


Abstract
Microsomal prostaglandin E synthase-1 (mPGES-1) is a terminal prostaglandin E2 (PGE2) synthase in the cyclooxygenase pathway. Inhibitors of mPGES-1 may block PGE2 production and relieve inflammatory symptoms. To test the hypothesis, we evaluated the antipyretic and analgesic properties of a novel and selective mPGES-1 inhibitor, MF63 [2-(6-chloro-1H-phenanthro-[9,10-d]imidazol-2-yl)isophthalonitrile], in animal models of inflammation. MF63 potently inhibited the human mPGES-1 enzyme (IC50 = 1.3 nM), with a high degree (>1000-fold) of selectivity over other prostanoid synthases. In rodent species, MF63 strongly inhibited guinea pig mPGES-1 (IC50 = 0.9 nM) but not the mouse or rat enzyme. When tested in the guinea pig and a knock-in (KI) mouse expressing human mPGES-1, the compound selectively suppressed the synthesis of PGE2, but not other prostaglandins inhibitable by nonsteroidal anti-inflammatory drugs (NSAIDs), yet retained NSAID-like efficacy at inhibiting lipopolysaccharide-induced pyresis, hyperalgesia, and iodoacetate-induced osteoarthritic pain. In addition, MF63 did not cause NSAID-like gastrointestinal toxic effects, such as mucosal erosions or leakage in the KI mice or nonhuman primates, although it markedly inhibited PGE2 synthesis in the KI mouse stomach. Our data demonstrate that mPGES-1 inhibition leads to effective relief of both pyresis and inflammatory pain in preclinical models of inflammation and may be a useful approach for treating inflammatory diseases.
Traditional nonsteroidal anti-inflammatory drugs (NSAIDs) and selective cyclooxygenase (COX)-2 inhibitors are widely used for pain relief in patients with rheumatoid and osteoarthritis. These drugs act by blocking COX activity and inhibiting the synthesis of prostaglandins (PGs). Since introduced on the market, selective COX-2 inhibitors have proved to have similar efficacy to traditional NSAIDs but an improved gastrointestinal tolerability due to reduced COX-1 inhibitory activity (Laine et al., 1999). More recently, however, randomized and placebo-controlled trials have shown an increased cardiovascular risk for myocardial infarction and stroke in patients after long-term use of selective COX-2 inhibitors (Cannon et al., 2006; Kearney et al., 2006). Such an effect has also been associated with some traditional NSAIDs (Couzin, 2005). In addition, the gastrointestinal tolerability of selective COX-2 inhibitors may be reduced in patients taking aspirin (Scheiman, 2003), possibly due to simultaneous inhibition of both COX-1 and COX-2. These new findings have sparked renewed interest in evaluating alternative targets for anti-inflammatory agents (Samuelsson et al., 2007).
PGE2 is one of the major proinflammatory prostaglandins derived from COX-2. Thus PGE2 synthase or E prostanoid (EP) receptors may be useful anti-inflammatory targets in the COX pathway. In support of this view, recent studies have shown that antagonism of EP4 receptor is sufficient for NSAID-like efficacy in animal models of inflammation (Nakao et al., 2007; Clark et al., 2008). The other target under evaluation in the COX pathway is microsomal prostaglandin E synthase-1 (mPGES-1). Among the three PGE synthases, namely cytosolic prostaglandin E synthase, mPGES-1, and mPGES-2, mPGES-1 is the major isozyme for producing PGE2 during inflammation (Kudo and Murakami, 2005). Similar to COX-2, mPGES-1 can be induced by proinflammatory stimuli in vitro (Matsumoto et al., 1997; Jakobsson et al., 1999; Stichtenoth et al., 2001) as well as in animal models of inflammation (Ek et al., 2001; Guay et al., 2004). In addition, deletion of mPGES-1 renders mice resistant to fever and chronic inflammation (Engblom et al., 2003; Trebino et al., 2003), similarly to COX-2 deletion (Li et al., 1999; Myers et al., 2000), suggesting that mPGES-1 inhibition may offer an alternative approach to COX-2 inhibitors for the treatment of inflammatory diseases. However, the hypothesis has not been validated because, until now, a selective mPGES-1 inhibitor suitable for studies in vivo has been unavailable.
We have recently identified a series of selective mPGES-1 inhibitors that are potent and bioavailable (Côté et al., 2007). In the present study, we determined the effects of the lead compound, MF63, on pyresis and inflammatory pain to examine whether selective inhibition of mPGES-1 is sufficient for efficacy. In addition, we also examined the gastrointestinal tolerability of the compound considering that the inhibition of mPGES-1, the major PGES isoform in the stomach, might be deleterious to the gastrointestinal tract.
Materials and Methods
Animals. All animal procedures were approved by the Animal Care Committee at the Merck Frosst Centre for Therapeutic Research and respected the guidelines established by the Canadian Council on Animal Care. mPGES-1 KI mice were custom-generated at TaconicArtemis (Köln, Germany) using ArteMice HUMANIZED technology (see below for details). The mice were then bred at Taconic Farms (Hudson, NY) to generate both KI and wild-type mice on pure C57Bl/6 background. In the present study, both male and female adult mice, approximately 10 to 12 weeks of age, were used. Young male Hartley guinea pigs (approximately 250 g) were obtained from Charles River Canada (Montreal, QC, Canada). Animals were allowed to acclimatize for at least 4 days before being used for experiments. In each experiment, animals were randomly assigned to different experimental groups. Mice and guinea pigs were fasted for approximately 4 and 14 h, respectively, before being administered with test compounds or vehicle. Otherwise, animals had free access to standard rodent feed and tap water. All of the animal experiments were performed strictly in a double-blind fashion.
Enzymatic Assay and Counterscreens. Recombinant microsomal PGES-1 from various species (human, 2 μg/ml; rat, 10 μg/ml, and guinea pig, 2.5 μg/ml) generated in-house was assayed as described previously (Riendeau et al., 2005). Compound or DMSO (final 1%) was added 20 min before the initiation of the reaction with PGH2 substrate (1 μM final concentration) at room temperature. After 30 s of incubation, the reaction was stopped by adding SnCl2 in 1 N HCl (1 mg/ml final concentration). The same protocol was used for assaying other prostaglandin synthases. Prostaglandins were measured by EIA by using commercial kits (Assay Design Inc., Ann Arbor, MI). The compound was also screened by MDS Pharma Services (Peitou, Taiwan) against a standard panel of drug targets, including COX-1, COX-2, 5-lipoxgenase, leukotriene, and prostanoid receptors using their standard assay protocols.
A549 Cell-Based Assay. The assay was performed as reported previously (Riendeau et al., 2005). A549 cells were seeded in 96-well dishes at a density of 25,000 cells/well and incubated for 20 h in RPMI 1640 medium supplemented with 2% fetal bovine serum (FBS). The cells were then incubated with 10 ng/ml IL-1β for 24 h in the presence of a vehicle or inhibitor from a 100-fold concentrated DMSO stock solution in a medium containing 2 or 50% FBS. Concentrations of PGE2 and PGF2α in the cell-free medium were measured by EIA (Assay Designs Inc.). IC50 values were derived from eight-point titrations.
Human Whole Blood Assay. Assays were performed as described previously (Brideau et al., 1996) to determine the effects of MF63 on COX-1-mediated TXB2 production and COX-2-mediated PGE2 formation. For measuring COX-1-mediated TXB2 production, 500 μl of fresh blood was allowed to clot in the presence of 2 μl of DMSO or a test compound at 37°C for 1 h before serum was obtained for analyzing TXB2. For measuring COX-2-mediated PGE2 formation, 500 μl of anticoagulated blood was incubated with 10 μl of LPS in PBS (100 μg/ml final concentration) for 24 h in the presence of 2 μl of vehicle (DMSO) or a test compound, which was added 15 min before the addition of LPS. After the incubation, plasma was isolated for analyzing PGE2. PGE2 and TXB2 were measured by EIA (Assay Designs Inc.). The dose titration of the compound was performed in triplicate in each experiment.
Generation of mPGES-1 Knock-In Mice. Mice expressing human mPGES-1 were generated by targeted disruption and replacement of part of the mouse ptges gene with a human minigene. In brief, a human ptges-targeting vector was constructed by using C57Bl/6J genomic DNA and human genomic DNA (Fig. 1). A 2.4-kb mouse genomic region starting immediately after the initiator ATG in exon 1 and ending with exon 2 (Fig. 1, top and middle panels) was replaced by a human minigene containing exon 1, intron 1, and fused exons 2 and 3. A synthetic polyadenylation signal and a flip recombinase targets-flanked-neo gene were inserted downstream of the human exon 3 (Fig. 1, middle panel). The targeting vector was designed, such that homologous recombination would result in the in-frame insertion of the human minigene immediately after the mouse mPGES-1 initiator ATG and in the deletion of exons 1 and 2 of the mouse gene (Fig. 1, middle panel). The targeting vector was electroporated into C57Bl/6N embryonic stem cells, and neomycin-resistant colonies were selected. Targeted insertion of the human minigene and deletion of the mouse exons 1 and 2 were verified by PCR. Three clones with the correct predicted gene organization were selected for injection in C57BL/6 blastocysts. Excision of the FRT-flanked neomycin resistant gene was performed by breeding chimeric mice with transgenic mice that express flippase (Fig. 1, bottom panel). One KI mouse line was bred at Taconic Farms to generate both KI and wild-type mice on pure C57Bl/6 background, which were used in the present study.

Targeted knock-in of human ptges in mice. A 2.4-kb portion of the mouse ptges starting immediately after the initiator ATG in exon 1 and ending with exon 2 (rectangles, top, WT allele) was replaced by a human minigene consisting of exon 1, intron-1, fused exons 2 and 3, and polyadenylation signal (pA) and an FRT-flanked neogene (middle, targ allele). The FRT-flanked neogene was deleted by breeding the chimeric mice with transgenic mice that express flippase (bottom, hum allele). Arrows and numbers indicate the sequence orientation of the PCR reactions. Data for the 103/104 reaction are not shown in the present study.
Genotyping by PCR. DNA was extracted and purified from frozen tail samples using a QIAGEN DNA purification system (QIAGEN, Inc., Valencia, CA). DNA yield and purity were determined by spectrophotometric DNA absorbance 260:280. Two PCR reactions were performed to detect the mouse mPGES-1 DNA with primers 106/SAB 29R and the human mPGES-1 DNA with 103/101 (Fig. 1A). The sequences for the primers are as follows: 106, 5′-AGG TTG ATT GTG ACG TCA CC-3′; AB29R, 5′-CA CTG ACA GGT ACT CAC C-3′; 101, 5′-CCC CAT CAA GGG GAC ATT TGC-3′, and 103, 5′-CCA GTT CTC TGT GGA AGC AAG G-3′.
LPS-Induced PGE2 Production in Peritoneal Macrophages. As previously reported (Boulet et al., 2004), peritoneal macrophages were collected 4 days after an intraperitoneal injection of 6% thioglycollate (1 ml/mouse) by lavaging the peritoneal cavity with PBS containing 10 U/ml heparin. Cells were harvested by centrifugation and resuspended in RPMI 1640 supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin and seeded on 96-well plates at a density of 2 × 105 cells per well. After a 1-h incubation at 37°C with 5% CO2 and the removal of nonadherent cells, adherent cells were incubated with fresh medium containing 2% FBS for 30 min with inhibitors. Next, they were given LPS (100 ng/ml, Salmonella enterica serotype minnesota Re-595; Sigma-Aldrich, St. Louis, MO) and incubated for 24 h. After the incubation, the medium was collected and assayed for PGE2 by EIA (Assay Design Inc.). Compounds were tested at seven concentrations using a 4-fold serial dilution. The activity of mPGES-1 was calculated as the difference in PGE2 levels between cells treated with LPS and untreated cells. IC50 values were derived using a four-parameter fit of all the titrations using the Prism software (GraphPad Software Inc., San Diego, CA).
Western Blot Analysis. Cell lysates (2 μg of protein) were analyzed by SDS-polyacrylamide gel electrophoresis on 10 to 20% Tris-glycine precast gradient gels (Novex, Inc., Wadsworth, OH) with recombinant human COX-2 and mPGES-1 proteins as standards. Immunodetection was performed using anti-COX-2 polyclonal antibody (MF243, 1:3000), anti-human mPGES-1 monoclonal antibody raised against the whole protein (MF137, 1:300) (custom-generated at Covance, Princeton, NJ) and anti-mPGES-1 polyclonal antibody (1:200) (Cayman Chemical, Ann Arbor, MI) in Tris-buffered saline supplemented with 0.5% Tween 20 and 5% nonfat skim milk. The secondary horseradish peroxidase-linked goat anti-rabbit and anti-mouse IgG antibodies (GE Healthcare, Piscataway, NJ) were used at a dilution of 1:3000. Chemiluminescence detection was performed using SuperSignal West Femto Maximum Sensitivity substrate (Pierce, Rockford, IL) with Fuji Film LAS-1000 charge-coupled device (Fuji Photo Film Co., Ltd., Tokyo, Japan).
Prostaglandins in the Mouse Air Pouch and the Brain. An air pouch was produced by subcutaneous injection of 5 ml of filtered air (0.2 μm filter) into the back of the knock-in mice anesthetized with 2% isoflurane. The air pouch was reinforced by injecting 2 ml of filtered air 2 days later. LPS (100 μg/mouse) was injected as a 1 mg/ml solution in saline into the pouch 5 days after the second air injection to induce inflammation. For prostaglandin analysis in the brain, the same amount of LPS was injected intraperitoneally. MF-tricyclic or MF63 were administered orally 1 h before the LPS injection. Brain or air pouch exudates were collected 3 h after LPS injection. Exudates were collected by lavage with 1 ml of ice-cold PBS.
Pharmacokinetics. MF63 was administered orally as a suspension in 0.5% methylcellulose (Methocel; Dow Chemical Company, Midland, MI) (30 mg/kg) to two guinea pigs. Plasma was prepared from venous blood samples collected at different times, and the concentration of the compound was determined by high-performance liquid chromatography with an ultraviolet detector.
LPS-Induced Thermal Hyperalgesia. LPS was injected intraplantarly in guinea pigs (10 μg/paw in 50 μl of saline) or mice (10 μg/paw in 50 μl of saline) to induce thermal hyperalgesia. MF63 was given orally 1 h before LPS injection. Thermal hyperalgesia was quantified by measuring paw withdrawal latency (PWL) upon stimulation with an infrared light beam at 1 (mice) or 4.5 (guinea pigs) h after LPS injection. The percentage of hyperalgesia was calculated by using the formula (ΔPWLcompound - ΔPWLnaive)/(ΔPWLveh - ΔPWLnaive) × 100, where ΔPWL is the decrease in paw withdrawal latency from the baseline and naives are animals not treated with LPS. Animals were euthanized by CO2 inhalation. Lumbar spinal cord of guinea pigs was removed and snap-frozen in liquid nitrogen for prostaglandin analysis.
Iodoacetate-Induced Osteoarthritic Pain. Under anesthesia with 2 to 3% isoflurane, a 50-μl monosodium iodoacetate (MIA) solution (7.5 mg/ml) or saline was injected into the left shoulder joint of a guinea pig. The right shoulder joint of the animal received 50 μl of saline. Weight distribution between the two forelimbs was measured by using an incapacitance meter before and at various time points after the injection to determine the weight bearing ratio between the left and right limb (L/R), an index of pain. For the measurement of weight distribution, each forelimb was placed on a weighing platform of the tester, and the weight on the limb was recorded. Under normal conditions, the L/R ratio is approximately 1.0. The ratio was significantly decreased to ∼0.5 on average on day 3 after MIA injection when compounds were tested. Animals with a L/R ratio of ≥0.7 were considered nonresponders and removed from the study. For assessing the effect of test compounds on inflammatory pain, the compounds were administered orally on day 3, and L/R was determined 6 h later. The percentage of incapacitance was calculated by using the formula (ΔL/Rcompound - ΔL/Rnaive)/(ΔL/Rveh - ΔL/Rnaive) × 100, where ΔL/R is the decrease in L/R from the baseline and naives are animals not treated with MIA.
LPS-Induced Pyresis. LPS was injected intraperitoneally as a 0.1 mg/ml solution in saline in guinea pigs (30 μg/kg) to induce pyresis. Body temperature was taken by using a digital rectal thermometer (Thermistor thermometer model 8502; Cole-Palmer Instrument Company, Chicago, IL). Animals were gently held in a supine position when the rectal temperature was taken. A saline-injected group was used to control for stress-induced changes in body temperature. The rectal temperature did not differ significantly between the baseline and 3 h after the saline injection. MF-tricyclic and MF63 were administered by oral gavage 1 h before LPS injection.
Stomach Prostaglandin and Erosions. Adult male- or female-humanized mPGES-1 mice were fasted for 4 h before oral administration of either vehicle (0.5% methylcellulose) or MF63. Animals were sacrificed 3 h after dose, and the stomach was harvested for prostaglandin analysis or assessment of mucosal erosions. For the assessment of mucosal erosion, a whole stomach was stretched and photographed under a microscope. The total length of lesions in each stomach was then quantified by using MCID Basic 7.0 from Imaging Resources Inc. (Saginaw, MI)
Measurements of Tissue Prostaglandins. Brain, lumbar spinal cord, or stomach tissues, weighing approximately 100 mg, were homogenized in 1 ml of PBS buffer containing 10 μM indomethacin and protease inhibitors (Complete cocktail tablet; Roche Diagnostics, Indianapolis, IN) using an automatic tissue homogenizer (Magna-Lyser; Roche Diagnostics). Supernatants were collected after centrifugation, and protein concentration was determined by dye-binding assay (Bio-Rad, Hercules, CA). Prostaglandin concentrations in the tissue homogenates and air pouch exudates were analyzed by using lipid chromatography/mass spectrophotometry as described previously (Guay et al., 2004).
Fecal Excretion of 51Cr in Squirrel Monkeys. Leakage of 51Cr from the blood to the feces through the gastrointestinal mucosa was assessed by measuring 51Cr fecal excretion as described previously (Riendeau et al., 2001). In brief, squirrel monkeys (Saimiri sciureus; 1–1.5 kg) were dosed orally with a test compound twice daily for 4 days. One hour after administration of the last dose, 51CrCl3 in sterile saline (1 ml/kg, 4 ± 5 μCi per animal) was injected via the saphenous vein. The monkeys were then housed individually in metabolism cages. Feces were collected for a 24-h period, and 51Cr fecal excretion was calculated as the percentage of total injected dose.
Statistical Analysis. Statistical analysis was performed by using one-way analysis of variance and Newman-Keuls test.
Results
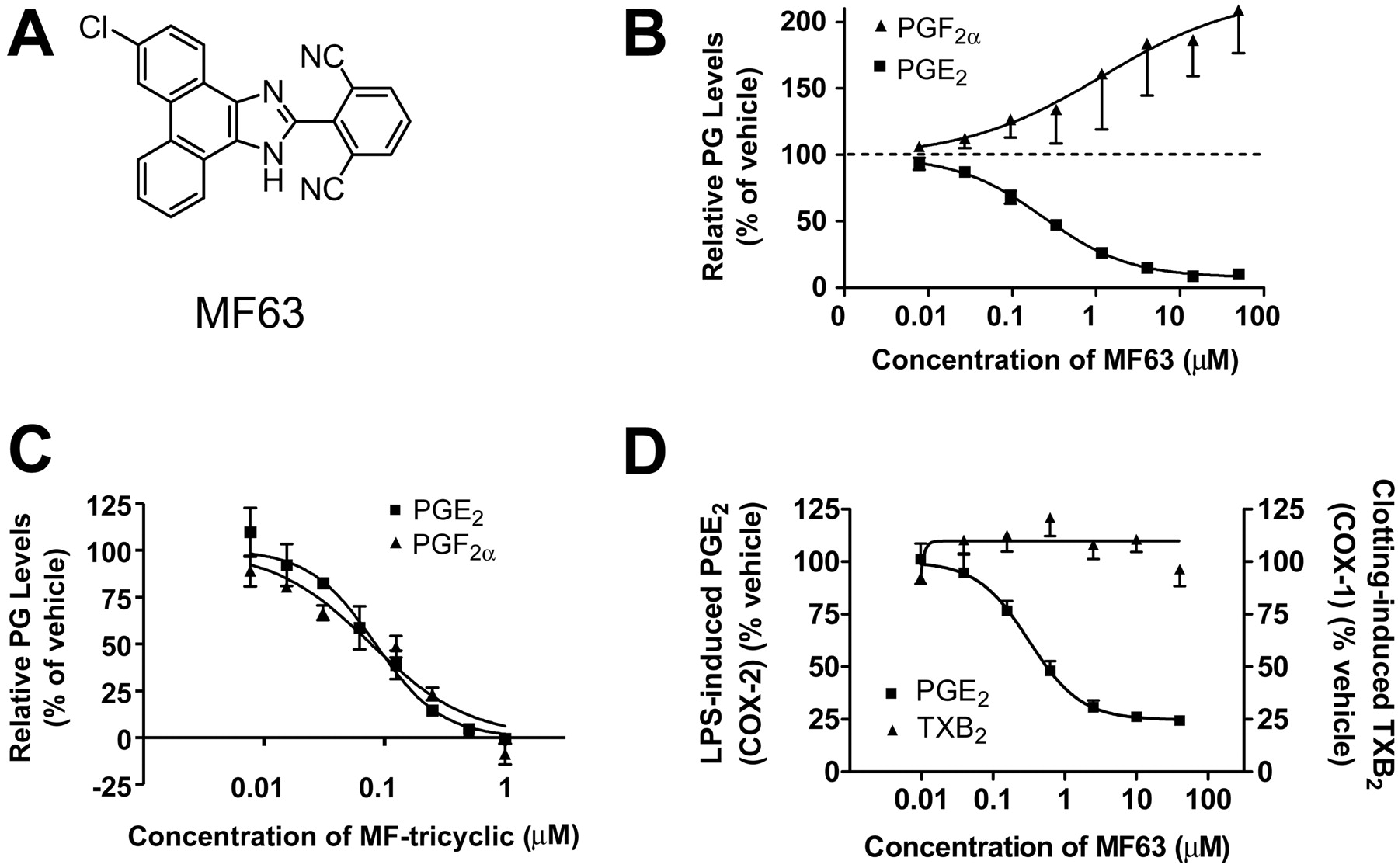
MF63 Is a Potent and Selective Inhibitor of mPGES-1. MF63 was developed through the optimization of a series of phenanthrene imidazole inhibitors (Fig. 2A) (Côté et al., 2007). The compound inhibited recombinant human mPGES-1 in a dose-dependent manner with an IC50 of 1.3 nM and at least a 1000-fold selectivity over other prostanoid synthases, such as mPGES-2, PGI2, PGD2, and thromboxane (TX) synthases. When tested against COX-1, COX-2, 5-lipoxygenase, and various prostanoid and leukotriene receptors, the compound also displayed a similar degree of selectivity. In A549 cells, MF63 selectively inhibited IL-1β-induced formation of PGE2 with IC50 values of 0.046 and 0.42 μM, respectively, in the presence of 2 and 50% serum, whereas it did not inhibit, but rather increased, the formation of PGF2α in a dose-dependent manner (Fig. 2B). In contrast, MF-tricyclic, a selective COX-2 inhibitor (Rowland et al., 2007), inhibited the production of both PGE2 and PGF2α with similar IC50 values (0.022 and 0.026 μM, respectively, at 2% serum; 0.084 and 0.077 μM, respectively, at 50% serum) (Fig. 2C, 50% serum). MF63 was also very potent in human whole blood, inhibiting COX-2-dependent production of PGE2 induced by LPS by a maximum of 75 to 80% with an IC50 of 0.8 μM. In contrast, the compound did not affect clotting-induced TXB2 production mediated by COX-1 (IC50 > 20 μM) (Fig. 2D).
Generation and Characterization of the KI Mouse. When tested against the mouse or rat mPGES-1, MF63 displayed little activity (IC50 > 30 μM). The finding prompted us to generate a KI mouse that could be used for in vivo proof-of-concept studies. To this end, we used a knock-in approach and replaced a 2.4-kb mouse genomic region starting immediately after the initiator ATG in exon 1 and ending with exon 2 of mPGES-1 with a human minigene containing exon 1, intron 1, and fused exons 2 and 3 (Fig. 1). The genotypes of the KI mice were confirmed by PCR analysis, which showed that the homozygote KI mice and wild-type littermates carried the human and mouse mPGES-1 DNA, respectively, whereas the heterozygotes had mPGES-1 DNA from both species (Fig. 3A). The expression of the knock-in mPGES-1 and its regulation at the protein level were examined in cultured peritoneal macrophages by Western blot. Similar to that of COX-2, the expression of mPGES-1 in macrophages of both wild-type and KI mice was low without LPS stimulation but was markedly up-regulated when stimulated with LPS (Fig. 3B). mPGES-1 protein was detected in both the wild-type and KI cells with a polyclonal antibody that recognizes both the mouse and human mPGES-1 (Fig. 3B, middle row), but exclusively in the KI cells with a monoclonal antibody that only reacts with the human protein (Fig. 3B, bottom row).
MF63 Inhibits PGE2 Synthesis and Hyperalgesia in the KI Mouse. The sensitivity of the KI mice to MF63 was assessed by examining the effect of the compound on LPS-induced PGE2 synthesis in cultured peritoneal macrophages and in vivo. MF63 attenuated PGE2 synthesis in a dose-dependent manner in KI but not in wild-type macrophages (Fig. 3, C and D). In contrast, MF-tricyclic was effective in both genotypes (Fig. 3, C and D). MF63 also displayed a similar PGE2 inhibitory effect in vivo, attenuating PGE2 accumulation in both the air pouches and brains of the KI mice at the dose of 100 mg/kg (Fig. 4, A and B). In the air pouch injected with LPS, MF-tricyclic (10 mg/kg) inhibited both PGE2 and PGI2 in a nondiscriminatory manner, whereas MF63 only suppressed the formation of PGE2 (Fig. 4A). MF63 also selectively inhibited PGE2 (Fig. 4B) over PGI2 (Fig. 4C) in the brain after an intraperitoneal injection of LPS. As expected from its lack of potency on the mouse mPGES-1, MF63 failed to inhibit PGE2 in the brains of wild-type mice (Fig. 4B).

Structure and whole-cell potency of MF63. A, structure of MF63. B, dose-dependent inhibition of PGE2 by MF63 in A549 cells with shunting to PGF2α. C, dose-dependent inhibition of both PGE2 and PGF2α by MF-tricyclic in A549 cells. PGE2 and PGF2α were analyzed in the medium of A549 cells stimulated with IL-1β for 24 h in the presence of various concentrations of test compound and 5% serum. D, inhibition of COX-2-mediated PGE2 but not COX-1-mediated TXB2 by MF63 in human whole blood. Serum TXB2 was analyzed after nonanticoagulated blood was allowed to clot for 1 h in the presence of various dose of MF63. Plasma PGE2 was analyzed after a 24-h incubation of anticoagulated blood with LPS in the presence of various doses of MF63. Data in B through D are reported as the mean ± S.E.M. of the relative prostanoid levels, with those in the vehicle group as 100%. Values were obtained from three to five representative experiments, each performed in triplicate.

Expression and activity of mPGES-1 in the KI mice. A, genotyping by PCR. Two PCR reactions were performed on each sample, taken from a homozygote (M), heterozygote (T), or wild-type (W) mouse, one using the human primer set (top) and the other the mouse primers (bottom). The 350-base pair human mPGES-1 PCR product was detected in both the homozygote and heterozygote KI mice (top, unfilled arrow), whereas the 750-base pair mouse mPGES-1 product was noted in the heterozygote KI and wild-type mice (bottom, filled arrow). B, Western blot. Western blot was performed with a polyclonal antibody for COX-2 (top row), a polyclonal (PcAb, middle row), or a monoclonal antibody specific for human mPGES-1 (McAb, bottom row). Both the COX-2 and mPGES-1 polyclonal antibodies detected an intense band, migrating with the corresponding recombinant protein standard (std) for COX-2 (top row) or mPGES-1 (middle row) in samples from both wild-type (WT) and KI macrophages treated with LPS (+LPS). By comparison, the monoclonal antibody detected a band in the KI but not WT sample. C, dose-dependent inhibition of PGE2 production by MF-tricyclic but not MF63 in LPS-stimulated WT macrophages (MΦ). D, dose-dependent inhibition of PGE2 production by both MF-tricyclic and MF63 in LPS-stimulated KI MΦ. Data in C and D are reported as the percentage of inhibition, with that in vehicle-treated cells as 0% (in triplicate).
Suppression of PGE2 in A549 cells resulted in the redirection of PGH2 to PGF2α. To examine whether such a phenomenon also occurred in vivo, we examined the dose-dependent effects of MF63 on PGE2 and other prostaglandins in the brains of LPS-injected KI mice. As in the cultured macrophages, MF63 inhibited PGE2 formation in a dose-dependent manner (Fig. 4D). However, the compound neither inhibited nor enhanced the levels of PGF2α (Fig. 4E) and other prostanoids, such as TXB2 (Fig. 4F).
To investigate whether a selective inhibition of PGE2 is sufficient for analgesia, we examined the effect of MF63 on LPS-induced hyperalgesia, a COX-2-dependent pain response (Abe et al., 2001). Intraplantar injection of LPS caused a significant hyperalgesic response, reducing the latency of heat-elicited paw withdrawal from a baseline of 14.8 ± 0.7 to 8.1 ± 0.7 s at 1 h after the injection. MF63 inhibited the hyperalgesic response in the KI mice in a dose-dependent manner, reducing the response by ∼50% at 10 mg/kg and 80% at 100 mg/kg but was without effect in the wild-type animals. By comparison, MF-tricyclic displayed similar efficacy in both genotypes (Fig. 5). The antihyperalgesic efficacy of MF63 in the KI mouse was comparable to that of MF-tricyclic.
MF63 Inhibits PGE2 Synthesis and Hyperalgesia in the Guinea Pig. To identify a second species for confirming the pharmacological effects of MF63, we tested the compound on the guinea pig mPGES-1 enzyme. The compound potently inhibited the guinea pig enzyme with an IC50 of 0.9 nM. Furthermore, MF63 showed desirable pharmacokinetics in the guinea pig, reaching average concentrations of 3.0, 4.1, and 3.2 μM, respectively, at 1, 2, and 6 h in the plasma and 20 μM in the brain at 6 h after oral administration at the dose of 30 mg/kg. The compound thus has a suitable potency and pharmacokinetic profile for efficacy studies in the guinea pig.
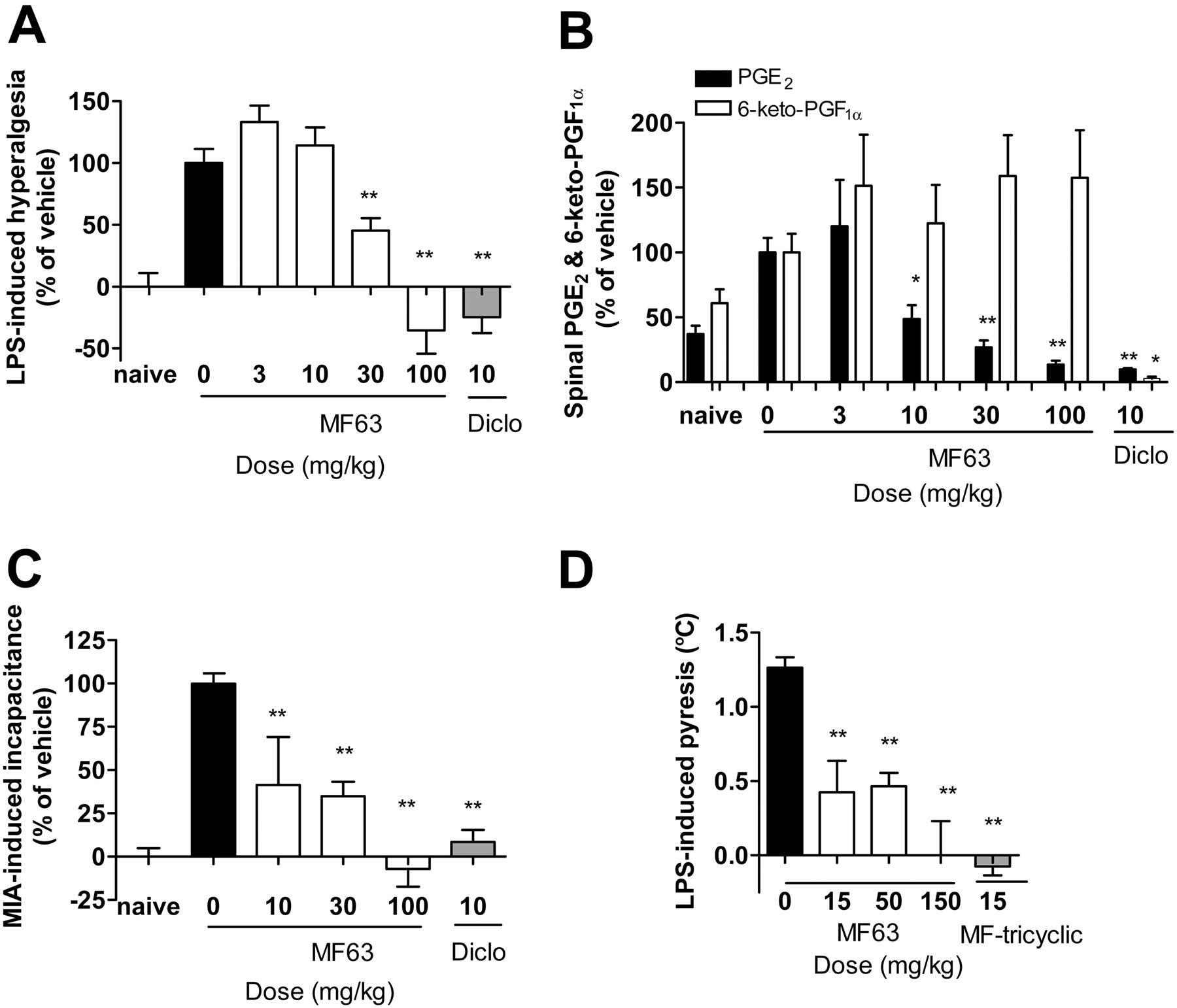
The antihyperalgesic effect of MF63 in guinea pigs was also assessed in the LPS-induced thermal hyperalgesia model. Injection of LPS into the plantar caused a significant thermal hyperalgesic response, reducing the latency of heat-elicited paw withdrawal from 7.7 ± 0.6 to 4.7 ± 0.6 s at 4.5 h after the injection. When administered orally at 1 h before LPS injection, the benchmark NSAID, diclofenac, completely inhibited the response at the dose of 10 mg/kg (Fig. 6A). Likewise, MF63 inhibited the hyperalgesic response in a dose-dependent manner with an ∼60% decrease at the dose of 30 mg/kg and a maximal analgesic effect comparable to that of diclofenac at 100 mg/kg (Fig. 6A).
The thermal hyperalgesic response is believed to be mediated by PGE2 in both the periphery and the central nervous system (Yaksh et al., 2006). In the spinal cord, PGE2 may block inhibitory glycinergic neurotransmission and hence facilitate nociception (Ahmadi et al., 2002). To examine whether the analgesic effect was due to selective PGE2 inhibition, we determined the effect of the selective mPGES-1 inhibitor on levels of PGE2 and 6-keto-PGF1α, the major metabolite of PGI2 in the spinal cord. Intraplantar injection of LPS resulted in a 2-fold increase in PGE2 levels in the lumbar spinal cord from 45 ± 8to120 ± 13 pg/mg protein (n ≥ 5/group). MF63 inhibited the increased formation of PGE2 in a dose-dependent manner but did not inhibit synthesis of PGI2, measured as 6-keto-PGF1α, in contrast to diclofenac, which suppressed the synthesis of both prostanoids (Fig. 6B).

Selective inhibition of LPS-induced PGE2 synthesis by MF63 in the KI mice. A, selective inhibition of PGE2 synthesis by MF63 in the air pouch. MF63, at the dose of 100 mg/kg, significantly reduced the levels of PGE2 but not those of 6-keto-PGF1α. In contrast, MF-tricyclic, at the dose of 10 mg/kg, reduced the levels of both prostanoids. B, inhibition of LPS-induced PGE2 synthesis by MF63 in the brains of the KI but not wild-type mice. C, lack of effect by MF63 on 6-keto-PGF1α formation in the brains of both the KI and wild-type mice. D to F, dose-dependent effects of MF63 on brain prostanoids. MF63 inhibited LPS-induced PGE2 synthesis in the brain in a dose-dependent manner (D) but was without effect on PGF1α (E) or TXB2 (F). For all experiments (A–F), test compound was administered orally at 1 h before LPS injection into the pouch (A, air pouch) or peritoneum (B–F, brain). Animals in the naive group were injected with saline, whereas those in the other groups were injected with LPS. Prostaglandins were analyzed 3 h after LPS or saline injection. Results are shown as mean ± S.E.M. (n = 5–10/group). *, p < 0.05; **, p < 0.01 for compound- versus vehicle-treated animals.
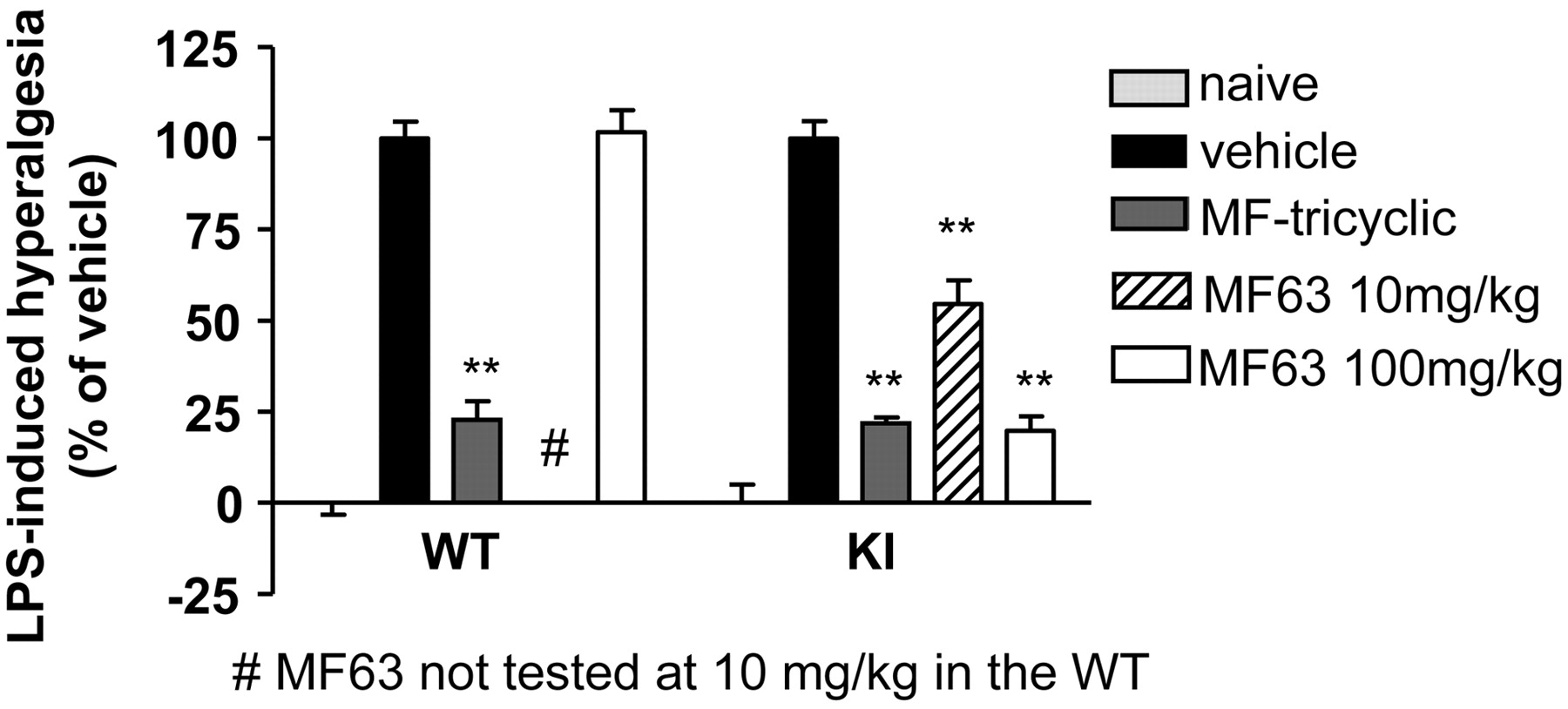
Effects of MF63 and MF-tricyclic on LPS-induced hyperalgesia in mice. MF-tricyclic, when administered orally at the indicated dose at 1 h before LPS injection, inhibited hyperalgesia in both wild-type and KI mice. In contrast, MF63 was only effective in the KI mice. Animals in the naive group were injected intraplantarly with saline, whereas those in the other groups were injected with LPS. Data are expressed as percentage of hyperalgesia, with the naive group (injected with saline) as 0% and the vehicle-treated LPS-injected group as 100%. Mean ± S.E.M. (n = 10/group); **, p < 0.01.
MF63 Relieves Chronic Osteoarthritic-Like Pain in the Guinea Pig. The analgesic properties of MF63 were further evaluated in a guinea pig model of osteoarthritic pain induced by intra-articular injection of MIA in the shoulder joint. In naive or saline-injected animals, weight distribution between the two forelimbs was comparable, yielding an L/R ratio between 1.0 and 1.2 (Fig. 6C, naive). Injection of MIA caused inflammation and pain, resulting a decreased weight bearing or incapacitance on the injected forelimb in a time-dependent manner, manifested as a gradual decrease in the L/R ratio reaching a minimum of approximately 0.5 by day 3. When tested on day 3, diclofenac nearly completely alleviated MIA-induced incapacitance at 6 h after dosing at 10 mg/kg (Fig. 6C). MF63 also reversed incapacitance by 60 to 70% at 10 and 30 mg/kg and 100% at the dose of 100 mg/kg compared with the vehicle (Fig. 6C).

Analgesic and antipyretic effects of MF63 in the guinea pig. A, inhibition of LPS-induced hyperalgesia by MF63 and diclofenac. Data are expressed as percentage of hyperalgesia, with the naive group (injected intraplantarly with saline) as 0% and the vehicle-treated LPS-injected group as 100%. B, selective inhibition PGE2 over 6-keto-PGF1α by MF63 in the lumber spinal cord. Spinal samples were collected from the experiment described in A for prostanoid analysis. Data are expressed as relative levels, with those for vehicle-treated animals as 100%. The absolute prostanoid levels in the vehicle group are 0.12 ± 0.01 ng/mg protein for PGE2 and 0.17 ± 0.02 ng/mg protein for 6-keto-PGF1α. MF63 reduced the levels of PGE2 but not those of 6-keto-PGF1α in a dose-dependent manner, whereas diclofenac reduced the levels of both prostanoids. C, dose-dependent reversal of MIA-induced incapacitance by MF63. Data are expressed as percentage of incapacitance with the naive group (injected intra-articularly with saline) as 0% and vehicle-treated MIA-injected group as 100%. MF63, when tested on day 3 after an injection of MIA into the shoulder joint, attenuated incapacitance in a dose-dependent manner at 6 h after an oral administration, with a similar efficacy as diclofenac. D, inhibition of LPS-induced pyresis by MF63 and MF-tricyclic. MF63, when administered 1 h before LPS injection, inhibited the increase in rectal temperature in a dose-dependant manner, with an efficacy similar to that of MF-tricyclic. Results are shown as mean ± S.E.M. (n = 5–10/group). *, p < 0.05; **, p < 0.01 for compound versus vehicle-treated animals.
MF63 Inhibits Pyresis in the Guinea Pig. The antipyretic property of MF63 was examined in a guinea pig model of LPS-induced pyresis. In this model, intraperitoneal injection of LPS caused a time-dependent increase in rectal temperature, reaching 1.2°C at 3 h after injection. Oral administration of MF63, given 1 h before LPS injection, prevented the increase in rectal temperature by ∼50% at the doses of 15 and 50 mg/kg and completely at the dose of 100 mg/kg, as achieved with MF-tricyclic (Fig. 6D).
Gastrointestinal Tolerability of MF63. The gastrointestinal tolerability of MF63 was assessed by examining the effects of the compound on stomach PGE2 and PGI2 as well as mucosal integrity in the KI mice. Under basal conditions, the stomach prostaglandin contents were comparable between the wild-type and KI mice (Table 1). MF63 reduced stomach PGE2 in the KI mice in a dose-dependent manner by a maximum of 85% at the highest dose 3 h after dose. MF63 was without any significant effects on 6-keto-PGF1α (Fig. 7A) and other prostanoids (data not shown), with the exception of TXB2, which was increased from 0.04 ± 0.01 to 0.08 ± 0.01 ng/mg protein at both 10 and 30 mg/kg and to 0.14 ng/mg protein at 100 mg/kg. Under these conditions, MF63 did not produce mucosal erosions as those noted with the NSAID indomethacin (Fig. 7, B and C).
Basal prostaglandin levels in the stomach of KI and wild-type mice (picogram/milligram protein) n = 5/group; mean ± S.E.M.
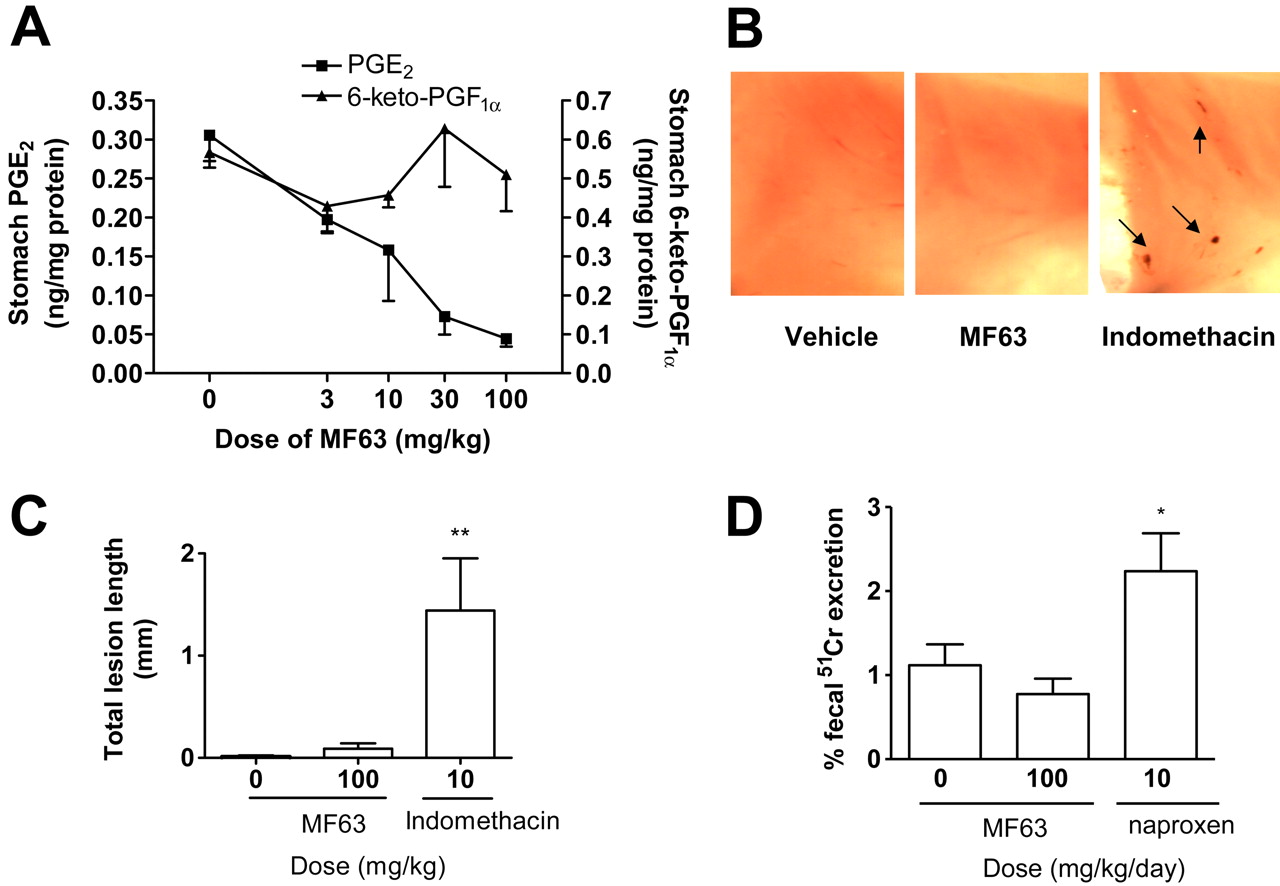
Lack of gastrointestinal toxic effects by MF63 in KI mice and nonhuman primates. A, PGE2 or 6-keto-PGF1α levels in the stomachs of mice at 3 h after oral administration of vehicle (n = 7) or MF63 (n = 4–5/group). B, representative images of mouse gastric mucosa. MF63 and indomethacin were administered orally at the dose of 100 and 10 mg/kg, respectively. Stomach was collected 3 h after the administration of compounds. Each image covers one-half of a mouse stomach. Arrows indicate erosions. C, quantitative analysis of the total lesion length of gastric erosions (mean ± S.E.M., n = 20/group). **, p < 0.01. D, fecal excretion of 51Cr expressed as a percentage of the total injected dose in squirrel monkeys treated with vehicle, MF63, or naproxen. Test compound was orally administered twice daily for 4 days. The total daily doses are shown. Radioactivity was counted in 24-h fecal samples collected after the last dose to obtain 51Cr excretion. Data are mean ± S.E.M. with n ≥ 4/treatment. *, p < 0.05.
The gastrointestinal tolerability of MF63 was also examined in squirrel monkeys, in which the compound had an IC50 of 2.2 μM in the whole blood, in a model for measuring the excretion of chromium from the blood into the feces (Riendeau et al., 2001). When dosed at 50 mg/kg twice daily for 4 days, MF63 did not increase 51Cr leakage compared with the vehicle. In contrast, the NSAID naproxen, at the dose of 5 mg/kg twice daily, significantly enhanced 51Cr leakage compared with the vehicle (p < 0.05) (Fig. 7D).
Discussion
Blockage of PGE2 activity with either a specific PGE2 antibody or EP4 antagonists produces NSAID-like anti-inflammatory and analgesic efficacy in preclinical models of inflammation (Portanova et al., 1996; Nakao et al., 2007; Clark et al., 2008), suggesting that selective targeting of PGE2 may provide an alternative approach to NSAIDs for treating inflammatory diseases. Here, we assessed the therapeutic potential of a different PGE2 targeting strategy, namely mPGES-1 inhibition, and demonstrated that MF63, an mPGES-1 inhibitor, selectively inhibited PGE2 synthesis and produced similar anti-pyretic and analgesic effects as NSAIDs or selective COX-2 inhibitors in rodent models of inflammation.
MF63 is a lead compound from a novel series of selective mPGES-1 inhibitors (Côté et al., 2007). Compared with a previous series of prototypic mPGES-1 inhibitors (Riendeau et al., 2005), MF63 had markedly improved potency and selectivity in cell-based assays. It is important that the compound was active in a clinically relevant human whole-blood assay, displaying a similar profile as selective COX-2 inhibitors at inhibiting PGE2 synthesis and sparing COX-1-mediated thromboxane production (Riendeau et al., 2001). The compound was also bioavailable in mice and rats but was limited by its lack of mPGES-1 inhibitory activity in these species.
The interspecies difference was overcome through the generation of a KI mouse expressing the human mPGES-1 and identification of the guinea pig as a sensitive species. In both species, MF63 selectively inhibited the synthesis of PGE2 over other prostaglandins. The effects of MF63 on PGE2 and other prostaglandins in both the brains and stomachs of the KI mice are similar to those produced by genetic deletion of mPGES-1 in the knockout mice (Boulet et al., 2004). In both types of mice, PGE2 inhibition in the stomach was associated with a marked increase in TXB2, possibly as a result of redirection or shunting of PGH2 into the synthesis of thromboxane. In contrast, no shunting was seen in the brain, either to TXB2 or to PGF2α. These results indicate that shunting is tissue-specific. The mechanism and the physiological impact of tissue-specific shunting are not clear. However, deletion of mPGES-1 in mice does not increase the urinary excretion of thromboxane metabolites or the susceptibility to thrombosis (Cheng et al., 2006), suggesting that tissue-specific shunting has little systemic impact.
At the doses that selectively suppressed PGE2 synthesis, MF63 inhibited acute inflammatory hyperalgesia and relieved chronic inflammatory pain with similar efficacy as NSAIDs in the KI mice or guinea pigs. MF63 failed to affect either PGE2 synthesis or hyperalgesia in wild-type mice, suggesting that the compound produces analgesia by selectively suppressing the synthesis of PGE2. These findings are in agreement with previous observations that blockage of PGE2 activity with either a specific PGE2 antibody or an EP4 antagonist results in similar analgesic efficacy as NSAIDs in rat models of inflammation (Portanova et al., 1996; Nakao et al., 2007; Clark et al., 2008). We did not assess the analgesic effects of MF63 in rats because the compound does not inhibit the rat enzyme. However, it is reasonable to expect an inhibitor of PGE2 formation to have similar efficacy to an antagonist of PGE2 activity.
PGE2 may produce hyperalgesia peripherally in inflamed tissues (Taiwo et al., 1987) and centrally in the spinal cord (Ahmadi et al., 2002) or in other brain regions (Heinricher et al., 2004). Previously, it has been shown that the expression of COX-2 and mPGES-1 and the synthesis of PGE2 are increased in both the inflamed tissues and the spinal cord during carrageenan-induced paw inflammation (Guay et al., 2004). Here, we show that inhibition of COX-2 or mPGES-1 attenuates the synthesis of PGE2 both at the inflammatory site and in the spinal cord. Together, these findings suggest that both the peripheral and central COX-2/mPGES-1 pathways are activated during inflammation and may jointly contribute to the induction of hyperalgesia and inflammatory pain.
In addition to having analgesic properties, MF63 also possesses a similar antipyretic efficacy to a selective COX-2 inhibitor at inhibiting LPS-induced fever in the guinea pig. The antipyretic efficacy is as predicted from the central role of PGE2 in mediating pyresis. Unlike inflammatory pain, in which PGI2 may also play an important role (Murata et al., 1997; Pulichino et al., 2006), pyresis is mediated primarily by PGE2 (Ushikubi et al., 1998) and mPGES-1 (Engblom et al., 2003). Together, these findings indicate that mPGES-1 inhibitors may be used for the treatment of fever.
Besides producing PGE2 during inflammation, mPGES-1 also contributes to the synthesis of gastric PGE2, a gastroprotective substance. Consequently, mPGE-1 inhibition may be as detrimental as NSAIDs to the stomach. This possibility was examined by testing MF63 in two models well characterized for assessing the gastrointestinal toxicities or tolerability of NSAIDs and selective COX-2 inhibitors (Hunt et al., 2000; Sigthorsson et al., 2000; Riendeau et al., 2001). As expected, the benchmark NSAIDs were toxic to the gastrointestinal tract, causing mucosal erosions or leakage. In contrast, MF63 was well tolerated. At the dose used, indomethacin was found to reduce stomach PGE2 and PGI2 by ≥95% in a pilot experiment on wild-type mice (unpublished data). By comparison, MF63 reduced stomach PGE2 in the KI mice by 85% without affecting PGI2, which is also a major gastroprotective prostanoid (Whittle and Vane, 1984). The remaining pool of gastroprotective prostanoids may be sufficient for protecting the gastric mucosa. It deserves mention that we used indomethacin in mice and naproxen in nonhuman primates as positive controls, both at doses that are predicted to produce maximal efficacy. At such doses, these compounds, but not selective COX-2 inhibitors, have been shown to cause lesions in rodents and nonhuman primates (Riendeau et al., 2001). We did not use diclofenac as the benchmark because, when tested in a pilot experiment on squirrel monkeys, the compound was not tolerated and caused gastrointestinal bleeding in some animals after oral dosing at 3 mg/kg. Collectively, these results suggest that mPGES-1 inhibitors may have better gastrointestinal tolerability than traditional NSAIDs.
An important question that is not within the scope of our study is whether selective mPGES-1 inhibitors are devoid of the cardiovascular risk as that noted with COX-2 inhibitors (Fitzgerald, 2004). Although the exact mechanism behind such a risk is not known, inhibition of PGI2, which has potent antithrombotic and vasodilatory properties, has been proposed as a contributing factor (Fitzgerald, 2004). mPGES-1 inhibitors, which do not suppress PGI2, may thus have an improved cardiovascular profile over COX-2 inhibitors. The view is supported by recent evidence that mPGES-1 deletion does not lead to an increased susceptibility to thrombosis in mice in contrast to COX-2 deletion (Cheng et al., 2006). Besides thrombosis, an increase in blood pressure is another cardiovascular risk factor. Although mPGES-1 deletion further increases blood pressure in mice with extremely high salt load (saline plus 3.5% of dietary NaCl) (Jia et al., 2006), it does not have any impact on hypertension under standard high salt conditions (6–8% dietary sodium) (Cheng et al., 2006; Francois et al., 2007) known to cause exaggerated hypertension in mice lacking COX-2 (Cheng et al., 2006).
In summary, we have demonstrated that a selective mPGES-1 inhibitor effectively relieves fever and inflammatory pain in rodent models of inflammation. In addition, the compound is well tolerated by the gastrointestinal tract and does not suppress the synthesis of PGI2. The lack of PGI2 suppression has been attributed to the diminished thrombotic and hypertensive responses in mice lacking mPGES-1 compared with mice having COX-2 inhibition or knockout (Cheng et al., 2006). Together, these findings suggest that mPGES-1 inhibitors have the potential to become a distinct class of novel anti-inflammatory agents that act by selectively suppressing PGE2 synthesis.
Acknowledgments
We thank Michel-Oliver Gratton, Diane Ethier, Lynn Dufresne, and Joel Rubin for technical support for the in vitro experiments, Marc Ouellet for characterizing the MF137 antibody, our colleagues from the Department of Comparative Medicine for excellent technical assistance with the in vivo experiments, and Kevin Clark for preparing the figures.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.108.138776.
-
ABBREVIATIONS: NSAID, nonsteroidal anti-inflammatory drug; COX, cyclooxygenase; EIA, enzyme immunoassay; EP, E prostanoid receptor; FBS, fetal bovine serum; kb, kilo base; KI, knock-in; LPS, lipopolysaccharide; L/R ratio, weight bearing ratio between left/right forelimbs; MF63, 2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)isophthalonitrile; MF-tricyclic, 3-(3,4-difluorophenyl)-4-(4-(methylsulfonyl)phenyl)-2-(5H)-furanone; MIA, monosodium iodoacetate; mPGES, microsomal prostaglandin E synthase; PG, prostaglandin; 6-keto-PGF1α, 6-keto-prostaglandin F1α; TXB2, thromboxane B2; WT, wild type; IL, interleukin; PBS, phosphate-buffered saline; DMSO, dimethyl sulfoxide; PCR, polymerase chain reaction; PWL, paw withdrawal latency.
- Received March 11, 2008.
- Accepted May 12, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}