


Abstract
Sphingosine kinase (SK) is an oncogenic sphingolipid-metabolizing enzyme that catalyzes the formation of the mitogenic second messenger sphingosine-1-phosphate (S1P) at the expense of proapoptotic ceramide. Thus, SK is an attractive target for cancer therapy because blockage of S1P formation leads to inhibition of proliferation, as well as the induction of apoptosis in cancer cells. We have recently identified novel SK inhibitors with nanomolar to low micromolar potencies toward recombinant human SK. This study describes the continuing analysis of these inhibitors through in vitro and in vivo experiments. All three structurally diverse SK inhibitors tested showed antitumor activity in mice without exhibiting toxicity. Blood and tumor inhibitor concentrations exceeded in vitro potency levels. Cell signaling analyses in vitro revealed mixed inhibition of mitogen-activated protein kinase kinase and Akt phosphorylation by the SK inhibitors. Importantly, 4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol (SKI-II) is orally bioavailable, detected in the blood for at least 8 h, and showed a significant inhibition of tumor growth in mice. These compounds are the first examples of nonlipid selective inhibitors of SK with in vivo antitumor activity and provide leads for further development of inhibitors of this important molecular target.
The sphingomyelin metabolism pathway is emerging as a promising target for rational cancer therapy. One of the most attractive sites of intervention in this pathway is the conversion of sphingosine to sphingosine-1-phosphate (S1P) by the enzyme sphingosine kinase (SK) (Spiegel and Milstien, 2003). SK catalyzes the final step of S1P formation from the proapoptotic lipid ceramide. A ceramide/S1P rheostat has been hypothesized to determine the fate of the cell, such that the relative cellular concentrations of ceramide and S1P determine whether a cell proliferates or undergoes apoptosis (Cuvillier et al., 1996). SK is an oncogene (Xia et al., 2000) and is tightly regulated by a number of growth factors (Olivera and Spiegel, 1993) and protein kinases (Shu et al., 2002), leading to rapid increases in the intracellular levels of S1P. The result is rescue of ceramide-dependent apoptosis by S1P, culminating in cell survival and proliferation.
Because the balance between the cellular concentrations of ceramide and S1P determines whether a cell proliferates or undergoes apoptosis, the enzymes in this pathway provide potential targets for the development of new anticancer drugs. Pharmacological studies to date have mainly used three compounds to inhibit SK activity: d-erythro-N,N-dimethylsphingosine, dl-threo-dihydrosphingosine, and N,N,N-trimethylsphingosine. However, these compounds are not specific inhibitors of SK as they are known to affect multiple lipid and protein kinases (Igarashi et al., 1989; Megidish et al., 1995). Previously, we used recombinant human SK1 to screen a library of synthetic compounds, resulting in the identification of a panel of inhibitors of this enzyme (French et al., 2003). These compounds are selective toward SK compared with other lipid and protein kinases. Furthermore, the compounds are antiproliferative and apoptotic toward a panel of human tumor cell lines. The compounds inhibit S1P formation in intact cells and maintain activity toward cells that express the drug transport proteins P-glycoprotein (Pgp) or MRP1. In continuing studies of these SK inhibitors as cancer therapeutic agents, we now report further in vitro and in vivo properties of three SK inhibitors (Fig. 1). The antitumor activities mostly correlated well with their concentrations in blood and tumors. One of the inhibitors, SKI-II, shows oral bioavailability and antitumor activity. These findings provide further validation for SK as a cancer therapeutic target, as well as evaluating these small molecule inhibitors for this enzyme.

Structures of SK inhibitors used in these studies.
Materials and Methods
Materials. Unless otherwise noted, all of the chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO). SKI-I and SKI-II were purchased from ChemBridge Corporation (San Diego, CA). SKI-V was synthesized as described previously (French et al., 2003). Antibodies against Akt, mitogen-activated protein kinase kinase (MEK) 1/2, Phospho-Akt (Ser473), and Phospho-MEK1/2 (Ser217/221) were purchased from Cell Signaling Technology (Beverly, MA).
Synthesis of 2-(4-Chloro-benylidene)-benzofuran-3-one. Benzofuran-3-one exists in equilibrium between the keto and enol tautomers and underwent an aldol condensation with p-chlorobenzaldehyde in aluminum oxide to form the target compound. The compound was confirmed using 1H NMR and mass spectral analyses. Purity was assessed via high-performance liquid chromatography (HPLC)/UV-visible and determined to be greater than 95%.
Cell Lines. NIH3T3, MDA-MB-231, and JC cell lines were obtained from American Type Culture Collection (Manassas, VA). The cells were cultured in either Dulbecco's modified Eagle's medium or RPMI 1640 medium containing 10% fetal bovine serum (Cool Calf Serum for NIH3T3) and 50 μg/ml gentamicin sulfate.
Cellular S1P Formation Assay. Cells were grown to near confluence in 10-cm culture plates and treated with 0.5% vehicle [dimethyl sulfoxide (DMSO)] or indicated concentrations of each inhibitor for 48 h. Cells were washed, harvested, and lysed via sonication in ice-cold phosphate-buffered saline (PBS) with 15 mM NaF and 1 mM sodium orthovanadate. Protein content of lysates was determined using the Bio-Rad (Hercules, CA) protein assay system. Sample preparation and analysis were performed according to the methods of Sullards and Merrill (2001). Cell lysates were combined with 0.5 ml of methanol, 0.25 ml of chloroform, and 375 pmol of C17-S1P as internal standard (Avanti, Alabaster, AL). After sonication, samples were incubated overnight 48°C in a water bath, followed by addition of 75 μl of 1 N potassium hydroxide in methanol. Next, samples were sonicated and incubated at 37°C for 2 h. Four-hundred microliters was then transferred to new tubes, dried over nitrogen gas, reconstituted in 250 μl of sphingolipid mobile phase, filtered, and transferred to vials. Analysis was performed using an Agilent 1100 binary pump HPLC system coupled to a Finnigan LCQ Classic ion trap quadrupole mass spectrometer running in electrospray ionization positive ion mode. Elution was performed at 0.45 ml/min with 35% mobile phase A (liquid chromatography/mass spectrometry grade water with 0.1% formic acid) and 65% mobile phase B ((liquid chromatography/mass spectrometry grade methanol with 0.1% formic acid) (JT Baker, Phillipsburg, NJ). Initial mobile phase ran for 2 min followed by a linear gradient to 100% phase B over 5 min. Ions for C17-S1P and S1P were monitored at m/z 366 (parent ion) - 250 (daughter ion) and 380 - 264, respectively.
Cell Lysate SK Activity Assay. Cells were grown to 90% confluence as described above in the presence of serum. Media were aspirated, and cells were washed three times with cold PBS and treated with 0.5 ml of SK lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM sodium chloride, 1% v/v Triton X-100, 10 mM β-glycerophosphate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 10 μM okadaic acid, and protease inhibitor mixture for mammalian tissues). Cells were scraped; lysates were isolated via centrifugation (11,000g, 4°C, 10 min); and total protein concentration was determined using the Bradford Bio-Rad Protein Assay. Seventy micrograms of protein was used per SK activity assay as described previously (French et al., 2003).
Cell Proliferation Assay. Cells were plated into 96-well tissue culture plates at approximately 15% confluence. After 24 h, cells were treated with varying concentrations of inhibitors. After an additional 48 h, cell survival was assayed as described previously (French et al., 2003).
Antitumor Evaluation. A syngenic mouse tumor model that uses a transformed murine mammary adenocarcinoma cell line (JC, ATCC no. CRL-2116) and BALB/c (Charles River) mice was performed as described previously (French et al., 2003). Animal care and procedures were in accordance with guidelines and regulations of the Institutional Animal Care and Use Committee of the Penn State College of Medicine. Animals were housed under 12-h light/dark cycles with food and water provided ad libitum. Cells were implanted s.c., and tumor volume was calculated using the equation (L × W2)/2. Treatment was then administered via two approaches: days 1, 5, 9, and 15 or day 1 and every odd day thereafter. Dosing consisted of i.p. administration of either 50 μl of vehicle (DMSO) or SK inhibitor at the indicated dose or p.o. dosing of vehicle (polyethylene glycol 400) or SKI-II. On the final day of the study, mice were dosed and 1 h later euthanized, and tumors were excised. Blood was removed via intracardial puncture, and all of the samples were stored at -80°C until analysis. Whole body weight and tumor volume measurements were performed each day of treatment. The p values were determined using one-way analysis of variance using GraphPad InStat (San Diego, CA).
Immunoblot Analysis of MEK1/2 and Akt. For cell culture experiments, JC cells were plated and grown to 90% confluence and serum-starved overnight. Next, cells were pretreated for 1 h with DMSO, positive controls (10 and 50 μM LY294002, a phosphoinositide 3-kinase inhibitor, and 20 μM BAY 43-9006, a raf kinase inhibitor), and each SK inhibitor at identical concentrations used in the S1P formation assay described above. Cells were then stimulated with 1% fetal bovine serum for 15 min, washed three times with ice-cold PBS, and harvested with SK lysis buffer. Lysates were prepared via centrifugation (11,000g, 4°C, 10 min), and determination of total protein concentration was performed using the Bradford Bio-Rad Protein Assay. Samples were combined with SDS-polyacrylamide gel electrophoresis sample buffer, heated at 95°C for 5 min, and loaded onto 10% Tris-glycine SDS-polyacrylamide gels at 50 μg/lane. Resolved gels were immunoblotted onto nitrocellulose membranes, and total and phosphorylated MEK1/2 or Akt levels were determined with antibodies according to the manufacturer's specifications. Quantification of the results was performed using a Fujifilm Intelligent Dark Box II. All of the measured intensities fell within the range of linearity for the instrument.
Extraction of Drug from Whole Blood Samples. Blood samples were collected at the indicated times and either frozen at -20°C or processed immediately. Frozen samples were thawed at 37°C, and 1.5 μg of the appropriate internal standard was added to each sample. Internal standards for SKI-I, -II, and -V were 5-P-tolyl-2H-pyrazole-3-carboxylic acid (2-HO-napthalen-1-YL methylene) hydrazide (C22H18N4O2), 4-methyl-N-(4-P-tolyl-thiazol-2-YL)-benzamide (C18H16N2OS), and 2-(4-chloro-benylidene)-benzofuran-3-one (C15H9ClO2), respectively. Samples were precipitated with 5 ml of ice-cold acetonitrile and vortexed to disperse large clumps. Whole blood extracts were then spun at 4°C at 4000g for 5 min, and supernatant was removed and evaporated using the N-Evap system with nitrogen gas and a 35°C water bath. Dried samples were reconstituted in 250 μl of methanol and analyzed by HPLC-UV as described below.
Extraction of Drug from Tumor. Tumors were excised at the indicated times and either frozen at -80°C or processed immediately. Frozen tumors were thawed and homogenized in PBS, and 1.5 μg of the appropriate internal standard described above was added in each sample. Proteins were precipitated with 20 ml of ice-cold acetonitrile and vortexed to disperse large clumps. Tumor extracts were then spun at 4°C at 5000g for 5 min, and supernatant was removed and evaporated using the N-Evap system with nitrogen gas and a 35°C water bath. Dried samples were reconstituted in 1 ml of methanol and analyzed by HPLC-UV.
Determination of SKI Concentrations in Whole Blood and Tumor. Samples were resolved using HPLC on a reverse-phase ZORBAX (Agilent) SB-C18 (5 μm, 80 Å, 4.6 × 250 mm) column with a flow rate of 1 ml/min. The HPLC unit consisted of the Beckman Coulter (Fullerton, CA) System Gold and Gold 166 Detector. For SKI-I, the initial mobile phase was 40% methanol 60%, 0.1% (trifluoroacetic acid) in HPLC grade H2O for 3 min, followed by a 25-min linear gradient to 100% methanol. For SKI-II and SKI-V, the initial mobile phase was 40% methanol 60%, 0.1% trifluoroacetic acid in water for 3 min, followed by a 20-min linear gradient from 40 to 100% methanol. The mobile phase was then maintained at 100% methanol for 8 min. The SKI compounds were detected using a UV-visible detector set at 254 nm. The retention times for the analytes and internal standards were as follows: SKI-I, 27.4 min; 5-P-tolyl-2H-pyrazole-3-carboxylic acid (2-HO-napthalen-1-YL methylene) hydrazide, 26.2 min; SKI-II, 20.1 min; 4-methyl-N-(4-P-tolyl-thiazol-2-YL)-benzamide, 23.1 min; SKI-V, 16.3 min; and 2-(4-chloro-benylidene)-benzofuran-3-one, 23.9 min. Each inhibitor was quantified based on standard curves, where the area ratios of inhibitor to internal standard fell within the range of linearity. The limits of detection for SKI-I, -II, and -V were 3, 45, and 50 ng/ml, respectively.
Oral Pharmacokinetics of SKI-II. Female Swiss-Webster mice (6-8 weeks old) were fasted overnight and administered 100 μl of SKI-II dissolved in polyethylene glycol 400 by gavage to a final dose of 100 mg/kg. After dosing, mice were anesthetized with halothane, and blood was removed via intracardial puncture at the indicated times. Blood samples were processed as described above. Noncompartmental pharmacokinetic analyses were performed using Win-Nonlin (Pharsight, Mountain View, CA).
Results
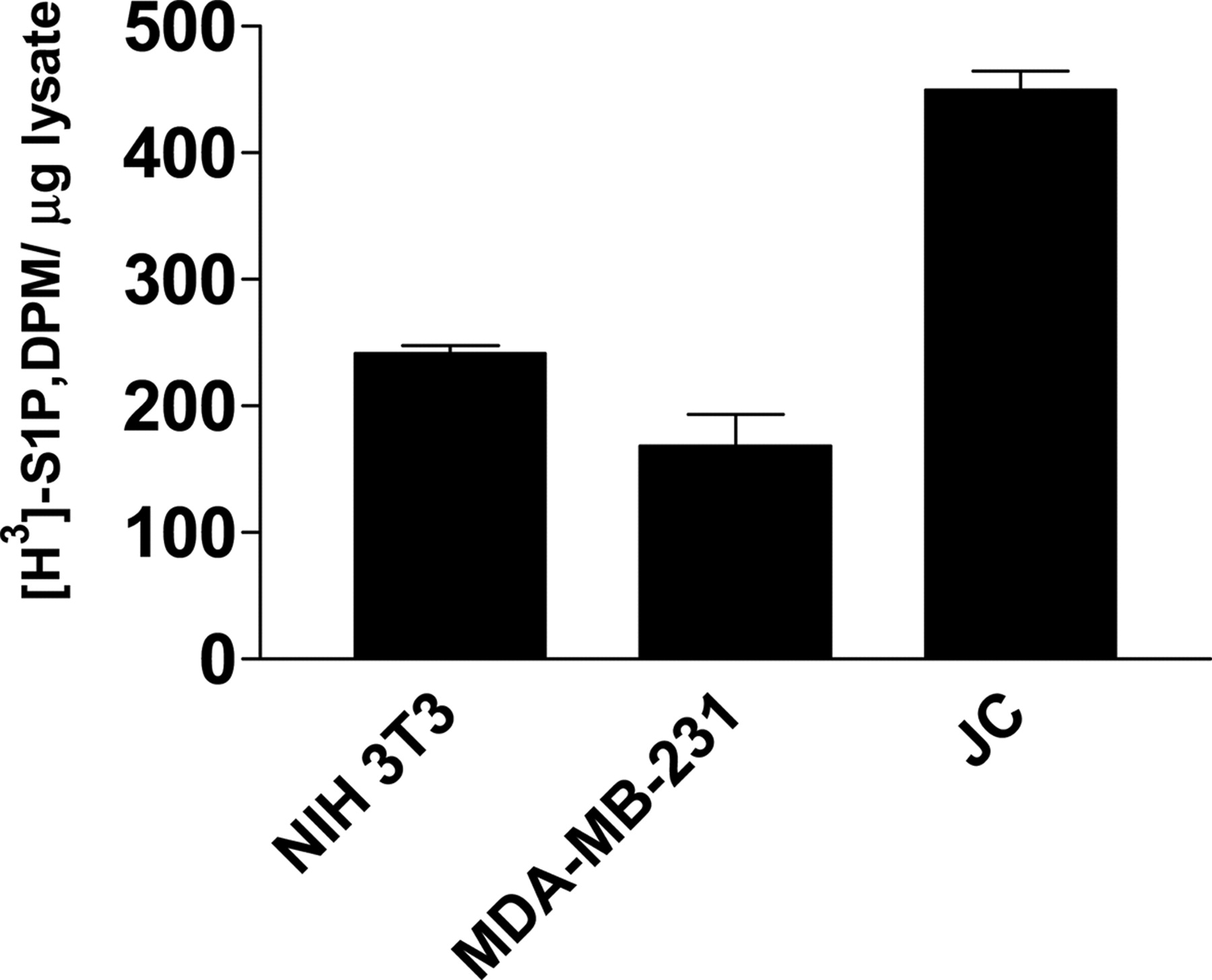
In Vitro Characterization of SK Inhibitors on JC Cells. We determined that the JC mouse mammary adenocarcinoma cell line was an appropriate model for in vitro characterization of SK inhibitors. As shown in Fig. 2, lysates isolated from growing cells had elevated SK activity compared with the nontransformed NIH3T3 cells. Furthermore, JC cells were higher in activity than the human MDA-MB-231 breast cancer cell line. In addition to increased SK activity, JC cells have shown overexpression of Pgp, a drug efflux transporter associated with drug resistance commonly seen in chemotherapy treatment (French et al., 2003). Thus, these novel findings indicate the JC cell line is a stringent model for evaluating the therapeutic potential of SK inhibitors.

Cytosolic SK activity from various cell lines. Lysates from subconfluent cell cultures were assayed for SK activity as described under Materials and Methods section. Seventy micrograms of lysate was used per sample. Data represent mean ± S.D. from triplicate samples in a typical experiment.
S1P Formation in JC Cells. Previously, we identified novel SK inhibitors from a small molecule screen using recombinant human SK1. To determine the effects of these inhibitors on intact cells, we pretreated JC cells with each SK inhibitor at varying concentrations and at similar exposure times as performed in the cell proliferation assay and determined endogenous S1P intracellular levels. The results are summarized in Fig. 3. As shown previously in MDA-MB-231 cells (French et al., 2003), all of the SK inhibitors decreased S1P formation in JC cells in a concentration-dependent manner, with IC50 concentrations below 1 μM for SKI-I and -II and approximately 5 μM for SKI-V. SKI-II was the most potent toward endogenous SK activity, followed by SKI-I and then SKI-V. Previous findings using MDA-MB-231 cells were performed by adding exogenous sphingosine and measuring intracellular S1P formation. However, the currently described studies did not use exogenously added sphingosine and measured endogenous S1P intracellular levels at various doses of each inhibitor, providing more physiologically relevant results. These novel findings show that all of the compounds are capable of inhibiting intracellular S1P formation in JC cells in a dose-dependent fashion.

Inhibition of S1P formation in JC cells by SK inhibitors. Triplicate cultures of JC cells were treated with 0.5% DMSO or the indicated concentrations of SKI-I, SKI-II, or SKI-V and analyzed for intracellular S1P levels as indicated under Materials and Methods section. Values represent the mean ± S.D. for these replicates and are typical of multiple experiments.
Modulation of Downstream Targets of S1P in Vitro. SK catalyzed S1P formation results in activation of proliferation and survival pathways (Pyne and Pyne, 2000). Most notably, S1P activates the Ras/mitogen-activated protein kinase (MAPK) proliferation pathway (Carpio et al., 1999; Rakhit et al., 1999; Sekiguchi et al., 1999), as well as the phosphatidylinositol 3-kinase (PI3K)/Akt survival pathway (Banno et al., 2001; Auge et al., 2002; Baudhuin et al., 2002). Therefore, we hypothesized that SK inhibition would result in decreased signaling via these pathways, leading to cell death and antitumor activity. JC cells were serum-starved, pretreated with similar concentrations of SKI-I, -II, and -V that decrease S1P formation, stimulated, and analyzed for MAPK and Akt pathway activation. As shown in Fig. 4, phospho-Akt levels were modulated by SKI-II and SKI-V but only minimally by SKI-I. Phospho-MEK levels were modulated by SKI-V, with minimal effects by SKI-II and no effect with SKI-I. LY294002 and BAY 43-9006, compounds that have previously shown PI3K and raf inhibition, respectively, blocked pathway activation. Thus, the SK inhibitors were varied in their abilities to affect downstream targets of S1P signaling, indicating the fact that the mechanism of cytotoxicity of certain compounds may not be linked to inhibition of these pathways. These are the first findings that describe modulation of proliferation and survival signaling pathways by these inhibitors.
Antitumor Activity of SK Inhibitors. To determine the in vivo activities of these inhibitors, we used a syngeneic BALB/c mouse solid tumor model that uses JC mammary adenocarcinoma cells (Lee et al., 2003). This system is attractive for several reasons: the mice maintain an intact immune system; there is a high tumor formation rate; and the cells show high SK activity and multidrug resistance. For the tumor studies described below, s.c. injected JC cells were grown to sizable tumors before treatment began (average tumor starting volume 250-1000 mm3). After confirmed tumor establishment, groups of mice were treated via i.p. administration of vehicle (DMSO) or one of the SK inhibitors. Preliminary toxicological studies revealed using the dosing regimen at 50 mg/kg for the inhibitors resulted in no overt toxicity or weight loss (data not shown). The lowest adverse effect level was detected with SKI-V, which was at 75 mg/kg. The results are depicted in Fig. 5. Each of the inhibitors decreased tumor growth relative to control groups, with maximal results observed at the end of the study. Interestingly, the inhibitors were similarly efficacious, as SKI-I, -II, and -V had strong inhibition of tumor growth from the start of treatment of 55, 65, and 79%, respectively. Importantly, no toxicity or weight loss was observed in any of the treatment groups. These findings strengthen the previous finding that SKI-V showed antitumor activity in the same in vivo tumor model while using a more frequent dosing regimen. In addition, these are the first results indicating that SKI-I and SKI-II show in vivo antitumor activity. Thus, the SK1 inhibitors show antitumor activity without overt toxicity.

Inhibition of downstream targets of S1P-mediated signaling. Near-confluent cultures of JC cells were serum-starved for 16 h, followed by pretreatment for 1 h with indicated inhibitors at identical concentrations used in the S1P formation assay. Cells were then exposed to 1% fetal bovine serum for 15 min, harvested, and analyzed for phospho-Akt and phospho-MEK levels. Total Akt and total MEK are also shown.
In Vivo SK Inhibitor Levels Correlate with Antitumor Activity. It was of interest to determine whether the antitumor activity described above correlated with SK inhibitor concentrations in both blood and tumor. Specifically, we wanted to compare in vivo concentrations with in vitro potencies. Thus, we determined the in vitro IC50 for JC cell cytotoxicity using the sulforhodamine B assay. As shown in Table 1, the cellular potencies of each inhibitor were in the low micromolar range. These potencies correlate with their respective potencies toward purified recombinant human SK1 (Table 1). As described previously with human cancer cell lines, the strong correlation between cytotoxicity and SK1 inhibition indicates that the mechanism is most likely SK-mediated (French et al., 2003). To determine the in vivo pharmacokinetics of these inhibitors, tumor-bearing mice were treated i.p. with each SK inhibitor similarly as in the antitumor studies, and mice were euthanized and tumors and blood removed 1 h later. The intratumoral and blood concentrations of the SK inhibitors were determined via liquid-liquid extraction and HPLC-UV-visible analysis. The values are listed in Table 1. Circulating SK inhibitor levels were above the effective concentration for JC cells in vitro for at least 1 h, indicating that all of the SK inhibitors reach the systemic circulation after i.p. administration. Furthermore, SKI-I and SKI-II were detected at therapeutic (i.e., above IC50 for JC cells in vitro) or higher concentrations in all of the tumors. With SKI-II, significant increases (up to 10-fold) in tumor levels were seen compared with blood, whereas SKI-V accumulated in tumors only 11% of blood levels. No metabolites of any of the SK inhibitors were detected in any of the blood or tumor samples. These novel findings reveal that the antitumor effects of these SK inhibitors mostly correlate with their circulating and intratumoral levels.
In vitro potencies of SK inhibitors against the JC cell line compared with both blood and tumor inhibitor concentrations The potencies of each SK inhibitor toward murine JC cells were determined using the sulforhodamine binding assay. (GST-hSK-1 inhibition data are provided for comparison.) Data represent the concentrations required to inhibit cell survival by 50% and are the mean ± S.D. of triplicate experiments. At the end of the i.p. tumor study outlined in Fig. 5 or in typical tumor-bearing mice, animals were i.p. dosed with an SK inhibitor and euthanized 1 h later. Blood and tumor samples were analyzed for SK inhibitor concentrations. Values represent the mean ± S.D. of each treatment group (n = 5).
Oral Activity and Pharmacokinetics of SKI-II. Although the SK inhibitors showed antitumor activity after i.p. administration, we were interested in determining whether our SK inhibitors were available following p.o. administration. Unfortunately, only SKI-II concentrations reached detectable levels at 1 h following a bolus dose of 100 mg/kg of each inhibitor. Therefore, we tested SKI-II in the BALB/c JC tumor model using p.o. dosing at 100 mg/kg every other day and monitored tumor growth and body weight. As shown in Fig. 6, the p.o. administration of SKI-II caused significant antitumor activity (p < 0.05 versus vehicle-treated mice) in well established tumors as early as day 5, with maximal response seen at the end of the study. SKI-II showed 79% inhibition of tumor growth from the start of treatment. As seen in the i.p. dosing study, no weight loss or overt signs of toxicity were observed with SKI-II treatment. At the end of the study, the mice were given a final treatment with SKI-II 1 h before euthanization, and blood and tumors were removed for analysis. The average concentration of SKI-II in the blood was determined to be 0.30 ± 0.29 μM, whereas tumor concentrations were 1.78 ± 0.46 nmol/g tissue. No metabolites were detected in either tissue, indicating that the parent compound is most likely the active agent. These are the first findings indicating that SKI-II is an orally active SK inhibitor.

Antitumor activity of SK inhibitors following i.p. administration. A, mice bearing JC tumors were treated with 50 mg/kg SKI-I (closed squares), SKI-II (closed triangles), SKI-V (closed circles), or vehicle (open squares) i.p. on odd days. Body weights and tumor volumes were measured on indicated days. B, average body weights of each treatment group. Values represent the mean ± S.E.M. for each group (n = 5). *, p ≤ 0.05, **, p ≤ 0.01 for differences from control.
The findings described above led to the determination of a pharmacokinetic profile of a p.o. dose of SKI-II at pharmacologically active concentrations. The results are shown in Fig. 6. At a dose of 100 mg/kg of SKI-II, a maximal concentration of 252 ng/ml (0.8 μM) in blood was achieved at 1 h. This concentration is sufficient to significantly inhibit the proliferation of JC cells (Table 1), indicating that therapeutically relevant levels are attained in vivo. Furthermore, significant levels of SKI-II were detected in the blood at 8 h, providing a favorable half-life (15.3 h) for the compound. These novel pharmacokinetic properties allow extended systemic exposure to the drug and alleviate the need for frequent dosing.
Discussion
Since the critical findings that S1P acts as a second messenger (Olivera and Spiegel, 1993) and inhibits ceramide-induced apoptosis (Cuvillier et al., 1996), the importance of the sphingolipid metabolites S1P and ceramide in tumor cell proliferation and apoptosis has become increasingly apparent. Phosphorylation of sphingosine by SK is the only known mechanism for the production of S1P in cells. Thus, we hypothesize that SK is a critical target in cancer therapeutics. We have shown previously that levels of mRNA encoding SK are approximately 2-fold higher in tumors of the breast, colon, lung, ovary, stomach, uterus, kidney, and rectum compared with normal tissue from the same patient (French et al., 2003). These increases in SK message have been correlated with increases in SK protein levels as determined in a panel of human paired cancer and normal tissue samples (J. Yun, unpublished data). SK1 has also been identified as an oncogene (Xia et al., 2000), capable of transforming 3T3 fibroblasts, which allows them to form tumors in mice. SK has also been linked to estrogen signaling (Sukocheva et al., 2003) and estrogen-dependent tumorigenesis in MCF-7 cells (Nava et al., 2002). Other pathways or targets to which SK activity has been linked in cancer include vascular endothelial growth factor signaling, including the Ras and MAPK pathway (Shu et al., 2002), protein kinase C (Nakade et al., 2003), tumor necrosis factor α (Vann et al., 2002), hepatocyte nuclear factor 1 and retinoic acid receptor α (Osawa et al., 2001b), intracellular calcium (Wheldon et al., 2001), and caspase activation (Edsall et al., 2001). Angiogenic factors and processes, such as cell motility, mitogenesis in smooth muscle cells, endothelial cell differentiation, and growth factor signaling, are also affected by SK and S1P (Lee et al., 1999). These findings, in conjunction with the in vitro findings described above, provide strong evidence that SK is an attractive target for cancer.
Despite the high level of interest in sphingolipid-derived signaling, there are very few established inhibitors of the enzymes of this pathway. In particular, the field suffers from a lack of potent and selective inhibitors of SK. Pharmacological studies to date have used sphingosine analogs, especially d-erythro-N,N-dimethylsphingosine. However, as indicated above, these lipids are known to inhibit several lipid and protein kinases. Therefore, selective and potent inhibitors of SK are required for both basic research and as lead compounds for developing novel anticancer agents. To this end, we initiated a program to identify and evaluate potent and structurally novel inhibitors of SK. We discovered a series of chemotypes that are potent inhibitors of recombinant human SK and that are cytotoxic toward a panel of human cancer cell lines. We showed that SKI-I, -II, and -V inhibited endogenous S1P formation in intact cells while inducing apoptosis. These inhibitors were also selective for SK versus other lipid and protein kinases. This study describes the continued characterization of these novel inhibitors as cancer therapeutics in both cell and animal models.

Antitumor activity of SKI-II following p.o. administration. A, tumor-bearing mice were treated p.o. with SKI-II at 100 mg/kg (open circles) or vehicle (closed circles) every other day starting at day 1. Body weights and tumor volumes were measured on days 1, 5, 9, 15, and 18. B, average body weights of each treatment group. Values represent the mean ± S.D. for each group. *, p ≤ 0.05 for differences from control; n = 4 for each group. C, pharmacokinetic profile of SKI-II after p.o. administration. Female Swiss-Webster mice were treated via p.o. gavage with 100 mg/kg of SKI-II, and blood was removed at the indicated times (n = 3 per time point). The concentrations of SKI-II were determined by HPLC-UV analysis.
We used the syngeneic BALB/c JC solid tumor model to further examine drug accumulation and pharmacodynamic studies. Previously, we have shown that the JC cell line was more sensitive to non-Pgp substrates such as cisplatin than Pgp substrates such as doxorubicin (Lee et al., 2003). Furthermore, in vivo studies revealed weak antitumor activity with doxorubicin treatment alone versus excellent activity with doxorubicin in combination with Pgp modulators cyclosporine A or Pgp-4008. Thus, the JC cell line is responsive to common chemotherapeutic drugs while also showing multi-drug resistance. JC cells were shown to be appropriate for examining SK inhibitor activity for several reasons. First, JC cells show elevated SK activity versus NIH3T3 and MDA-MB-231 cells. Previous studies showed S1P formation inhibition in MDA-MB-231 cells at a single dose (French et al., 2003). The rank order and magnitude of potency were similar versus JC cells. Although S1P formation studies were not performed in 3T3 cells, proliferation studies showed that the nontransformed cells were less sensitive to the inhibitors than other cancer cell lines (data not shown). Second, all of the SK inhibitors tested were cytotoxic to JC cells and inhibited intracellular S1P formation in a dose-response fashion. Finally, JC cells exhibit multidrug resistance and are therefore a stringent system for studying SK inhibitors. All SK inhibitors administered via i.p. injection had antitumor activity against the solid tumor model without any signs of toxicity. The fact that all of the inhibitors showed activity without toxicity reinforces targeting SK in cancer therapy. After the study was concluded, we showed that effective concentrations of each SK inhibitor were present in both the tumor and the circulation. No metabolites were detected, indicating that the parent compounds are responsible for their activity. IC50 values of the SK inhibitors toward decreasing intracellular S1P levels are very similar to the IC50 values for purified human recombinant SK and JC cytotoxicities. Thus, we believe the antiproliferative properties and SK inhibitory properties of the compounds are closely linked, lending strong evidence toward SK1 as an exciting chemotherapy target. In addition, the strong correlation of decreased S1P formation with down-regulation of proliferation and survival pathways at similar exposure times, which has been previously shown to induce apoptosis (French et al., 2003), provides further evidence that the mechanism of inhibition is through SK.
SK is an important target for cancer therapy because its product, S1P, has been linked to numerous proliferative and survival pathways. For example, S1P stimulates proliferative signaling through the MAPK pathway (Carpio et al., 1999; Shu et al., 2002), calcium mobilization (Meyer zu Heringdorf et al., 2003; Villullas et al., 2003), and store-operated calcium entry (Itagaki and Hauser, 2003). S1P also stimulates cell survival pathways, including nuclear factor κB activation (Xia et al., 2002) and the Akt/PI3K pathway (Banno et al., 2001; Osawa et al., 2001a; Yamada et al., 2004). S1P has also been identified as a ligand for the S1P receptor family, a group of G protein-coupled receptors that have multiple downstream effectors (Lee et al., 1998; Hobson et al., 2001; Paik et al., 2001; Mandala et al., 2002). These findings led us to examine whether pharmacological inhibition of SK would result in modulation of downstream proliferative and survival targets of sphingolipid signaling. Modulation of these pathways in vitro using similar concentrations for S1P inhibition had encouraging results. When inhibitor concentrations were normalized to equal S1P formation inhibition, the rank order of modulation for both pathways was SKI-V > SKI-II >> SKI-I. The reason for differences in pathway modulation is unknown. From our previous kinase selectivity assays, it was observed that SKI-V was the most promiscuous, potently inhibiting PI3K (French et al., 2003). Therefore, SKI-V may potently inhibit SK but most likely inhibits other kinases as well. This finding, in conjunction with the fact that SKI-I and SKI-V showed extremely poor oral bioavailability (data not shown), led to the conclusion that this inhibitor may not be ideal for targeted therapeutics. Like SK, it is well known that these pathways are modulated by many different effectors. Further studies need to be done to delineate these mechanisms. Because all three inhibitors had similar antitumor activity, correlation of activity with inhibition of MAPK and Akt pathways would be tenuous at best, leaving open the possibility that the link between SK inhibition and cell death is not through these pathways. It would not be surprising because SK activation occurs through a wide range of stimuli. We observed that antitumor activity of the SK inhibitors correlated with mostly decreased phosphorylation of MEK and Akt. SKI-dependent Akt pathway modulation correlated well between in vivo and in vitro findings, whereas correlation with MAPK pathway activation was more variable. Perhaps intracellular S1P formation has a more direct effect on Akt than MAPK signaling and therefore may serve as a more accurate pharmacodynamic indicator.
Evidence for a link between SK and angiogenesis has also been steadily progressing, specifically with observed associations of SK activity with the proangiogenic factors vascular endothelial growth factor (Shu et al., 2002; Tanimoto et al., 2002; Sanchez et al., 2003), tumor necrosis factor α (Xia et al., 1998), and platelet-derived growth factor (Boguslawski et al., 2002). Thus, it is possible that these SK inhibitors are also having an antiangiogenic effect on the tumor. These links provide an impetus to examine pharmacological inhibition of angiogenesis through these compounds.
In conclusion, SK and S1P are critical in the regulation of tumor cell proliferation and survival, and so represent important targets for the development of new anticancer drugs. We have further characterized several novel low molecular weight compounds that inhibit SK. The identification of multiple SK inhibitors with antitumor activity in vivo further substantiates the hypothesis that SK is an attractive target for new therapeutics. The correlation with pharmacokinetic and pharmacodynamic properties, in addition to oral bioavailability and antitumor activity of SKI-II, provides strong validation toward this approach to cancer therapy.
Footnotes
-
This work was supported by National Institutes of Health Grant R43 CA097833.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.101345.
-
ABBREVIATIONS: S1P, sphingosine-1-phosphate; SK, sphingosine kinase; Pgp, P-glycoprotein; SKI-II, 4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol, CAS 312636-16-1; SKI-I, 5-naphthalen-2-yl-2H-pyrazole-3-carboxylic acid (2-hydroxy-naphthalen-1-ylmethylene)-hydrazide, CAS 306301-68-8; SKI-V, 2-(3,4-dihydroxy-benzylidene)-benzofuran-3-one; MEK, mitogen-activated protein kinase kinase; HPLC, high-performance liquid chromatography; DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline; LY294002, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one; BAY 43-9006, 4-(4-(3-(4-chloro-3-trifluoromethylphenyl)ureido)phenoxy)pyridine-2-carboxyllic acid methyamide-4-methylbenzenesulfonate; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3-kinase.
- Received January 13, 2006.
- Accepted April 20, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}