent metal cations (Jinsmaa et al., 2014). In this regard, DOPAL again stands out compared with DA and other catechols.
AS contains a relatively large proportion of lysine residues (15 in a 140–amino acid protein, or 11%), and it appears that interactions with lysine residues contribute to DOPAL-induced AS oligomerization (Follmer et al., 2015; Plotegher et al., 2017). In the present study, we found that citraconic anhydride, which renders lysine residues unavailable, prevented DOPAL-induced oligomerization and quinonization of AS, confirming that lysine residues are the likely target of attack for products of oxidation of DOPAL.
Although a linkage between DOPAL and DOPAL-induced AS oligomerization is established by now, the pathogenetic consequences of the oligomerization are the subject of different points of view (Winner et al., 2011; Follmer et al., 2015; Kang et al., 2017; Plotegher et al., 2017; Werner-Allen et al., 2017). Also unsettled are the putative pathogenic roles of AS fibrils (Ardah et al., 2014; Chen et al., 2015; Khalife et al., 2015; Cox et al., 2018) and phosphorylated AS (Chau et al., 2009; Oueslati, 2016). DOPAL-induced AS quinonization has not been described previously, and so the functional significance of the quinonization has yet to be explored.
Recently, a specific mechanism has been proposed relating DOPAL oxidation to AS oligomerization. Superoxide, which is generated pari passu with the oxidation of DOPAL, propagates a chain reaction oxidation resulting in a dicatechol pyrrole adduct with lysine [dicatechol pyrrole lysine (DCPL)] (Werner-Allen et al., 2017). The same investigators have reported that auto-oxidation of the catechol rings in DCPL produces an intermediate dicatechol isoindole lysine (DCIL) product formed by an intramolecular reaction of the two catechol rings, yielding an unstable tetracyclic structure. DCIL then reacts with a second DCIL to give a dimeric, di-DCIL. Therefore, DOPAL-catalyzed formation of AS oligomers may be separable into two steps, the first involving generation of DCPL and the second cross-linking of AS molecules via the interadduct reaction. Since our results indicate that DOPAL quinonizes even monomeric AS (Fig. 3, A and B), the exact relationship between DOPAL-induced quinonization and oligomerization of AS requires further study. We hypothesize that DOPAL-induced protein quinonization is an intermediate step in the formation of protein oligomers.
LAAAD activity is decreased in the putamen in two forms of synucleinopathy, PD and MSA (Goldstein et al., 2017b). Here we report for the first time that DOPAL both quinonizes (Fig. 5) and oligomerizes (Fig. 8A) LAAAD. We found that DOPAL also inhibits activity of the enzyme (Fig. 8C) with a potency (IC50 53 μM) about half that of the well established LAAAD inhibitor carbidopa (IC50 29 μM). Since NAC improved the LAAAD activity (albeit not to the control activity) and prevented DOPAL-induced quinonization and oligomerization of the enzyme, we propose that the decrease in LAAAD activity in PD and MSA partly reflects effects of DOPAL oxidation.
We also found that DOPAL quinonizes VMAT2 (Fig. 6H). So far, there is no information about whether DOPAL inhibits VMAT2 activity, although DOPAL-induced AS oligomers interfere with vesicular functions (Plotegher et al., 2017). Similarly, we found that DOPAL quinonizes GBA, and although there is growing evidence of GBA deficiency in PD, even in the absence of GBA mutation (Gegg and Schapira, 2018), whether DOPAL affects GBA activity is unknown.
The UBQ proteasome system is the primary cytosolic proteolytic machinery for the selective degradation of damaged proteins and, together with the autophagic lysosome system and the molecular chaperone network, contributes importantly to cellular protein homeostasis (Hochstrasser, 1996; Hershko and Ciechanover, 1998). In principle, ubiquitination tags misfolded or damaged proteins for proteasomal degradation, so that they do not interfere with vital cellular processes (Hershko and Ciechanover, 1998; Jung et al., 2009). The present finding that DOPAL evokes striking ubiquitination of multiple intracellular proteins fits with the concept that DOPAL causes a variety of cellular proteins to misfold (Rees et al., 2009). Moreover, since selegiline if anything augments spontaneous oxidation of cytoplasmic DA (Goldstein et al., 2016), our finding that selegiline prevents DA-induced protein ubiquitination suggests that protein misfolding evoked by cytoplasmic DA depends on enzymatic rather than spontaneous oxidation of DA.
We also report that DOPAL both quinonizes (Fig. 6A) and oligomerizes (Fig. 8A) UBQ itself. Other catechols, including DA, failed to produce this effect. The functional consequences of DOPAL-induced quinonization and oligomerization of UBQ are unknown.
At physiologically relevant concentrations, DOPAL triggers intracellular AS aggregation in cultured neuroblastoma cells (Burke et al., 2008). We replicated this finding in MO3.13 oligodendrocytes (Fig. 3, A and B). Since GCIs within oligodendrocytes are a neuropathologic characteristic of MSA (Wakabayashi et al., 1998), the MO3.13/DOPAL/AS preparation may provide a useful cellular model of the GCIs in MSA.
One may question the physiologic relevance of the 100 µM DOPAL concentration in the incubation medium that was used to evoke intracellular AS aggregation. Therefore, we assayed the intracellular concentrations of DOPAL over time (Fig. 11F). The initial peak intracellular DOPAL concentration was orders of magnitude lower than the concentration in the medium and declined further over hours as its metabolite DOPAC increased. After 24 hours of incubation, the intracellular DOPAL concentration, about 5 nM, was similar to the endogenous DOPAL level attained during blockade of vesicular uptake and metabolizing enzymes in PC12 cells (Goldstein et al., 2012). Therefore, DOPAL at physiologically attainable intracellular concentrations seems able to aggregate AS.
Consistent with previous reports that a variety of antioxidants inhibit DOPAL reactivity with proteins (Rees et al., 2009; Follmer et al., 2015), treatment with NAC was remarkably effective in mitigating all the protein modifications exerted by DOPAL—quinonization, oligomerization, ubiquitination, and aggregation—as well as the inhibitory effect of DOPAL on LAAAD. The finding that NAC prevented DOPAL-induced AS oligomerization replicates the report by Anderson et al. (2016).
Our experiments did not formally test whether endogenous aldehyde-metabolizing enzymes prevent DOPAL-induced oligomerization and formation of quinones. One may speculate that this is the case based on previous reports that blockade of ALDH and AR increases endogenous DOPAL levels and augments apoptosis evoked by incubating PC12 cells with DA (Goldstein et al., 2012).
The present results fit well with data from clinical post-mortem studies and animal genetic and chemical-induced models related to the catecholaldehyde hypothesis. Putamen DOPAL concentrations are increased in PD (Goldstein et al., 2011), probably due to a “double hit” of decreased vesicular sequestration of cytoplasmic dopamine and decreased DOPAL metabolism by ALDH (Goldstein et al., 2017b). A mouse genetic model of low VMAT2 activity is associated with a shift from vesicular uptake to oxidative deamination of cytoplasmic catecholamines (Goldstein et al., 2014b) and with aging-related nigrostriatal neurodegeneration (Caudle et al., 2007), whereas increased VMAT2 expression is preventive (Lohr et al., 2014). Double knockout of the genes encoding ALDH1A1 and ALDH2 produces another mouse genetic model of PD (Wey et al., 2012), and ALDH2 overexpression is also preventive (Chiu et al., 2015). Both the pesticide rotenone and the fungicide benomyl decrease ALDH activity and build up endogenous DOPAL (Casida et al., 2014; Goldstein et al., 2015a). No study to date, however, has assessed experimentally whether blocking both components of the double hit prevents catecholaminergic neurodegeneration in animal models of PD.
Therapeutic Implications.
Our data support the view that DOPAL exerts multiple potentially harmful protein modifications, including quinonization, oligomerization, aggregation, and misfolding, and that antioxidation with NAC attenuates or prevents these modifications. Based on the present results, a novel target for future experimental therapeutics may be the two-step sequence of enzymatic oxidation of DA by MAO to form DOPAL and subsequent spontaneous oxidation of DOPAL to form DOPAL-quinone (Goldstein et al., 2014a). Specifically, combining an MAO inhibitor (to decrease DOPAL production) with NAC (to decrease DOPAL oxidation) merits further consideration in the treatment or prevention of diseases resulting from catecholaminergic neurodegeneration.
Authorship Contributions
Participated in research design: Jinsmaa, Sharabi, Goldstein.
Conducted experiments: Jinsmaa, Sullivan, Isonaka.
Performed data analysis: Jinsmaa.
Wrote or contributed to the writing of the manuscript: Jinsmaa, Sharabi, Goldstein.
Footnotes
- Received February 27, 2018.
- Accepted April 19, 2018.
This work was supported by the Intramural Research Program of the National Institute of Neurologic Disorders and Stroke.
The authors have no conflicts of interest to disclose.
Abbreviations
- AL-1576
- spiro-(2,7-difluoro-9H-fluoren 9,4′-imidazoline) 2′,5′-dione
- ALDH
- aldehyde dehydrogenase
- AR
- aldehyde/aldose reductase
- AS
- α-synuclein
- DA
- dopamine
- DCIL
- dicatechol isoindole lysine
- DCPL
- dicatechol pyrrole lysine
- DOPA
- 3,4-dihydroxyphenylalanine
- DOPAC
- 3,4-dihydroxyphenylacetic acid
- DOPAL
- 3,4-dihydroxyphenylacetaldehyde
- FCS
- fetal calf serum
- GBA
- glucocerebrosidase
- GCI
- glial cytoplasmic inclusion
- LAAAD
- l-aromatic-amino-acid decarboxylase
- MAO
- monoamine oxidase
- MSA
- multiple system atrophy
- NAC
- N-acetylcysteine
- nIRF
- near-infrared fluorescence
- PD
- Parkinson’s disease
- UBQ
- ubiquitin
- VMAT
- vesicular monoamine transporter
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}