


Abstract
Capacitative Ca2+ entry (CCE) in vascular smooth muscle cells contributes to vasoconstrictor and mitogenic effects of vasoactive hormones. In A7r5 rat aortic smooth muscle cells, measurements of cytosolic free Ca2+ concentration ([Ca2+]i) have demonstrated that depletion of intracellular Ca2+ stores activates CCE. However, there is disagreement in published studies regarding the regulation of this mechanism by the vasoconstrictor hormone [Arg8]-vasopressin (AVP). We have employed electrophysiological methods to characterize the membrane currents activated by store depletion [store-operated current (ISOC)]. Because of different recording conditions, it has not been previously determined whether ISOC corresponds to CCE measured using fura-2; nor has the channel protein responsible for CCE been identified. In the present study, the pharmacological characteristics of ISOC, including its sensitivity to blockade by 2-aminoethoxydiphenylborane, diethylstilbestrol, or micromolar Gd3+, were found to parallel the effects of these drugs on thapsigargin- or AVP-activated CCE measured under identical external ionic conditions using fura-2. Thapsigargin-stimulated ISOC was also measured in freshly isolated rat mesenteric artery smooth muscle cells (MASMC). Members of the transient receptor potential (TRP) family of nonselective cation channels, TRPC1, TRPC4, and TRPC6, were detected by reverse transcription-polymerase chain reaction and Western blot in both A7r5 cells and MASMC. TRPC1 expression was reduced in a stable A7r5 cell line expressing a small interfering RNA (siRNA) or by infection of A7r5 cells with an adenovirus expressing a TRPC1 antisense nucleotide sequence. Thapsigargin-stimulated ISOC was reduced in both the TRPC1 siRNA- and TRPC1 antisense-expressing cells, suggesting that the TRPC1 channel contributes to the ISOC/CCE pathway.
Vasoconstrictor hormones cause contraction of vascular smooth muscle (VSM) cells by increasing cytosolic free Ca2+ concentration ([Ca2+]i), which in turn activates the contractile apparatus of the cells. A well characterized signal transduction pathway leading to elevation of [Ca2+]i involves inositol trisphosphate-mediated release of Ca2+ from the sarcoplasmic reticulum and Ca2+ entry via nonselective calcium-permeable cation channels. This pathway may also contribute to the mitogenic actions of vasoactive hormones (Wang et al., 2001).
The A7r5 rat aortic smooth muscle cell line is among the most extensively characterized vascular smooth muscle preparations in terms of agonist-stimulated Ca2+ signaling pathways. Using the Ca2+-sensitive fluorescent indicator, fura-2, the vasoconstrictor hormone [Arg8]-vasopressin (AVP) was found to activate both capacitative (also known as store-operated) and noncapacitative Ca2+ entry (CCE and NCCE, respectively) in A7r5 cells (Byron and Taylor, 1995; Broad et al., 1999). Several subsequent studies (Moneer and Taylor, 2002; Moneer et al., 2003, 2005) have utilized fura-2 fluorescence measurements to examine the regulation of these Ca2+ entry pathways by AVP, with some disagreement among the published results (Brueggemann et al., 2005). It remains unclear whether CCE contributes to a sustained elevation of [Ca2+]i while AVP is present.
CCE and NCCE in A7r5 cells probably involve different Ca2+-permeable channels, because they appear to have different permeabilities to Sr2+ and Mn2+ (Byron and Taylor, 1995) and are differentially sensitive to pharmacological blockers, such as 2-aminoethoxydiphenylborane (2-APB), LOE 908, and Gd3+ (Broad et al., 1999; Iwamuro et al., 1999; Moneer and Taylor, 2002). However, the earlier fura-2 fluorescence studies did not characterize the electrophysiological properties of the CCE pathway, and the channel proteins that are the presumed targets of the pharmacological blockers remain unidentified.
A number of earlier electrophysiological studies (Nakajima et al., 1996; Iwasawa et al., 1997; Iwamuro et al., 1999; Jung et al., 2002) failed to detect store-operated currents in A7r5 cells despite evidence from fura-2 fluorescence studies that Ca2+ influx is stimulated following depletion of Ca2+ stores. In a recent study (Brueggemann et al., 2005), we reported conditions under which store-operated currents can be detected in A7r5 cells, but the resulting current differs in its apparent cation selectivity and voltage dependence from ISOC recorded from other vascular smooth muscle preparations. Conditions used for recording isolated currents are generally much different from the ionic conditions used to detect Ca2+ entry using fura-2. These conditions may affect the behavior of the channels and their responses to pharmacological agents. Thus, there is a paucity of information related to the electrophysiological or molecular nature of the CCE pathway in vascular smooth muscle cells and disagreements among different groups regarding the mechanisms regulating its activity.
Molecular candidates for the channels involved in capacitative Ca2+ entry pathways may be emerging from the discovery in the mid-1990s of the Drosophila transient receptor potential (TRP) channels and seven canonical mammalian TRP homolog [TRPC1-TRPC7; reviewed by Montell (2005)]. The primary structure of these proteins indicates that they have six membrane-spanning domains reminiscent of the voltage-gated K+ channel family. In analogy to K+ channels, the TRP channels are believed to form as tetramers of individual TRP homolog protein subunits. The TRPC proteins are ubiquitously expressed in mammalian tissues, with many cell types, including VSM expressing multiple TRPC homologs. When expressed heterologously, they generally form functional channels that exhibit electrophysiological properties of nonselective cation channels.
In the present study, we use electrophysiological techniques to record store-operated currents (ISOC) in A7r5 cells and mesenteric artery smooth muscle cells (MASMC). The pharmacological characteristics of ISOC are compared with CCE recorded with fura-2 under similar ionic conditions. The findings suggest that CCE is active during the sustained phase of AVP Ca2+ signaling. We have also identified several TRPC homologs expressed in A7r5 cells and MASMC. Evidence is presented to support a role of TRPC1 as a component of the store-operated Ca2+ entry pathway.
Materials and Methods
A7r5 Cell Culture. A7r5 cells were cultured as described previously (Byron and Taylor, 1993). Cells were subcultured onto rectangular (9 × 22-mm, number 1½) glass coverslips or plastic tissue culture dishes (Corning, Acton, MA). For fura-2 fluorescence measurements, confluent cell monolayers grown on glass coverslips were used 2 to 5 days after plating. For electrophysiological recordings, cells grown on plastic tissue culture dishes were trypsinized (0.25% trypsin and 1% EDTA) and replated on 12-mm round glass coverslips. Cells were allowed to adhere for approximately 15 min and then transferred to external solution. Cells were then used for patch-clamp recording within 30 min or maintained at 4°C for up to 2 h prior to recording.
Mesenteric Artery Smooth Muscle Cell Isolation. Male Sprague-Dawley rats weighing approximately 300 g were anesthetized with isoflurane (4% by inhalation), and a section of the mesenteric arcade from the small intestine was surgically excised. The rats were then humanely killed by thoracotomy and cardiac extirpation. All experiments were conducted in accordance with the policies of the Public Health Service and the Animal Welfare Act. Ten to 15 segments (6–10 mm in length) of mesenteric artery were quickly dissected free of connective tissue in ice-cold solution containing 145 mM NaCl, 4.7 mM KCl, 1.2 mM NaH2PO4, 1.17 mM MgSO4, 2 mM pyruvic acid, 0.02 mM EDTA, 3 mM MOPS, 0.1 mM CaCl2, and 5 mM d-glucose, with 10 mg/ml bovine serum albumin (BSA). MASMC were isolated essentially as described by Plane et al. (2005). Arteries were placed in an isolation solution: 120 mM NaCl, 25 mM NaHCO3, 4.2 mM KCl, 1.2 mM MgCl2, 0.6 mM KH2PO4, and 11 mM d-glucose, pH 7.4, when bubbled with 5% CO2 and 21% O2 (at room temperature) with BSA (1 mg/ml) and 0.1 mM CaCl2. The arteries were cut into 4- to 5-mm segments, which were then digested in isolation solution containing papain (1.0 mg/ml), dithiothreitol (1 mg/ml), and BSA (1 mg/ml) for 10 min at 37°C. The artery segments were then transferred to isolation solution containing collagenase (0.3 mg/ml collagenase, type H and 0.7 mg/ml collagenase, type F), 0.01 mM CaCl2, dithiothreitol (1 mg/ml), and BSA (1 mg/ml) for 10 min at 37°C. The tissue was then washed 10 times in ice-cold isolation solution without collagenase and triturated gently with polished Pasteur pipettes to separate the individual cells. Freshly isolated cells with characteristic elongated morphology of vascular myocytes were added directly to the chamber and allowed to adhere for 10 to 15 min for patch-clamp electrophysiology or collected for preparation of RNA for RT-PCR (see below).
Whole Cell Voltage Clamp. The whole cell ruptured and perforated (200 μg/ml amphotericin B) patch configurations were used to measure membrane currents under voltage-clamp conditions in single A7r5 cells or MASMC. All experiments were performed at room temperature with continuous perfusion of bath solution. Resistances of patch pipettes were 1.5 to 2.5 MΩ after filling with internal solution. Series resistance was 2 to 4 MΩ in ruptured path experiments and was not compensated.
Solutions for recording ISOC contained 100 mM sodium aspartate (or 100 mM NMDG aspartate as indicated in figure legends), 20 mM calcium aspartate, 1 mM MgCl2, 10 mM HEPES, pH 7.3 (external), 90 mM Cs aspartate, 20 mM CsCl, 4.6 mM MgCl2, 5 mM HEPES, and 10 mM BAPTA-Cs4, pH 7.2 (internal). For divalent cation selectivity experiments, external solutions contained 100 mM NMDG aspartate, 20 mM CaCl2, and 10 mM HEPES, pH 7.3, or 20 mM CaCl2 was replaced with equimolar concentrations of SrCl2, BaCl2, or MnCl2.
Osmolality was adjusted to 265 to 270 mOsm/l for A7r5 cells and 290 mOsm/l for MASMC with d-glucose. Verapamil (10 μM) and 100 μM spermine chloride were added to all bath solutions to block L-type Ca2+ channels and Mg2+-inhibitable currents, respectively, as described previously (Brueggemann et al., 2005). Currents were recorded with a 100-ms voltage ramp protocol (from +85 to –115 mV) from –15-mV holding potential every 10 s; at least 1 min (six time points) of stable current recording was used for current density measurements. Liquid junction potentials were calculated using junction potential calculator provided by pCLAMP8 software and subtracted off-line. Experiments in whole-cell perforated patch configuration were started with series resistance (Rs) below 30 MΩ; cells with an abrupt decrease in Rs were discarded. Recordings of ISOC in ruptured patch configuration were started 30 s after break-in. Voltage-clamp command potentials were generated using an Axopatch 200B amplifier under control of pCLAMP8 software. Whole-cell currents were digitized at 10 kHz, filtered at 5 kHz, and analyzed off-line. Currents were normalized to membrane capacitance. Data are presented as mean ± S.E.M.
A well accepted characteristic of store-operated currents is that they are inhibited by lanthanides, such as Gd3+. CCE in A7r5 cells is also reportedly inhibited by low concentrations of Gd3+ (<5 μM; Broad et al., 1999; Moneer and Taylor, 2002; Moneer et al., 2003, 2005). As described previously (Brueggemann et al., 2005), the Gd3+ sensitivity of ISOC in A7r5 cells was evaluated by the addition of 100 μM GdCl3 to the external solution. We estimate that the final concentration of free Gd3+ is in the 1 to 5 μM range (calculated with WinMaxC software using Gd-aspartate binding constants kindly provided by Chris Patton, Stanford University, Palo Alto, CA). For some experiments, 7 mM GdCl3 was added to provide a free [Gd3+] of approximately 100 μM (also calculated using WinMaxC software).
Thapsigargin was used in several experiments to passively empty intracellular Ca2+ stores. Its effectiveness in this regard has been demonstrated previously using fura-2 fluorescence techniques (Byron and Taylor, 1995). In control experiments, we have confirmed that sustained thapsigargin-activated store-operated currents persist after its removal from the bathing medium and are not different if thapsigargin is continuously present.
[Ca2+]i Measurements with Fura-2. Essentially as described previously (Fan and Byron, 2000), A7r5 cells were grown to confluence on glass coverslips. Coverslips were washed twice with control medium (135 mM NaCl, 5.9 mM KCl, 1.5 mM CaCl2, 1.2 mM MgCl2, 11.5 mM glucose, and 11.6 mM HEPES, pH 7.3) and then incubated in the same medium with 5 μM fura-2-AM, 0.1% bovine serum albumin, and 0.02% Pluronic F127 detergent for 90 min at room temperature (22–25°C) in the dark. The cells were then washed twice and incubated in the dark in control medium (or pretreated with drugs) for 0.5 to 5 h prior to the start of the experiment. All experiments were performed at room temperature. Fura-2 fluorescence (excitation alternating between 340 and 380 nm, emitted fluorescence detected at 509 nm) was measured in cell populations with a PerkinElmer LS50B fluorescence spectrophotometer (PerkinElmer Life and Analytical Sciences, Boston, MA) or a BioTek fluorescence plate reader [used only for 2-APB, Gd3+, and diethylstilbestrol (DES) concentration-response determinations; BioTek Instruments Inc., Winooski, VT]. For the latter, A7r5 cells were plated on 96-well plates, grown to confluence, and loaded with fura-2 as described above. Immediately prior to each recording, the control medium was replaced with 100 μl of nominally Ca2+-free control medium (CaCl2 was omitted) ± varying concentrations of 2-APB, Gd3+, or DES. Two injectors were used, first to inject thapsigargin (±2-APB, Gd3+, or DES in a volume of 100 μl of Ca2+-free medium at time = 60 s) and then CaCl2 (plus thapsigargin ±2-APB, Gd3+, or DES; 100 μl of a 4.5 mM CaCl2 solution to restore external [Ca2+] to 1.5 mM at time 13 min); fura-2 fluorescence was recorded at 5-s intervals. Each concentration of 2-APB, Gd3+, or DES was tested in triplicate in three to four separate experiments. All [Ca2+]i recordings were made in the continuous presence of 10 μM verapamil to eliminate any contributions of voltage-gated L-type Ca2+ channels.
RT-PCR Detection and Quantification of TRPC Homolog Expression. Total RNA was prepared from cultured A7r5 cells or freshly isolated MASMC by the guanidinium method (Chomczynski and Sacchi, 1987). Reverse transcription was performed using 1 μgof RNA and Moloney murine leukemia virus-reverse transcription according to the manufacturer's protocol (Life Technologies Inc., Gaithersburg, MD). One-tenth of the reverse transcription reaction product was used for PCR using Platinum PCR SuperMix (Invitrogen, Carlsbad, CA) and 10 pmol of each primer according to the manufacturer's protocol. Primer pairs for TRPC1-TRPC7 were as published by McDaniel et al. (2001). Negative controls were minus reverse transcriptase, and positive controls were total rat brain RNA (or in the case of TRPC2, total rat testes RNA). For MASMC PCR, individual freshly isolated arterial myocytes (∼500) were selected based on their morphology using a micropipette and placed directly into guanidinium buffer for RNA isolation (Chomczynski and Sacchi, 1987). RNA was divided into eight separate portions and then used in RT-PCR using SuperScript One-Step RT-PCR System (Invitrogen) and primer pairs for TRPC1 through TRPC7. One-tenth of the PCR was run on 0.8% agarose/Tris-buffered EDTA gels. For real-time RT-PCR, RNA was prepared from cultured A7r5 cells using RNeasy Minikit (QIAGEN, Valencia, CA), and reverse transcription was performed using i-Script cDNA synthesis kit (Bio-Rad, Hercules, CA). cRNA was subjected to real-time PCR using Platinum Quantitative PCR SuperMix-UDG w/ROX (Invitrogen) and TaqMan Gene Expression Assays and 18S RNA endogenous controls with a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA). Results were analyzed using RQ Relative Quantitation software (Applied Biosystems).
Western Blot Analysis of TRPC Proteins. Confluent cultures of A7r5 cells or MASMC were lysed in a solution containing 1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, 100 mM NaF, 10 mM sodium pyrophosphate, 1 mM EGTA, 1.5 mM MgCl2, 10% glycerol, 150 mM NaCl, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM Na3VO4, and 50 mM HEPES, pH 7.4. Following centrifugation to remove particulate material, protein concentration in supernatants was determined using a bicinchoninic acid protein assay kit (Pierce Chemical, Rockford, IL), and equal amounts of protein (100 to 400 μg) were separated by SDS-polyacrylamide gel electrophoresis on a 7.5% acrylamide gel. The proteins were then transferred to nitrocellulose and blotted for TRPC1, TRPC4, and/or TRPC6 using commercially available antibodies from Alomone Laboratories (1:500 dilution). In additional experiments (results not shown), duplicate samples blotted using antibodies preadsorbed with immunogenic peptide served as negative controls to confirm correct identification of the bands of interest. Chemiluminescent detection of proteins and densitometric analysis of blots was performed as described previously (Byron and Lucchesi, 2002).
TRPC1 siRNA Construct. A vector containing a neomycin resistance gene and expressing rat TRPC1 siRNA was generously provided by Dr. M. L. Villereal (University of Chicago, Chicago, IL). This construct was previously demonstrated to selectively reduce TRPC1 expression in H19-7 hippocampal neuronal cells (Wu et al., 2004) and human embryonic kidney 293 cells (TRPC3, TRPC4, TRPC6, and TRPC7 levels were not affected; Zagranichnaya et al., 2005). A7r5 cells were transfected with the rat siTRPC1 construct and grown in selection medium containing G418 (500 μg/ml) to establish a stable cell line.
Transfection. Proliferating A7r5 cells were grown in 150-mm cell culture dishes to 50% confluence and transfected the next day with 20 μg of plasmid DNA using Lipofectamine transfection reagent (Invitrogen) according to standard transfection protocol. Forty-eight hours after transfection, 500 μg/ml G418 was added. Parallel cultures of nontransfected A7r5 cells were also treated with G418 to verify its lethality in the absence of the selection marker; these cultures died within 5 days. Surviving transfected cells were maintained in the presence of G418 for several weeks.
TRPC1 Antisense Adenovirus. We constructed an adenoviral vector for expression of antisense TRPC1 DNA and coexpression of green fluorescent protein (AS-TRPC1-GFP). A 1694-base pair portion of the human TrpC1 cDNA (generously provided by Dr. I. Ambudkar, National Institutes of Health, Bethesda, MD) was inserted in the antisense direction into the adenovirus GFP-tagged shuttle vector pAdTrack-cytomegalovirus (generously provided by Dr. B. Vogelstein, Johns Hopkins University, Baltimore, MD). Recombinant virus was generated using the AdEasy system essentially as described by He et al. (1998), using electrocompetent BJ5183-AD-1 cells (Stratagene, La Jolla, CA). A7r5 cells were infected with AS-TRPC1-GFP at a multiplicity of 30:1, and cells were analyzed 4 to 6 days later by electrophysiology and RT-PCR as described above.
Immunohistochemical Detection of TRPC1. A7r5 cells were subcultured on 12-mm round glass coverslips and infected with AS-TRPC1-GFP at a multiplicity of infection of 100. After 5 days, the cells were fixed with 2% paraformaldehyde in phosphate-buffered saline (PBS: 138 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, and 1.2 mM KH2PO4, pH 7.4) for 15 min, washed twice with PBS, and permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, St. Louis, MO) in PBS for 15 min. After washing with 0.1% Triton X-100 in PBS, coverslips were blocked with Image-iT FX Signal Enhancer (Molecular Probes Inc., Eugene, OR) according to the manufacturer's instructions for 30 min. An additional blocking step with 10% goat serum for 2 h was used for staining with anti-TRPC1 (H-105) rabbit polyclonal IgG antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Coverslips were washed three times with 0.1% Triton X-100 in PBS and incubated with anti-TRPC1 antibodies, either generously provided by Dr. M. L. Villereal or commercially available (Santa Cruz Biotechnology, Inc.) at a 1:100 dilution overnight at +4°C. Coverslips incubated without primary antibody were used to control for nonspecific background staining. After three washing steps with 0.1% Triton X-100 in PBS for 15 min each, coverslips were incubated for 2 h at room temperature in the dark with Alexa Fluor 594 goat anti-rabbit secondary antibody (1:400 dilution in 0.1% Triton X-100 plus 10% goat serum in PBS). Three additional washing steps for 15 min with 0.1% Triton X-100 in PBS were performed before mounting coverslips on glass slides using SlowFade Light Antifade Kit (Molecular Probes Inc.). Cell images were acquired using C Imaging System (Compix Inc.) with an Olympus IX71 inverted epifluorescence microscope (10× fluorescent objective) and Simple PCI software (version 5.3.1.). Two images of each field were captured at 490-nm excitation wavelength for GFP fluorescence and at 595-nm excitation wavelength for Alexa Fluor 594 fluorescence, respectively.
Statistics. Data are expressed as means ± S.E.M. Statistical comparisons were performed using SigmaStat 3.1 (Systat Software, Inc.). Differences between two groups were evaluated with Student's paired or unpaired t test as appropriate. Concentration-response relationships were analyzed by one-way repeated measures analysis of variance and Holm-Sidak method for comparisons with controls. Differences were considered to be significant when p < 0.05.
Materials. Rabbit polyclonal anti-TRPC1, TRPC4, and TRPC6 antibodies were from Alomone Laboratories (Jerusalem, Israel). PCR primers were from Integrated DNA Technologies, Inc. (Coralville, IA). Cyclopiazonic acid and 1-oleoyl-2-acetyl-sn-glycerol were from Biomol Research Laboratories, Inc. (Plymouth Meeting, PA). Collagenase types H and F, diethylstilbestrol, gadolinium chloride, flufenamic acid, [Arg8]-vasopressin, verapamil, and thapsigargin were from Sigma-Aldrich. Fura-2-AM, Alexa Fluor 594 anti-rabbit IgG, SlowFade Light Antifade Kit, and Pluronic F127 were from Molecular Probes, Inc. Papain was from Worthington Biochemical Corporation (Lakewood, NJ). Anti-TRPC1 rabbit polyclonal IgG was from Santa Cruz Biotechnology, Inc.
Results
In a previous study (Brueggemann et al., 2005), we identified patch-clamp recording conditions for detection of ISOC in A7r5 cells. Under these recording conditions (perforated patch configuration in 0 Na+ and 20 mM Ca2+ external solution), ISOC is activated rapidly in response to emptying of intracellular Ca2+ stores with 100 nM AVP, reaching a peak within 1.5 min followed by partial inactivation (Fig. 1A). The current declines slowly after washout of AVP. As described previously (Brueggemann et al., 2005), ISOC has an inwardly rectifying current-voltage (I-V) relationship and reversal potential near +40 mV (Fig. 1B). Under the same recording conditions, ISOC was also detected in freshly isolated MASMC (Fig. 1, C and D). Small inwardly rectifying currents were detected in five of seven cells exposed to thapsigargin (1 μM); mean current amplitude at –115 mV was estimated as –0.37 ± 0.14 pA/pF (n = 4).
We reported previously (Brueggemann et al., 2005) that ISOC developed in A7r5 cells as a result of passive emptying of Ca2+ stores with 1 μM thapsigargin (irreversible inhibitor of the sarcoplasmic reticulum Ca2+ ATPase) or by dialyzing cells with 10 mM BAPTA in the ruptured patch configuration. A number of pharmacological agents have been used to characterize store-operated currents or Ca2+ entry pathways in a variety of cell types (reviewed by Parekh and Putney, 2005). We previously demonstrated (Brueggemann et al., 2005) that 100 μM GdCl3 in aspartate-based external solution (approximately 5 μM free Gd3+) completely inhibited thapsigargin-activated ISOC. Here we show that 5 μM free Gd3+ inhibits ISOC in A7r5 cells reversibly. A significant recovery to 88 ± 7% of its original level was recorded after washout of Gd3+ [n = 6, p < 0.001, paired Student's t test (Fig. 2A)]. 2-APB also significantly inhibited Gd3+-sensitive ISOC (by 95 ± 2%, n = 4, p < 0.001, paired Student's t test), and this effect was partially reversed after washout (current returned to 56 ± 17% of its original level, n = 4). At a concentration of 10 μM, inhibition by 2-APB was reproducibly slower than by Gd3+ (Fig. 2B). We also found that DES, a drug recently reported to inhibit capacitative Ca2+ entry in smooth muscle cells (Zakharov et al., 2004), was a very effective inhibitor of ISOC in A7r5 cells (Fig. 2C). DES (10 μM) applied externally for 10 to 20 min significantly inhibited 96 ± 3% (p < 0.001, paired Student's t test) of Gd3+-sensitive ISOC with 93 ± 15% recovery during washout for 15 to 25 min (n = 4; Fig. 2C).

ISOC in A7r5 and mesenteric artery smooth muscle cells. A, representative time course of ISOC development in a A7r5 cell [perforated patch, capacitance (C) = 95.5 pF] during application of 100 nM AVP (5 min, black box) followed by washout of AVP. Vasopressin was applied after stable recording of resting current for 5 min. Current was recorded with a ramp voltage protocol (from +85 to –115 mV) from –15-mV holding potential every 10 s. Inward currents measured at –115 mV (open circles) and outward current measured at +85 mV (filled circles) are presented. Dotted line indicates 0 current level. B, current-voltage relationship of AVP-activated ISOC recorded at time point b on panel A after subtraction of resting current (time point a on panel A). Results are representative of four similar experiments. C, representative time course of ISOC development in a MASMC cell in perforated patch configuration (C = 25.4 pF) during application of 1 μM thapsigargin (10 min, white box). D, current-voltage relationship of thapsigargin-activated ISOC recorded with the ramp voltage protocol [8 ramps recorded at the end of thapsigargin application (time point labeled b on C) was averaged, and mean resting current (at time point a on C) was subtracted]. Results are representative of four similar experiments.
Potential activators of nonselective cation channels were also tested for effects on ISOC. Flufenamate has been used to distinguish among members of the TRP channel family, and diacylglycerol analogs have been reported to activate nonselective cation channels, including store-operated channels in some cases (see below). ISOC activated by passive depletion of Ca2+ stores, either with thapsigargin (n = 3) or with BAPTA (n = 4, Fig. 2D), was partially inhibited by 100 μM flufenamate (80 ± 16% inhibition of thapsigargin-activated ISOC, p < 0.05, and 44 ± 7% inhibition of BAPTA-activated ISOC, p < 0.05, paired Student's t test). Inhibition of thapsigargin-activated ISOC by flufenamate was not significantly different from inhibition of BAPTA-activated ISOC by flufenamate (p > 0.05, Student's t test). ISOC was not activated by the soluble diacylglycerol analog 1-oleoyl-2-acetyl-sn-glycerol (OAG; 100 μM, n = 4) in perforated patch recordings (Fig. 2E).
We previously found ISOC to be highly selective for Ca2+ over Na+ (Brueggemann et al., 2005). In the present study, the selectivity of ISOC was evaluated for Ca2+, Ba2+, Sr2+ and Mn2+, divalent cations commonly used for fura-2 fluorescence measurements of CCE. In response to equimolar substitution of 20 mM Ba2+, Sr2+, or Mn2+ for Ca2+ in NMDG-containing external solution, the ISOC amplitudes measured at –115 mV were in the following order relative to Ca2+: Sr2+ (1.7 ± 0.2, n = 6) > Ba2+ (1.3 ± 0.1, n = 5) ≥ Ca2+(1) ≫ Mn2+ (0.13 ± 0.10, n = 4) (Fig. 3). The mean amplitude of the current in 20 mM Ca2+ was 0.35 ± 0.07 pA/pF (n = 15).
Fura-2 Fluorescence Studies. To further investigate whether ISOC measured by whole-cell patch clamp corresponds to the CCE pathway described previously (Byron and Taylor, 1995; Broad et al., 1999; Moneer and Taylor, 2002; Moneer et al., 2003, 2005; Brueggemann et al., 2005), we used fura-2 fluorescence techniques to compare the pharmacological properties of the Ca2+ entry pathway under the same ionic conditions used for ISOC measurements.
In A7r5 cells pretreated with 1 μM thapsigargin for 30 to 60 min to deplete intracellular Ca2+ stores and then using the same external solutions used for electrophysiological detection of ISOC, we found that CCE was reversibly blocked by 2-APB (10 μM; Fig. 4A). The decline in [Ca2+]i was reproducibly slower and to a slightly lesser extent than that induced by Gd3+, mimicking the pattern of inhibition of ISOC by these same treatments (Fig. 2B). In several experiments, a transient increase in [Ca2+]i preceded the decline in [Ca2+]i upon the addition of 2-APB (for examples see Figs. 5B and 6A, representative of five of 13 experiments). A similar pattern was occasionally observed during measurements of 2-APB effects on ISOC (data not shown). The concentration dependence for 2-APB was evaluated in cells exposed to thapsigargin in Ca2+-free medium followed by re-exposure to external Ca2+ (Fig. 4, B and C). 2-APB had no effect on basal [Ca2+]i or release of intracellular Ca2+, but it inhibited the sustained phase of the [Ca2+]i response after re-exposure to external Ca2+ with an IC50 of ≈2.2 μM (Fig. 4C).

Pharmacology of ISOC. A, reversibility of GdCl3 block. Representative time course of ISOC activation by 1 μM thapsigargin (15 min) recorded in a single A7r5 cell (C = 269.5 pF) with the perforated patch configuration is described under Materials and Methods and measured at –115 mV. Drug applications are indicated by boxes. External solution contained 100 mM Na+ and 20 mM Ca2+; 100 μM GdCl3 (∼5 μM free Gd3+) was applied for 5 min followed by washout. Dotted lines indicate 0 current level. Similar results were obtained in six experiments. B, 2-APB reversibly inhibited ISOC. Application of 10 μM 2-APB for 10 min reversibly inhibited ISOC activated with 1 μM thapsigargin (20 min) in perforated patch configuration (C = 467.5 pF, representative of four similar experiments) followed by washout and reversible inhibition of current by 100 μM GdCl3. C, reversible inhibition of ISOC by DES. Thapsigargin-activated ISOC (1 μM thapsigargin for 15 min in perforated patch recording, C = 177.2 pF, representative of four similar experiments) was reversibly blocked by external application of 10 μM DES for 10 min. D, inhibition of ISOC by flufenamic acid. Representative time course of ISOC activation by dialyzing a cell (C = 195 pF) with 10 mM BAPTA in ruptured patch configuration (external solution contained 100 mM NMDG+ and 20 mM Ca2+) followed by application of 100 μM flufenamate. Similar results were obtained in four experiments with BAPTA dialysis and in three experiments when thapsigargin was used to activate ISOC. E, OAG did not activate ISOC. No measurable inward current developed at –115 mV in perforated patch recording upon application of 100 μM OAG for 10 min in external solution containing 100 mM NMDG+ and 20 mM Ca2+ (C = 186 pF, representative of four similar experiments).

Divalent selectivity of ISOC. A and B, representative time courses of ISOC development at –115 mV (ruptured patch configuration) recorded in 20 mM CaCl2 followed by replacement of CaCl2 with an equimolar concentration of SrCl2 (A, C = 102 pF) or MnCl2 (B, C = 114 pF). C, relative current densities of Gd3+-sensitive ISOC measured at –115 mV in 20 mM CaCl2, 20 mM SrCl2 (n = 6), 20 mM BaCl2 (n = 5), and 20 mM MnCl2 (n = 4) normalized by current densities in CaCl2 as described under Materials and Methods. Data are presented as mean ± S.E. *, p < 0.05 versus current densities in CaCl2.
Gd3+ (approximately 5 μM free Gd3+ in the aspartate-based solution; see Materials and Methods) inhibited CCE reversibly (Fig. 4A). The concentration dependence for inhibition of thapsigargin-stimulated CCE by Gd3+ is shown in Fig. 5A (IC50 ≈600 nM). The effects of Gd3+ on CCE were previously reported to be irreversible (Broad et al., 1999). The latter studies utilized a chloride-based external solution for recording fura-2 fluorescence. When we tested the effects of 2.5 μMGd3+ in a chloride-based external solution (control medium used for loading cells with fura-2; see Materials and Methods), we also found that its effects on both ISOC (data not shown) and CCE were very poorly reversible (Fig. 5B). Reversibility of Gd3+ block in the aspartate-based solution may be due to chelation of Gd3+ by aspartate.
DES is an estrogen analog, which has been recently reported to potently block CCE in vascular smooth muscle cells (Zakharov et al., 2004). DES also effectively inhibited CCE in A7r5 cells (Fig. 6A), with an IC50 of ≈800 nM (Fig. 6B).
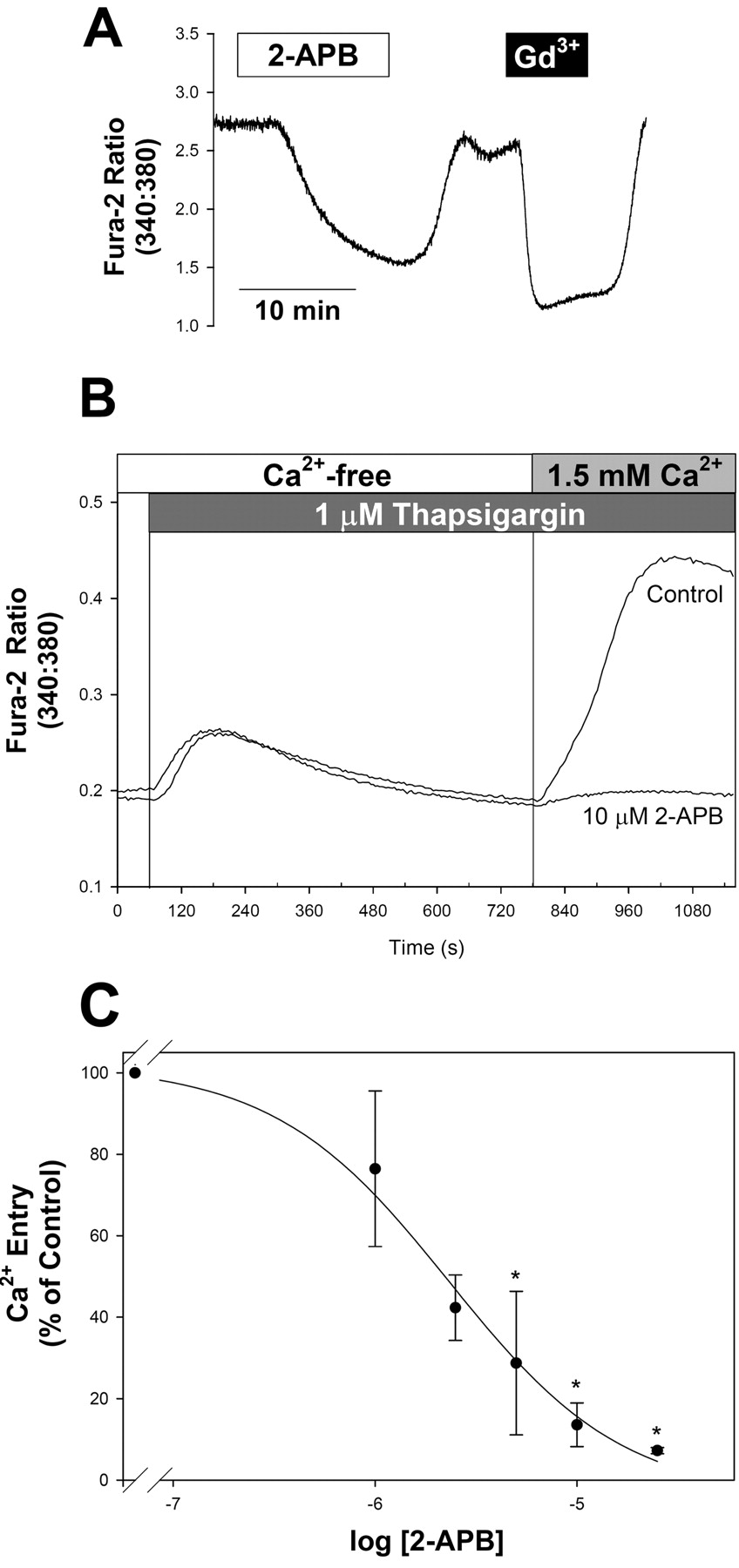
Inhibition of CCE by 2-APB. A, reversible inhibition of CCE by 2-APB and Gd3+. Fura-2-loaded A7r5 cells were pretreated with 1 μM thapsigargin and exposed to the same external solution used for recording ISOC (see Materials and Methods) until the fluorescence ratio stabilized (approximately 40 min). 2-APB (10 μM) was added as indicated by the white box, followed by washout of 2-APB and application of Gd3+ (black box; 100 μM total Gd3+ to give a free concentration of approximately 5 μM). Results are representative of 13 similar experiments. B, the concentration dependence for 2-APB inhibition of CCE was evaluated in A7r5 cells exposed to thapsigargin (1 μM) in Ca2+-free medium followed by readdition of extracellular Ca2+ (final concentration, 1.5 mM). Traces are representative of four experiments performed in triplicate and demonstrate control responses (in the absence of 2-APB) and responses recorded with 10 μM 2-APB in all solutions. C, concentration dependence for 2-APB inhibition of Ca2+ entry. Ca2+ entry was estimated by calculating the maximal change in fura-2 fluorescence ratio on the addition of extracellular Ca2+. Results from four experiments, presented as mean ± S.E., are normalized to the control responses in the absence of 2-APB recorded from cells on the same 96-well plate. *, p < 0.05 versus control.
There is some controversy related to the contributions of different Ca2+ entry pathways to AVP-activated Ca2+ signals (Brueggemann et al., 2005). Therefore, we evaluated the effects of various ISOC/CCE inhibitors on the sustained phase of the Ca2+ response to 1 nM AVP under the same external ionic conditions used for recording ISOC. As shown in Fig. 7, 2-APB, Gd3+, and DES were similarly effective in reducing the AVP-stimulated Ca2+ signals as they were for CCE activated by thapsigargin pretreatment (Fig. 6A). These results suggest a substantial contribution of CCE to the AVP-induced Ca2+ response.

Inhibition of CCE by Gd3+. A, concentration dependence for inhibition of CCE by Gd3+ was evaluated as for 2-APB (see legend for Fig. 4). Data from four experiments performed in triplicate are presented as mean ± S.E. *, p < 0.05 versus control. B, in chloride-based external solution (control medium; see Materials and Methods), the steady-state Ca2+ plateau in thapsigargin-pretreated cells was reversibly inhibited by 2-APB (10 μM, white box), whereas the inhibition by Gd3+ (2.5 μM, black box) was poorly reversed on washout of Gd3+. Results are representative of four similar experiments.
The decrease in the sustained phase of the AVP-induced Ca2+ signal by DES was transient. In the continued presence of DES, [Ca2+]i gradually rebounded to greater than its initial level (Fig. 7A). This did not appear to be due to transient block of CCE, because the same effect was observed when DES was applied in the presence of Gd3+ at a concentration (5 μM free Gd3+) that stably blocks both ISOC and CCE (Fig. 7B). Increasing external Gd3+ concentration to achieve a free concentration of approximately 100 μM (7 mM total GdCl3) was also ineffective in preventing the DES-induced increase in [Ca2+]i (Fig. 7C). A similar slow rebound of [Ca2+]i was apparent when DES was added to thapsigargin-pretreated cells in the presence of Gd3+ (data not shown).
TRP Channel Expression. The identities of the channels responsible for nonselective cation currents in A7r5 cells may be related to expression of TRPC homologs. Using RT-PCR, we have detected TRPC1, TRPC4, and TRPC6 mRNA in A7r5 cells. As shown in Fig. 8A, total RNA prepared from A7r5 cells (top), freshly isolated rat MASMC (middle), and adult rat brain/testis (bottom) was reverse-transcribed and subjected to PCR using primers specific for TRPC1 through TRPC7 (lanes 1–7). Using rat testis as a positive control for TRPC2 or rat brain as a positive control for the other TRPC homologs, TRPC2, TRPC3, TRPC5, and TRPC7 were undetectable in A7r5 cells, whereas TRPC3 and TRPC5 were faintly detected in MASMC. Western blot analysis using commercially available anti-TRPC antibodies (Fig. 8B) revealed that TRPC1, TRPC4, and TRPC6 proteins are detected in both A7r5 cell and MASMC lysates.

Inhibition of CCE by DES. A, effects of DES on CCE were evaluated in fura-2-loaded A7r5 cells pretreated with thapsigargin in external solution used for recording ISOC. A representative recoding is shown demonstrating sequential inhibition of CCE by Gd3+ (5 μM, black box), 2-APB (10 μM, white box), and DES (10 μM, gray box). Similar results were obtained in five experiments. B, concentration dependence for inhibition of thapsigargin-stimulated CCE by DES was determined as that for 2-APB and Gd3+ (see legend for Fig. 4). Data from three experiments performed in triplicate are presented as mean ± S.E. *, p < 0.05 versus control.
TRPC1 has previously been implicated in store-operated Ca2+ entry in vascular smooth muscle cells (Golovina et al., 2001; Xu and Beech, 2001; Sweeney et al., 2002). We used an siRNA strategy previously demonstrated by Wu et al. (2004) to selectively depress TRPC1 expression in H19-7 hippocampal neuronal cells. In A7r5 cells selected with G418 for stable expression of the TRPC1 siRNA construct (see Materials and Methods), expression of TRPC1 mRNA was significantly decreased (by 42 ± 4%, n = 3, p = 0.01, Student's t test; Fig. 9A), and TRPC1 protein levels based on Western blot analysis were significantly reduced (by 62 ± 5%, p < 0.001, n = 6, Student's paired t test compared with control cells maintained in parallel cultures without G418). In three experiments, Western blotting for all three TRPC homologs (TRPC1, TRPC4, and TRPC6; Fig. 9B) revealed no significant reductions in TRPC6 or TRPC4 expression (reductions of 14 ± 27 and 14 ± 13%, respectively, compared with control cultures). In the same samples, there was a consistent and significant reduction in TRPC1 protein expression (56 ± 6% less than controls, p < 0.05). ISOC was also significantly decreased (by 51 ± 6%, n = 8, p < 0.001, Student's t test; Fig. 9C). Likewise, A7r5 cells infected with an adenovirus to express AS-TRPC1-GFP exhibited significantly depressed ISOC compared with non-GFP-expressing cells in the same cultures (Fig. 9, D–F). Infected A7r5 cells (20–30%) for 4 to 6 days with AS-TRPC1-GFP (30 m.o.i.) exhibited green fluorescence (Fig. 9D). TRPC1 immunostaining (detected using either a commercial anti-TRPC1 antibody or anti-TRPC1 antibodies obtained from Dr. M. L. Villereal; see Materials and Methods) was reduced in the GFP-positive cells (Fig. 9D), and ISOC was significantly reduced (by 44%) in cells with GFP fluorescence versus nonfluorescent cells (n = 15 and 16, respectively, p < 0.001, Student's t test; Fig. 9, E and F).
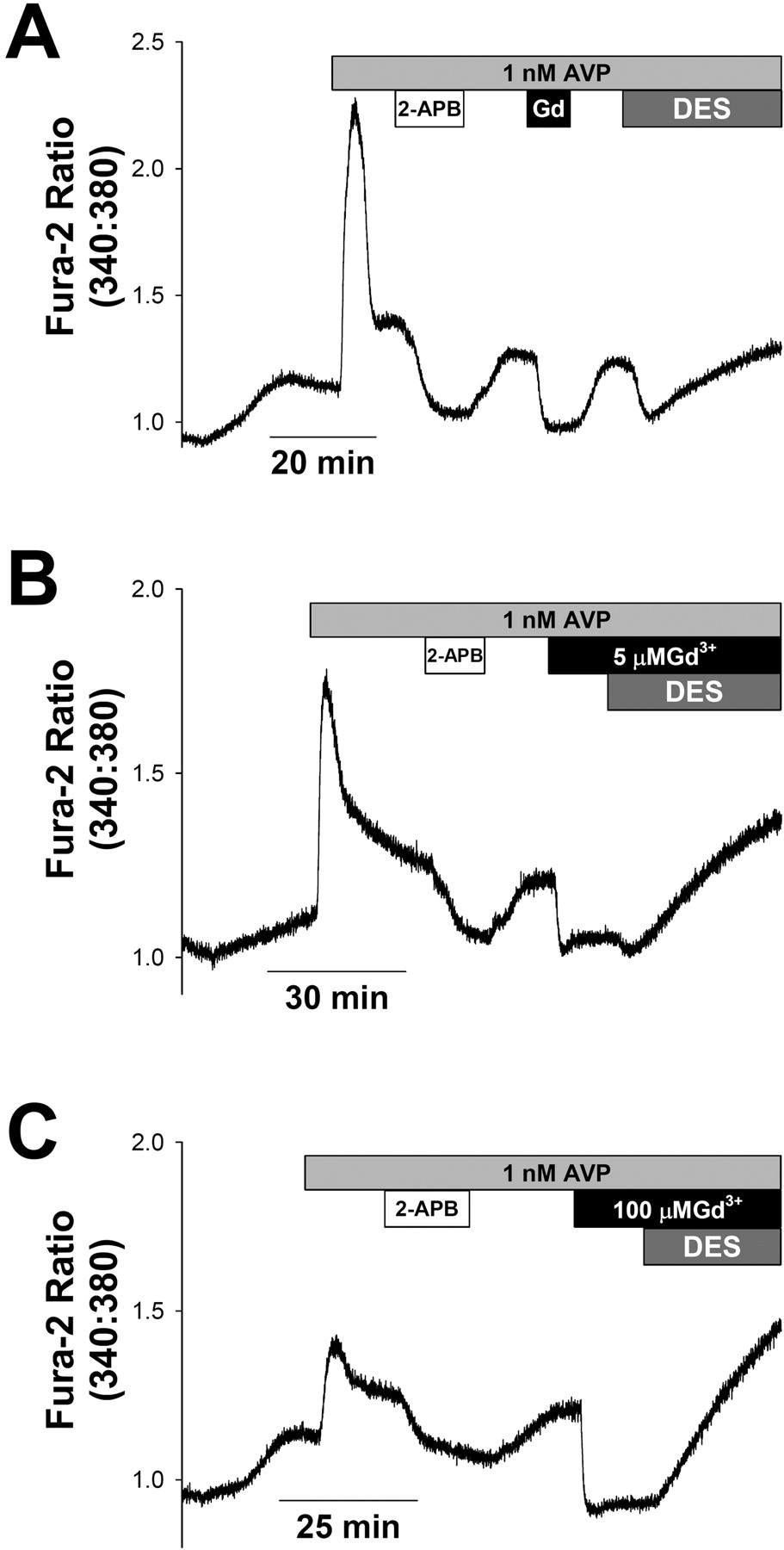
AVP-induced Ca2+ entry: inhibition by 2-APB, Gd3+, and DES. A7r5 cells were loaded with fura-2 in control medium (see Materials and Methods). At the start of recording, the cells were exposed to the external solution used for recording ISOC. All subsequent recordings were in this solution. A, after [Ca2+]i stabilized in the external solution, the cells were exposed to 1 nM AVP (light gray box). 2-APB (10 μM, white box), Gd3+, (5 μM, black box), and DES (10 μM, dark gray box) were applied sequentially. Responses are representative of six similar experiments. Note that, after inhibition of the Ca2+ response by DES, the Ca2+ signal begins to rebound in the continued presence of DES. B, application of DES in the presence of low [Gd3+] (5 μM) does not prevent the rebound of [Ca2+]i. Trace is representative of three similar experiments. C, a similar rebound of [Ca2+]i is observed on application of DES in the presence of high [Gd3+] (100 μM) (representative of two similar experiments).
Discussion
Our electrophysiological and pharmacological studies suggest that ISOC, activated either by AVP or by passive depletion of Ca2+ stores, corresponds to CCE in A7r5 cells. ISOC is a relatively Ca2+-selective current, which is insensitive to LOE 908 but blocked by micromolar Gd3+ (Brueggemann et al., 2005). These properties are consistent with the CCE pathway previously measured in A7r5 cells using fura-2 fluorescence techniques (Broad et al., 1999; Moneer and Taylor, 2002) or in cerebral arteriolar myocytes using fura-PE3 or fluo-4 fluorescence (Flemming et al., 2003). Additional pharmacological agents 2-APB and DES, which have been used as selective blockers of store-operated currents or Ca2+ entry in A7r5 or other cell systems, were also effective blockers of both ISOC and CCE measured under identical external ionic conditions in A7r5 cells.

TRPC homolog expression in A7r5 and MASMC. A, total RNA prepared from A7r5 cells (top), freshly isolated MASMC (middle), and adult rat brain (bottom) was reverse-transcribed and subjected to PCR using primers specific for TRPC1 through TRPC7 (lanes 1–7). Molecular weight marker (M) is a 100-base pair ladder, and 800 base pair is indicated by the arrowhead on the left. B, Western blot analysis of TRPC1, TRPC4, and TRPC6 protein expression in A7r5 cells and cultured MASMC.
ISOC in A7r5 Cells Compared with Other VSMC. ISOC recorded in A7r5 cells displays inward rectification of its I-V relationship in high-external Ca2+ and a positive reversal potential (Fig. 1B) like that recorded in choroidal arteriolar myocytes (Curtis and Scholfield, 2001) but unlike the linear I-V relationship and 0-mV reversal of thapsigargin-activated ISOC from mouse or rabbit aortic smooth muscle cells (Trepakova et al., 2001) or pulmonary artery myocytes (Golovina et al., 2001; McDaniel et al., 2001). Differences in I-V relationships may be attributable to different natures of ISOC from different VSM preparations or to different recording conditions used by different groups. Ca2+-selective and nonselective store-operated channels may even exist within the same cells (Bolotina, 2004; Vanden Abeele et al., 2004; Bugaj et al., 2005), although in A7r5 cells, we find that depletion of Ca2+ stores activates only an inwardly rectifying, relatively Ca2+-selective current measured by whole-cell voltage clamp (Brueggemann et al., 2005). We have also detected a similar current in freshly isolated mesenteric artery smooth muscle cells using the same recording conditions (Fig. 1C).

Knockdown of TRPC1 expression and ISOC in A7r5. A, level of TRPC1 mRNA measured with real-time PCR as described under Materials and Methods was significantly decreased by 42 ± 4% (white bar, n = 3, *, p < 0.05 versus control, paired Student's t test in A7r5 cells stably expressing TRPC1 siRNA), compared with control cells maintained in parallel cultures (black bar). B, representative Western blot of TRPC expression in control or TRPC1 siRNA stable cell line. A single blot was probed simultaneously with antibodies for TRPC1, TRPC4, and TRPC6. On average, the density of the TRPC1 band was reduced by 56 ± 6% in the TRPC1 siRNA stable cell line compared with controls (p < 0.05, paired Student's t test, n = 3). The TRPC4 and TRPC6 bands were not significantly different. C, current densities of Gd3+-sensitive ISOC measured at –115 mV with ruptured patch configuration in control-untransfected cells (–0.49 ± 0.06 pA/pF, n = 8, black bar) and in A7r5 cells stably transfected with TRPC1 siRNA (–0.22 ± 0.02 pA/pF, n = 8, white bar). *, p < 0.001 versus control. D, immunostaining for TRPC1 (left) or GFP fluorescence (right) in A7r5 cells infected with AS-TRPC1-GFP adenovirus (100 m.o.i., 5 days). Two different anti-TRPC1 antibodies were used (I, Villereal antibody; III, Santa Cruz antibody) to demonstrate that TRPC1 immunoreactivity was reduced in GFP-positive cells (II and IV, cells outlined in red) compared with non-GFP-expressing cells in the same field. E, representative time courses of ISOC development in A7r5 cells treated with AS-TRPC1-GFP adenovirus (30 m.o.i., 4–6 days). Currents were measured at –115 mV with ruptured patch configuration in nonfluorescent cells (filled circles), and fluorescent cells expressing TRPC1 antisense (open circles). F, summary of current densities of Gd3+-sensitive ISOC in control (nonfluorescent) cells (–0.55 ± 0.04 pA/pF, n = 16, black bar) and fluorescent cells expressing TRPC1 antisense (–0.31 ± 0.04 pA/pF, n = 15, white bar). *, p < 0.001 versus control.
In rabbit portal vein myocytes, a moderately Ca2+-selective ISOC (Helliwell and Large, 1997; Albert and Large, 2002a,b) is activated by OAG, and pharmacological studies have implicated protein kinase C as an essential step in its activation by either passive store depletion or by noradrenaline (Albert and Large, 2002a). In contrast, ISOC in A7r5 cells is neither activated by OAG (Fig. 2E), nor do protein kinase C inhibitors prevent store-operated Ca2+ entry (Broad et al., 1999), suggesting that distinct channels or regulatory mechanisms may be involved.
ISOC Identification and Relationship to CCE. Considering that currents identified as “store-operated” in various vascular smooth muscle preparations vary significantly in their electrophysiological characteristics, it is important to consider the basis for their identification as store-operated currents. In A7r5 cells, ISOC is activated by both physiological and pharmacological treatments that lead to store depletion and its activation is reversed under conditions in which the stores are allowed to refill (Brueggemann et al., 2005). Furthermore, pharmacological agents that distinguish store-operated from nonstore-operated divalent cation influx measured with fura-2 similarly distinguish ISOC from other cation currents. Few other studies have met these criteria using vascular smooth muscle cells.
In A7r5 cells, CCE activation has been demonstrated for both passive and active depletion of Ca2+ stores using fura-2 fluorescence to examine Ca2+, Ba2+, Mn2+, and Sr2+ influx (Byron and Taylor, 1995). Its activation is reversed by refilling of Ca2+ stores (Byron and Taylor, 1995; Brueggemann et al., 2005). Cyclopiazonic acid (CPA) is a reversible inhibitor of the sarcoplasmic reticulum Ca2+-ATPase, and we have shown that ISOC in A7r5 cells is activated by CPA and deactivates after washout of CPA (Brueggemann et al., 2005). Other studies have used CPA to activate ISOC in vascular myocytes but failed to demonstrate that this effect reverses upon removal of CPA (Ng and Gurney, 2001; Albert and Large, 2002a,b; Sweeney et al., 2002; Bergdahl et al., 2005).
In some cases, pharmacological inhibition of CPA- or thapsigargin-stimulated Ca2+ entry characterized using fluorescent indicators (Flemming et al., 2003) does not match the effects of the same agents on store-operated currents in cells from the same tissue (Bergdahl et al., 2005). In other cases (e.g., McDaniel et al., 2001; Ng and Gurney, 2001), CCE and inward current were similarly inhibited by agents that may not distinguish the store-operated influx pathway from other cation entry pathways that may be active in the vascular myocytes.
Physiological Activation of ISOC. It is generally accepted that Ca2+ influx via the store-operated pathway may contribute to the refilling of intracellular stores following inositol 1,4,5-trisphosphate-mediated Ca2+ release. Therefore, it is important to confirm that ISOC is induced by agonists that activate phospholipase C and that this current corresponds to currents activated by other treatments that deplete intracellular Ca2+ stores.
In rabbit portal vein myocytes, noradrenaline was found to activate a current with properties similar to a CPA-activated current (Albert and Large, 2002b), but the current was activated under conditions that were not consistent with store depletion, and the authors concluded that the effects of noradrenaline were store-independent. In pulmonary artery myocytes, phenylephrine and CPA activated similar currents (McDaniel et al., 2001). However, the conditions used by McDaniel et al. (2001) for recording CPA- and phenylephrine-activated currents would favor development of magnesium-inhibited currents (IMIC), which, as noted by Bakowski and Parekh (2002), had likely been a contaminant of store-operated currents in some earlier studies.
DES has been recently described as an effective inhibitor of store-operated currents and CCE at concentrations that have no effect on IMIC mediated by TRPM7 (Zakharov et al., 2004). Our results suggest a somewhat less potent inhibitory effect of DES on CCE in A7r5 cells and indicate that, at concentrations that maximally inhibit CCE (10 μM), DES has additional effects on Ca2+ transport that may complicate its use as a CCE inhibitor. DES was also previously reported to inhibit L-type Ca2+ currents in A7r5 cells with an IC50 < 10 μM, and at a concentration of 30 μM, DES inhibited AVP-activated nonselective cation currents and K+ currents (Nakajima et al., 1995), providing additional evidence that the effects of DES are not particularly selective.
ISOC, ICAT, and AVP-Stimulated Ca2+ Entry in A7r5 Cells. In A7r5 cells, AVP was previously found to activate both CCE and NCCE (Byron and Taylor, 1995; Broad et al., 1999). Previous electrophysiological studies identified a nonselective cation current (ICAT) in A7r5 cells activated by high [AVP] (100 nM; Van Renterghem et al., 1988; Krautwurst et al., 1994; Nakajima et al., 1996; Iwasawa et al., 1997; Jung et al., 2002), which may relate to AVP-activated NCCE. ICAT is clearly distinct from ISOC in several ways. 1) OAG was reported to directly activate ICAT in A7r5 cells (Jung et al., 2002), but OAG did not activate ISOC (Fig. 2C). 2) ICAT activated by 100 nM AVP in A7r5 cells was inhibited by LOE 908 (Krautwurst et al., 1994), but LOE 908 did not inhibit ISOC (Brueggemann et al., 2005). 3) AVP-activated ICAT is enhanced by flufenamate (Jung et al., 2002), whereas ISOC is inhibited by flufenamate (Fig. 2B). 4) ICAT is a nonselective cation current (Iwasawa et al., 1997; Jung et al., 2002), whereas ISOC appears to be highly selective for Ca2+ over Na+ (Brueggemann et al., 2005).
Using fura-2 fluorescence to examine divalent cation entry pathways in A7r5 cells, Byron and Taylor (1995) found that both thapsigargin- and AVP-stimulated Ca2+ entries were inhibited by other divalent cations (Be2+ > Zn2+ > Ni2+ > Sr2+), with the same relative effectiveness. They concluded that either CCE was the predominant contributor to the sustained phase of the AVP-activated Ca2+ response or both CCE and NCCE have similar sensitivities to these cation blockers. In the present study, we have found that both thapsigargin- and AVP-stimulated Ca2+ entries are inhibited similarly by pharmacological agents that are reportedly selective for the store-operated pathway, leading us to conclude that CCE contributes substantially to the sustained phase of the AVP-induced Ca2+ response. These findings are consistent with our earlier results (Brueggemann et al., 2005) but directly challenge the model put forward by Moneer and Taylor (2002), which suggests that AVP inhibits CCE in A7r5 cells.
Divalent Cation Selectivity: Electrophysiology (ISOC) versus Fura-2 Fluorescence (CCE). Initial studies of the multiple divalent cation entry pathways in A7r5 cells using fura-2 fluorescence suggested that the CCE pathway was relatively less permeable to Sr2+ but more permeable to Mn2+ compared with the NCCE pathway (Byron and Taylor, 1995). More recent publications have extended the original interpretation to assert that the CCE pathway is impermeable to Sr2+ and that the NCCE pathway is impermeable to Mn2+ (Moneer and Taylor, 2002; Moneer et al., 2005). However, fura-2 fluorescence cannot be used to assess absolute conductance. A poorly detectable Sr2+ signal compared with a large Ca2+ signal when CCE is active [e.g., Fig. 2 of Byron and Taylor (1995)] may reflect the large differences in affinities of Sr2+ and Ca2+ for fura-2 (Schilling et al., 1989; Kwan and Putney, 1990). Our ionic selectivity measurements for ISOC suggest that this pathway conducts Sr2+ at least as well as it does Ca2+ (Fig. 3, A and C). Mn2+ currents, on the other hand, were barely detectable by whole-cell patch clamp with 20 mM external Mn2+ (Fig. 3, B and C), whereas Mn2+ entry is readily detected by fura-2 fluorescence, even with 200-fold less Mn2+ in the external medium (Byron and Taylor, 1995). The greater sensitivity of fura-2 compared with electrophysiology may be attributable to the very high affinity of fura-2 for Mn2+ (KD = 8.64 nM; Kwan and Putney, 1990). These differences in sensitivity unfortunately prevent conducting fura-2 Mn2+ quench and electrophysiological measurements of ISOC under the same ionic conditions.
TRPC Channels. Several TRPC homologs are expressed in A7r5 cells. Jung et al. (2002) detected TRPC1 and TRPC6 by Northern blot. Our own results using RT-PCR and Western blotting (Fig. 8) confirm that finding and identify TRPC4 as another homolog expressed in this cell line and in MASMC. A recent study by Moneer et al. (2005) has used quantitative RT-PCR to evaluate TRPC expression in several strains of A7r5 cells and reported expression of TRPC1 and TRPC6, with less expression of TRPC2, TRPC3, TRPC4, and TRPC5; TRPC7 expression was undetectable.
Given the potential for heterotetrameric assembly of TRPC homologs (Hofmann et al., 2002; Strübing et al., 2003), we should be cautious in assigning a particular current or Ca2+ entry pathway to a particular TRPC homolog. Nonetheless, previous studies have suggested that TRPC1 may be involved in capacitative Ca2+ entry in VSM (Golovina et al., 2001; Xu and Beech, 2001; Sweeney et al., 2002; Bergdahl et al., 2005). TRPC1 has also been reported to form heterotetramers with TRPC4 (Hofmann et al., 2002), and studies in vascular endothelial cells [reviewed by Cioffi et al. (2003)] suggest that TRPC4 may be required for normal targeting of TRPC1 to the plasma membrane and formation of functional store-operated channels. Interestingly, store-operated currents recorded from those cells exhibit inward rectification and Ca2+ selectivity similar to ISOC recorded in A7r5 cells (Cioffi et al., 2003). Our results using siRNA or antisense constructs to reduce expression of TRPC1 (Fig. 9) support a role of TRPC1 in store-operated Ca2+ entry in A7r5 cells.
Enhancement of an AVP-stimulated nonselective ICAT by flufenamate, its activation by OAG (Hofmann et al., 1999), and evidence of TRPC6 mRNA expression have been cited as evidence that TRPC6 proteins may form the channels through which this current passes in A7r5 cells (Jung et al., 2002). The differences in pharmacological profiles (activation by flufenamate and OAG) and cation selectivity suggest that TRPC6 may form channels that are distinct from those that mediate ISOC/CCE in A7r5 cells. Nonetheless, it seems likely that TRPC6 mediates ICAT and contributes to NCCE in A7r5 and other vascular smooth muscle cells (Inoue et al., 2001). These results emphasize the importance of establishing electrophysiological recording conditions and pharmacological profiles that distinguish among different agonist-stimulated Ca2+ entry pathways in vascular smooth muscle cells.
Potential Functional Significance of Ca2+-Permeable Cation Channel Activation by AVP. It is increasingly apparent that Ca2+-permeable cation channels may be important targets for development of new therapeutic agents for the treatment of cardiovascular diseases (Stevens, 2001). Store-operated Ca2+ currents were recently found to be correlated with TRPC1 expression in cerebral arteries, and store depletion-induced contractile responses were inhibited by anti-TRPC1 antibodies (Bergdahl et al., 2005). In the same study, TRPC1 expression was found to be up-regulated in human internal mammary arteries after dilation by a balloon used for angioplasty. Several studies have suggested a role for TRPC1 in store-operated Ca2+ entry and growth of vascular smooth muscle cells (Golovina et al., 2001; Xu and Beech, 2001; Sweeney et al., 2002). Plasticity of TRPC expression and the resulting alterations of cation permeability may relate to pathological conditions, such as the proliferation of vascular smooth muscle cells that contributes to restenosis following angioplasty. Pharmacological, electrophysiological, and molecular characterization of store-operated or receptor-activated nonselective cation channels may facilitate the development of more effective therapeutic regimens for the prevention or reversal of cardiovascular diseases.
Acknowledgments
We thank Patrycja Galazka, John Barakat, and Beverly Martin for technical assistance.
Footnotes
-
This work was supported by the John and Marian Falk Trust for Medical Research and the National Heart, Lung, and Blood Institute, National Institutes of Health Grant R01HL70670 (to K.L.B.).
-
A preliminary account of this work has been presented and accepted for publication as an abstract: Brueggemann LI, Markun DR, Cribbs LL, and Byron KL (2006) Electrophysiological and pharmacological characterization of store-operated currents and capacitative Ca2+ entry in rat vascular smooth muscle cells. J Physiol (Lond), in press.
-
doi:10.1124/jpet.105.095067.
-
ABBREVIATIONS: VSM vascular smooth muscle; TRP, transient receptor potential; TRPC, TRP nonselective cation channel; [Ca2+]i, cytosolic free Ca2+ concentration; 2-APB, 2-aminoethoxydiphenylborane; AS-TRPC1-GFP, adenoviral vector for expression of antisense TRPC1 DNA and coexpression of green fluorescent protein; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; BSA, bovine serum albumin; CCE, capacitative Ca2+ entry; CPA, cyclopiazonic acid; ISOC, store-operated current; LOE 908, (R,S)-(3,4-dihydro 6,7-dimethoxy-isoquinoline-1-yl)-2-phenyl-N,N-di-[2-(2,3,4-trimethoxyphenyl)ethyl]-acetamide; MASMC, mesenteric artery smooth muscle cell(s); m.o.i., multiplicity of infection; NCCE, noncapacitative Ca2+ entry; OAG, 1-oleoyl-2-acetyl-sn-glycerol; RT, reverse transcription; PCR, polymerase chain reaction; siRNA, small interfering RNA; SOCE, store-operated Ca2+ entry; DES, diethylstilbestrol; MOPS, 4-morpholinepropanesulfonic acid; NMDG, N-methyl-d-glucamine; PBS, phosphate-buffered saline.
- Received September 1, 2005.
- Accepted January 12, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}