


Abstract
Nicotine is the major addictive agent in tobacco. The primary catalyst of nicotine metabolism in humans is CYP2A6. However, the closely related enzyme CYP2A13 is a somewhat better catalyst. CYP2A13 is an extrahepatic enzyme that is an excellent catalyst of the metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone (NNK). Here we report that both CYP2A6 and CYP2A13 were inactivated during nicotine metabolism. Inactivation of both enzymes was dependent on NADPH and increased with time and concentration. Alternate substrates for CYP2A6 and CYP2A13 protected these enzymes from inactivation. Inactivation of CYP2A13 was irreversible upon extensive dialysis and seems to be mechanism-based. The KI of CYP2A13 inactivation by nicotine was 17 μM, the rate of inactivation, kinact, was 0.1 min-1, and the t1/2 was 7 min. However, the loss in enzyme activity occurred after nicotine metabolism was complete, suggesting that a secondary or possible tertiary metabolite of nicotine may be responsible. [5-3H]Nicotine metabolism by CYP2A13 was monitored by radioflow high-pressure liquid chromatography during the course of enzyme inactivation; the major product was the Δ1′(5′)iminium ion. However, cotinine was a significant metabolite even at short reaction times. The metabolism of the nicotine Δ1′(5′)iminium ion to cotinine did not require the addition of aldehyde oxidase. CYP2A13 catalyzed this reaction as well as further metabolism of cotinine to 5′-hydroxycotinine, trans-3′-hydroxycotinine, and N-(hydroxymethyl)-norcotinine as enzyme inactivation occurred. Studies are on-going to identify the metabolite responsible for nicotine-mediated inactivation of CYP2A13.
Nicotine is the major addictive agent in tobacco, and individuals continue to smoke and use tobacco products to maintain plasma nicotine levels (Benowitz, 1988; Hukkanen et al., 2005). The plasma concentration of nicotine varies among tobacco users, due in part to differences in nicotine metabolism. The major catalyst of nicotine metabolism in humans is CYP2A6 (Hukkanen et al., 2005). However, the closely related enzyme CYP2A13 is a better catalyst (Bao et al., 2005; Murphy et al., 2005). Whether or not CYP2A13, an extrahepatic P450 expressed in the respiratory tract (Su et al., 2000), plays a significant role in nicotine metabolism in smokers is unknown. However, CYP2A13 is an excellent catalyst of the metabolic activation of the tobacco carcinogen NNK (Jalas et al., 2005; Wong et al., 2005b). NNK is only present in tobacco and tobacco smoke; therefore, exposure to NNK is always concurrent with nicotine exposure. NNK requires metabolic activation to exert its carcinogenic potential, and CYP2A13 is likely a key catalyst of activation in smokers. Therefore, any inactivation of CYP2A13 by nicotine will influence the metabolic activation of NNK, a potent lung carcinogen.
Nicotine is primarily metabolized by 5′-oxidation, but 2′-oxidation and methyl-oxidation also occur (Fig. 1) (Hecht et al., 2000; Hukkanen et al., 2005; Murphy et al., 2005). The latter two pathways are minor ones, each accounting for 10% or less of total nicotine metabolism in smokers (Hecht et al., 1999; Hukkanen et al., 2005). 5′-Oxidation typically accounts for 70 to 80% of metabolism (Hukkanen et al., 2005). Each of these pathways results in the formation of a reactive iminium ion that exists in equilibrium with the hydroxy-nicotine metabolite. The Δ1′(5′)iminium ion is further metabolized to cotinine (Fig. 1). One catalyst of this reaction is the cytosolic enzyme aldehyde oxidase (Brandänge and Lindblom, 1979). The major metabolite of cotinine in smokers is trans-3′-hydroxycotinine, which with its glucuronide conjugate accounts for greater than 50% of the nicotine dose received by a smoker (Davis and Curvall, 1999).
CYP2A6 catalyzes all three oxidation pathways of nicotine metabolism, and CYP2A13 catalyzes both 5′-oxidation and methyl oxidation (Hecht et al., 2000; Murphy et al., 2005). CYP2A6 also catalyzes the conversion of cotinine to trans-3′-hydroxycotinine (Nakajima et al., 1996). However, it is neither a very good catalyst nor a specific catalyst of this reaction (K. M. Brown, L. B. von Weymarn, and S. E. Murphy, manuscript submitted for publication; Murphy et al., 1999). The major product of CYP2A6-catalyzed cotinine metabolism is N-(hydroxymethyl)norcotinine. The most abundant metabolite of N-(hydroxymethyl)-norcotinine in smokers urine is norcotinine, but it accounts for <5% of the nicotine dose (Benowitz et al., 1994). Therefore, other P450s must contribute to the 3′-hydroxylation of cotinine in vivo. We recently reported that CYP2A13 is both a more efficient and more specific catalyst of cotinine 3′-hydroxylation (KM Brown, LB von Weymarn, and SE Murphy, manuscript submitted for publication). These data have led us to suggest that both CYP2A6 and CYP2A13 may contribute to nicotine metabolism in smokers.

Nicotine metabolism.
As noted above, the oxidation of nicotine results in the formation of three reactive iminium ions, any of which might contribute to some of the toxic effects of nicotine. Previously, it was reported that, during nicotine metabolism by rabbit liver microsomes, covalent modification of proteins occurs (Shigenaga et al., 1988). It was suggested that the Δ1′(5′)iminium ion was the reactive species responsible for this modification. More recently, Denton et al. (2004) reported that nicotine, but not the nicotine Δ1′(5′)iminium ion, inhibited CYP2A6-catalyzed coumarin 7-hydroxylation. They referred to the inhibition as mechanism-based. However, the only characteristic of inhibition they reported was that it was time-dependent. No other details of CYP2A6 inactivation by nicotine were described (Denton et al., 2004).
In carrying out experiments to characterize the products of nicotine metabolism by CYP2A6 and CYP2A13, we observed a significant loss of enzyme activity. CYP2A13 is 94% identical to CYP2A6, and nicotine metabolism by these enzymes is qualitatively similar. Therefore, we hypothesized that the observed nicotine-mediated inactivation of CYP2A13 and CYP2A6 occurs by a similar mechanism. We present data here comparing CYP2A6 and CYP2A13-mediated inactivation by nicotine and confirm that CYP2A13 inactivation is mechanism-based.
Materials and Methods
Chemicals and Enzymes. [5-3H]-(S)-Nicotine was prepared from (S)-5′-bromonicotine by Chemsyn (Lenexa, KS) and purified by HPLC as described previously (Murphy et al., 2005). Nicotine Δ1′(5′)iminium ion was synthesized as previously described (Peterson et al., 1987). (S)-Nicotine, cotinine, nornicotine, 7-hydroxycoumarin, coumarin, dilauroyl-l-α-phosphatidylcholine, NADPH, bovine serum albumin, catalase, and all other biochemical reagents were obtained from Sigma-Aldrich (St. Louis, MO) and were of analytical grade. Trifluoroacetic acid (TFA) and dialysis cassettes were obtained from Pierce Chemical (Rockford, IL). Human liver cytosol was prepared in our laboratory by a previously established method (Fowler et al., 1994). Cytosol protein concentration was determined using Coomassie Blue Plus protein assay reagent kit (Pierce Chemical). The enzymes used in this study were expressed and purified from Escherichia coli according to previously published methods (Hanna et al., 1998; Kent et al., 1999; Soucek, 1999; von Weymarn et al., 2005) and were a kind gift from Dr. Paul Hollenberg (Department of Pharmacology, University of Michigan, Ann Arbor, MI). CYP2A6 (10.1 nmol/mg protein) and CYP2A13 (1.1 nmol/mg protein) are full-length enzymes. The CYP2A13 used in this study is the same preparation as that used by von Weymarn et al. (2005). At saturating conditions of coumarin, the rate of coumarin 7-hydroxylation for both CYP2A13 and CYP2A6 is comparable with that reported previously (Soucek, 1999; von Weymarn et al., 2005).
Nicotine Metabolism by CYP2A6 and 2A13. The purified P450 enzymes were reconstituted with rat NADPH-cytochrome P450 oxidoreductase (reductase) and dilauroyl-l-α-phosphatidylcholine (0.2 μg/pmol P450) for 45 min at 4°C. The ratio of P450 to reductase was 1:2. Tris buffer (100 mM, pH 7.4) and catalase (60 U/μl) were added to the reconstituted enzyme to achieve a P450 concentration of either 0.5 or 1 pmol/μl. Aliquots of the reconstituted enzyme solution containing 10 to 15 pmol of P450 for 2A6 or 4 to 20 pmol of P450 for 2A13 were added to incubation mixtures containing 100 mM Tris buffer (pH 7.5), an NADPH-generating system (0.4 mM NADP, 10 mM glucose 6-phosphate, and 0.4 units/ml glucose phosphate dehydrogenase) and nicotine (specific activity, 0.02 to 2.0 Ci/μmol, 2 to 500 μM). The final incubation volume was 200 μl. The samples were incubated for 3 to 30 min (CYP2A13) or 5 to 45 min (CYP2A6) at 37°C. Reactions were terminated by the addition of 20 μl of 10% TFA and centrifuged at 1500g (10 min), and the supernatant was removed and analyzed by HPLC on the same day. The nicotine metabolites were separated as described previously (Murphy et al., 2005). The metabolites were identified by coelution with standards (UV detection at 254 nm). The Δ1′(5′)iminium ion was also collected and quantitatively converted to cotinine upon the addition of cytosol as described previously (Murphy et al., 2005). Michaelis-Menten constants were calculated for nicotine 5′-oxidation using the EZ-Fit 5 software (Perrella Scientific, Amherst MA).
Effects of Nicotine on CYP2A6 and CYP2A13 Activity. CYP2A6 or CYP2A13 (0.1–1 nmol) was reconstituted with reductase and lipid as described above. Catalase and Tris buffer were added to the reconstituted enzymes to give final concentrations of 0.5 pmol/μl P450, 1 pmol/μl reductase, 0.1 μg/μl lipid, 35 U/μl catalase, and 50 mM Tris buffer, pH 7.4. The molar ratio of P450 to reductase was 1:2 unless otherwise noted.
Time-, Concentration-, and NADPH-Dependent Inactivation of P450s 2A6 and 2A13 by Nicotine. Nicotine (0–500 μM) and 1 mM NADPH were added to the reconstituted enzyme solution described above to generate the primary reaction mixture. The primary reaction mixtures were incubated for 5 min at 30°C prior to the addition of NADPH (1 mM). Upon the addition of NADPH, aliquots (5 μl = 2.5 pmol P450) were removed at the indicated times (0–90 min) and added to a secondary reaction mixture containing 40 μg/ml bovine serum albumin, 20 μM coumarin, and 0.2 mM NADPH in 50 mM Tris buffer (pH 7.4) in a total volume of 300 μl. The secondary reactions were incubated for 10 min at 30°C and then terminated by the addition of trichloroacetic acid, and the formation of 7-hydroxycoumarin was analyzed by HPLC with fluorescence detection as described previously (von Weymarn et al., 1999).
Partition Ratio. CYP2A13 was reconstituted, and the primary reaction mixtures were prepared as described above. Primary reaction mixtures containing 0 to 500 μM nicotine were incubated for 30 min at 30°C in the presence of 1 mM NADPH. Aliquots containing 2.5 pmol of CYP2A13 were removed at 0 and 30 min and added to the secondary reaction mixture for the 7-hydroxycoumarin assay described above. The partition ratio was obtained by plotting percent activity remaining versus [nicotine]/[2A13]. The partition coefficient was estimated from the intercept of the regression line obtained at low [nicotine]/[2A13] ratios and the line obtained at saturating nicotine concentrations.
Substrate Protection. Nicotine inactivation experiments were carried out in the presence of alternate substrates, testosterone (CYP2A13), or S-NNN (CYP2A6). The primary reaction and secondary reactions were carried out as described above. CYP2A6 reactions contained 100 μM nicotine and were incubated for 30 min. CYP2A13 reactions were incubated with 25 μM nicotine for 15 min. The concentrations of S-NNN in the CYP2A6 primary reactions were 0, 50, or 100 μM, and the concentrations of testosterone in the CYP2A13 reactions were 0, 10, or 100 μM. The control sample did not contain either nicotine or the alternate substrate. Testosterone was dissolved in ethanol; the same final concentration of ethanol (0.8%) was included in the control reactions
Effect of Exogenous Nucleophiles. The primary reaction mixture prepared as described above contained 25 μM nicotine and 0, 1, or 10 mM glutathione or 0, 50 μM, 100 μM, or 1 mM KCN. The control incubations did not contain any nicotine or exogenous nucleophile. At 0 and 16 min, aliquots were removed and added to the secondary reaction mixture for determination of coumarin 7-hydroxylation activity. The effect of GSH (10 mM) or KCN (20, 50, 100, 500, or 1000 μM) on 2A13-mediated coumarin 7-hydroxylation activity was determined independently.
Irreversibility of Inactivation. CYP2A13 was reconstituted as described above. Each primary reaction mixture (exposed control and inactive) contained 1 pmol/μl P450, 2 pmol/μl reductase, 0.2 μg/μl lipid, 35 U/μl catalase, 25 μM nicotine, and 1 mM NADPH (inactive) or water (exposed control) in 50 mM Tris buffer (pH 7.4). Samples were preincubated for 5 min without NADPH at 30°C. At 0 and 16 min following NADPH addition, 5 μl of the primary reaction mixture was removed (in duplicate) and analyzed for coumarin 7-hydroxylation activity in a secondary reaction as described above. A second aliquot (70 μl) was removed and added to 430 μl of an ice-cold quench buffer containing 40% glycerol and 0.6% Tergitol in 50 mM potassium phosphate (pH 7.4). The reduced CO spectra of the quenched samples were measured on a DW2 UV-visible spectrophotometer (SLM Aminco, Urbana, IL) with an OLIS spectroscopy operating system (On-Line Instrument Systems, Inc., Bogart, GA) using the method of Omura and Sato (1964). At 16 min, 90 μl was removed and used to measure the loss in native heme by HPLC. The native heme and any modified heme were separated from the apoprotein using a previously published method (von Weymarn et al., 2004) with minor modification. The mobile phase consisted of 0.1% TFA in water (A) and 0.05% TFA in acetonitrile (B). The column Zorbax 300SB-C3 (150 × 2.4 mm; Agilent Technologies, Palo Alto, CA) was eluted isocratically at 70% A:30% B for 10 min followed by a linear gradient to 80% B in 15 min and then to 95% B in 10 min. The flow rate was 0.3 ml/min. The elution of native heme and any heme-containing products was monitored at 405 nm.
The remainder of the control and inactivated samples (200–250 μl) were either applied to a G50 Sephadex spin-column (von Weymarn et al., 2004) or subjected to 24-h dialysis. Dialysis was performed in 50 mM Tris (pH 7.4) containing 20% glycerol at 4°C. The spin column-treated and dialyzed samples were reconstituted with lipid and then assayed for coumarin 7-hydroxylation activity, the formation of a reduced CO spectrum and native heme loss.
Inactivation Experiments with [5-3H]-(S)-Nicotine. Two parallel experiments were carried out with reconstituted CYP2A13 prepared as described above, except that the ratio of reductase to P450 was reduced to 1:1. For each experiment, the primary reaction contained 1 pmol/μl P450, 1 pmol/μl reductase, 0.1 μg/μl lipid, 35 U/μl catalase, 25 μM [5-3H]-(S)-nicotine (0.09 μCi/nmol), and 1 mM NADPH (inactive) or water (exposed control) in 50 mM Tris buffer (pH 7.4). In one experiment, which was to determine the extent of nicotine metabolism relative to enzyme inactivation, two 3-μl aliquots and a 25-μl aliquot were removed at 0, 0.5, 1, 2, 5, 10, and 16 min following the addition of NADPH. The 3-μl aliquots were assayed for coumarin 7-hydroxylation activity in a secondary reaction. The 25-μl aliquot was added to TFA (0.5% final concentration), and the extent of nicotine metabolism was determined by radioflow HPLC as described for the nicotine metabolism experiments, except that a shallow methanol gradient was used. The mobile phase was kept constant at 100% A (0.2% TFA in water) for 20 min followed by a linear gradient to 10% B (methanol) in 5 min, holding at 10% B for 10 min, and then to 20% B in 5 min and to 25% B in 3 min. The same experiment was carried out with the addition of 1 mM KCN to the primary reaction.
The second experiment with [5-3H]nicotine was conducted to determine possible tritium binding to the heme or apoprotein after inactivation. After a 20-min incubation of the primary reaction, 3 μl (3 pmol of P450) was removed (in duplicate) and assayed for coumarin 7-hydroxylation activity in a secondary reaction. The remaining sample (120 μl) was analyzed by reverse phase HPLC with radioflow and UV detection (260 nm for protein or 405 nm for heme). The UV detection was used to establish the retention times of the different proteins as well as the retention time of the native heme to determine whether there was any radioactivity associated with the proteins or the heme molecule in the inactivated samples. The mobile phase and column were the same as described for the experiments on irreversibility described above, and the gradient was slightly modified from a previously published protocol (von Weymarn et al., 2004). The proteins were eluted by holding the mobile phase at 35% B for 10 min followed by a linear gradient to 90% B in 25 min and then to 100% B in 5 min. The flow rate was 0.3 ml/min. The scintillant (Monoflow X; National Diagnostics, Atlanta, GA) flow rate was 1 ml/min.
Results
The products of nicotine metabolism by partially purified full-length CYP2A6 and 2A13 expressed in E. coli were characterized by radioflow HPLC (Fig. 2). The primary nicotine metabolite generated by both enzymes was the Δ1′(5′)iminium ion (tR, 8.1). This metabolite was quantitatively converted to cotinine when reactions were carried out in the presence of cytosol as a source of aldehyde oxidase. Interestingly, cotinine was detected as a product of CYP2A13-mediated nicotine metabolism in the absence of cytosol (Fig. 2, tR, 22.8 min). Cotinine was also detected as a product of nicotine metabolism by CYP2A6, but only at higher enzyme concentrations and longer reaction times. Nornicotine was identified as a product of both CYP2A6- and 2A13-mediated nicotine metabolism. A striking characteristic of CYP2A13-catalyzed nicotine metabolism was its lack of linearity with time. At reactions times as short as 5 min, the rate of CYP2A13-catalyzed nicotine 5′-oxidation, either in the presence or absence of cytosol, began to slow down. This is in contrast to the rate of CYP2A6-catalyzed 5′-oxidation, which was linear for up to 45 min.

Radioflow HPLC analysis of 100 μM nicotine metabolism by E. coli expressed and purified 20 pmol CYP2A6 (A) and 2A13 (B). The arrows indicate the retention time of standards. The incubation times were 5 and 30 min for CYP2A6 and 2A13, respectively.
The kinetic parameters for nicotine 5′-oxidation by CYP2A6 were determined under conditions at which reaction rates were linear with time; the Km was 234 ± 26 μM, and the Vmax was 9.3 ± 0.5 nmol/min/nmol P450. The kinetic parameters for CYP2A13 were also determined. However, because of the limits of detection of the assay, the kinetic parameters for CYP2A13 were determined with 5-min reaction times when rates were already decreased by about 20% compared with 3 min. Therefore, the Km and Vmax values, 40 ± 2 μM and 34.7 ± 5.4 nmol/min/nmol P450 for CYP2A13, should be considered as estimates.
The significant loss of enzyme activity that was detected during CYP2A13-catalyzed nicotine metabolism led us to investigate the inactivation of CYP2A13 by nicotine. Possible nicotine-mediated inactivation of CYP2A6 was also determined. Because of the significant loss (32%; Table 1) in CYP2A6 activity upon incubation at 37°C with reductase, NADPH, and no nicotine, it was necessary to carry out these experiments at 30°C. At the lower temperature, there was no loss in either CYP2A6 or CYP2A13 activity in control samples (Table 1). After 15 min, in the presence of 25 μM nicotine, reductase, and NADPH, CYP2A13 lost approximately 60% of its catalytic activity. CYP2A6 was also sensitive to inactivation by nicotine metabolism. However, a higher concentration of nicotine was required and the extent of inactivation was less (Table 1).
Temperature effects on inactivation of CYP2A6 and 2A13 by nicotinea
For both CYP2A6 and CYP2A13, the nicotine-mediated loss of activity was dependent on NADPH and increased with time. Inactivation was also concentration-dependent for both enzymes. However, in the case of CYP2A6, the inactivation was not linear over the range of concentrations used (Fig. 3A). This is clearly illustrated when plotting 1/kinact (rate of inactivation) versus 1/[nicotine] (Fig. 3A, inset). There was a concentration-dependent loss in enzyme activity among 0, 25, and 50 μM nicotine. However, at higher concentrations, 100 and 500 μM, there was little or no increase in the extent of enzyme inactivation, and at 10 μM nicotine, there was no detectable inactivation. Nicotine inactivation of CYP2A13 was clearly concentration-dependent (Fig. 3B). The kinetic rate constants were determined from the slopes of the lines when the logarithm of the percent activity remaining was plotted against time (Fig. 3B). The concentration of nicotine required to achieve half the maximum rate of inactivation (KI) was 17 μM. The kinact was 0.1 min-1, and the t1/2 was 7 min. The same approach was used to obtain an approximation of the kinetic parameters for the inactivation of CYP2A6 by nicotine. The KI was estimated to be 21 μM, the kinact was 0.021 min-1, and the t1/2 was 33 min.

Time- and concentration-dependent inactivation of CYP2A6 (A) and CYP2A13 (B) by nicotine. Activity remaining refers to coumarin 7-hydroxylation activity determined in a secondary reaction after the indicated time of nicotine metabolism. Details of the experiments are as described under Materials and Methods. The nicotine concentrations used were 0 (▪), 25 (▴), 50 (▾), 100 (♦), and 500 μM (•) for CYP2A6 and 0 (□), 5 (▪), 10 (▵), 25 (▴), and 50 μM (○) for CYP2A13. Values are the mean ± S.D. from three independent experiments performed in duplicate. The insets represent the double-reciprocal plot generated from the slopes of the lines at the various concentrations.
The partition ratio, a measure of the efficiency of inactivation, which is defined as the number of molecules of the inactivator metabolized per molecule of enzyme inactivated (Silverman, 1996), was determined to be 33 for CYP2A13 inactivation by nicotine (Fig. 4). The partition ratio was determined by letting the inactivation of the enzyme at different nicotine concentrations go to completion. The complete inactivation of CYP2A13 occurred within 30 min. In contrast, the inactivation of CYP2A6 was not complete by 60 min, at which time significant CYP2A6 activity was lost in the absence of nicotine. Therefore, the partition ratio for CYP2A6 was not determined.
Inactivation by nicotine required enzymatic turnover; therefore the ability of alternate substrates to protect the enzyme from inactivation was investigated. The presence of S-NNN [Km = 2.3 μM (Wong et al., 2005a)] significantly decreased nicotine-mediated inactivation of CYP2A6 (Table 2). The alternate substrate, testosterone [Km = 22 μM (von Weymarn et al., 2005)], protected CYP2A13 from nicotine inactivation by nicotine. A testosterone concentration that was 4-fold higher than that of nicotine was required for significant protection of CYP2A13.
Effect of alternate substrates on nicotine-mediated inactivation of CYP2A6 and CYP2A13a
To further characterize the inactivation of CYP2A13 by nicotine, the effect of inactivation on the integrity of the heme moiety of the enzyme was determined. Under conditions where the loss in activity was 60%, the ability of the inactivated CYP2A13 to form a reduced CO complex was decreased by 31% relative to a non-NADPH-treated control (Table 3). A similar level of reduction in the native heme was detected by HPLC analysis with UV detection at 405 nm. The possible reversibility of this loss in heme and enzyme activity was determined using two methods, spin-column gel filtration and dialysis. The inactivation of CYP2A13 by nicotine was not affected by either method (Table 3). In addition, there was no significant change in either the amount of reduced CO complex or the amount of native heme detected after spin-column gel filtration or dialysis treatment of the inactivated enzyme.
Effect of CYP2A13-catalyzed nicotine metabolism on coumarin 7-hydroxylation, reduced CO spectrum, and HPLC-detected hemea

Partition ratio determination for CYP2A13 with nicotine. The inactivation was allowed to go to completion, and then coumarin 7-hydroxylation activity was determined in a secondary reaction as described under Materials and Methods. Values are the mean ± S.D. of three independent experiments performed in duplicate. The arrow indicates the intercept used to determine the partition ratio.
The formation of adducts to the heme and/or apoprotein was investigated using [5-3H]-(S)-nicotine. Under conditions at which the enzyme was 60% inactivated (as measured by the remaining coumarin 7-hydroxylation activity), the enzyme was analyzed by radioflow HPLC. No tritium binding to the heme, the P450 apoprotein, or the reductase was detected (Data not shown).
To potentially trap reactive intermediates formed during nicotine-mediated inactivation of CYP2A13, experiments were carried out in the presence of the two nucleophiles, GSH and cyanide. The addition of 10 mM GSH to nicotine metabolism reactions had no effect on the inactivation of CYP2A13 (Table 4), whereas the presence of 1 mM KCN during the inactivation of CYP2A13 by 25 μM nicotine resulted in a significant protection from inactivation. In the absence of KCN or in the presence of 50 or 100 μM KCN, CYP2A13-catalyzed nicotine metabolism resulted in a 64% loss in activity, but if 1 mM KCN was included in this reaction only a 25% loss in enzyme activity occurred (Table 4). However, a significant amount of the 25% loss may be due to direct inhibition of the enzyme by KCN, because the addition of 1 mM KCN to control incubations that contain no nicotine resulted in a 14% loss in activity (i.e., decreased coumarin 7-hydroxylation activity in the secondary reaction). This was not surprising because KCN had previously been shown to inhibit P450-catalyzed reactions (Kitada et al., 1977; Shigenaga et al., 1988). In a series of experiments that directly measured the effect of KCN on CYP2A13-catalyzed coumarin 7-hydroxylation, we observed a 53% inhibition of CYP2A13 activity by 1 mM KCN and 15% inhibition by 50 μM KCN (data not shown).
Effects of exogenous nucleophiles on inactivation of CYP2A13 by nicotinea
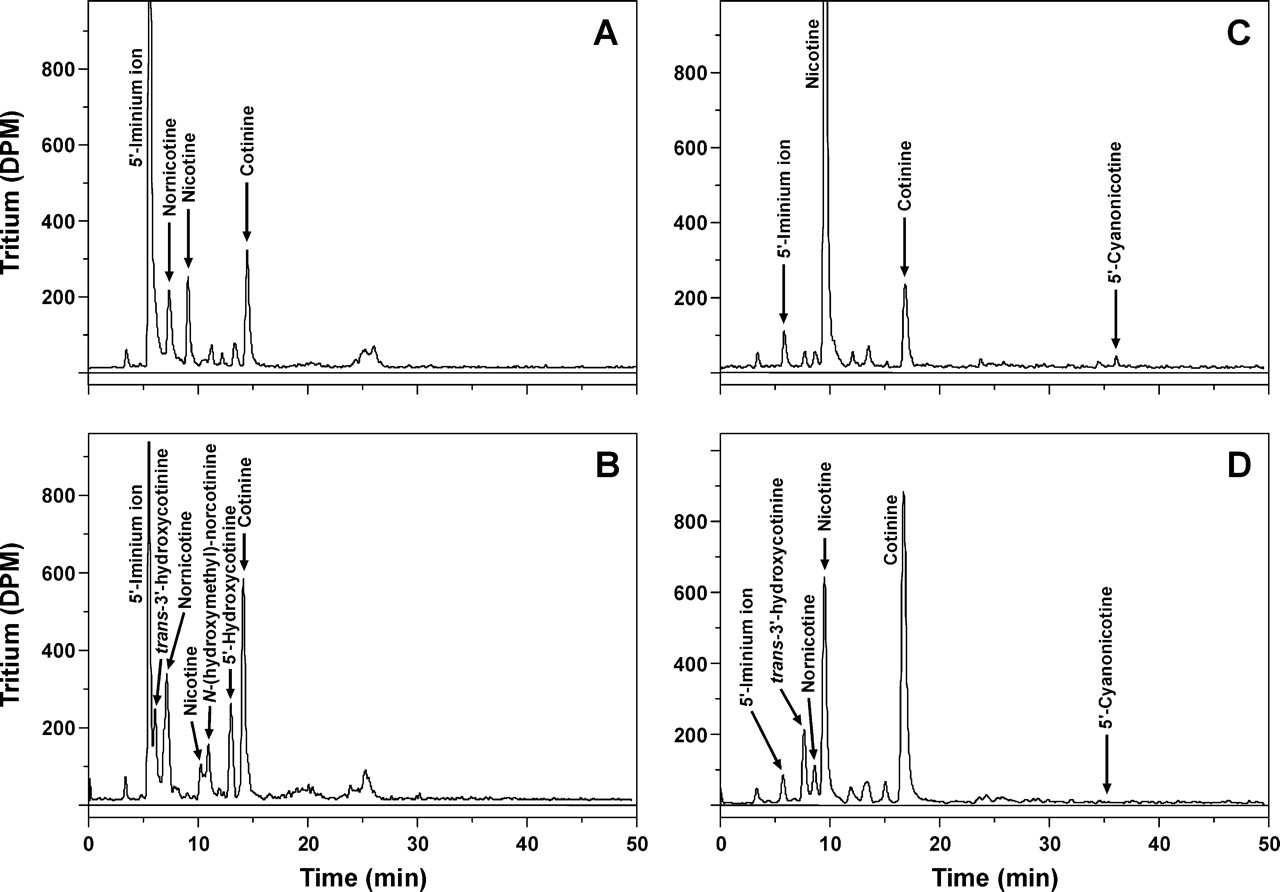
The significant enzyme inhibition by 1 mM KCN suggests that the observed KCN protection of CYP2A13 from nicotine-mediated inactivation is most likely due to decreased nicotine metabolism. To investigate the effect of KCN on the relationship between CYP2A13-catalyzed nicotine metabolism and enzyme inactivation, experiments were carried out with [5-3H]nicotine. Nicotine metabolism was quantified by radioflow HPLC analysis over the time period that inactivation occurred, both in the absence and presence of KCN. The experimental conditions were those used to determine the kinetics of inactivation (Fig. 3B) with a [5-3H]-(S)-nicotine concentration of 25 μM. Radioflow HPLC analyses of the products of nicotine metabolism after a 2- and 10-min reaction are presented in Fig. 5. The time course of inactivation compared with nicotine metabolism is presented in Table 5. In the first 0.5 min of the reaction, 36% nicotine was metabolized; however, no detectable inactivation was observed. After 2 min, 89% nicotine was converted to products but only 22% of the enzyme was inactivated (Fig. 5A; Table 5). The major nicotine metabolite detected at this time was the Δ1′(5′)iminium ion, which accounted for 68% of the total products. The secondary metabolite, cotinine, accounted for another 12%, and nornicotine was detected at a level of 9% of total metabolism (Fig. 5A). Within 5 min, essentially all of the nicotine was metabolized, whereas enzyme inactivation continued to occur until 16 min. During this time, the Δ1′(5′)iminium ion was further metabolized to cotinine. In addition, cotinine was further metabolized to a number of products, including 5′-hydroxycotinine, trans-3′-hydroxycotinine, and N-(hydroxymethyl)-norcotinine. The identities of these tertiary metabolites are based on retention time and coelution with standards (Fig. 2B).
Extent of CYP2A13-catalyzed nicotine (CN) metabolism and loss of enzyme activity in the absence and presence of cyanidea

Radioflow HPLC analysis of nicotine metabolites generated during nicotine-mediated inactivation of CYP2A13 in the absence (A and B) or presence (C and D) of 1 mM KCN. CYP2A13 (200 pmol) was incubated with 25 μM (0.09 μCi/nmol) [5-3H]-(S)-nicotine at 30°C for 2 (A and C) and 10 min (B and D). Arrows indicate the retention times of coinjected standards.
In the presence of cyanide, there was a significant reduction in nicotine metabolism and a parallel decrease in enzyme inactivation (Table 5; Fig. 5). For example, only 28% of the nicotine was metabolized after a 2 min reaction in the presence of KCN compared with 89% in the absence of KCN. In the absence of KCN, at 2 min, 22% inactivation of the enzyme had occurred; however, a similar level of inactivation in the presence of KCN was not detected until 10 min when >50% nicotine was metabolized. In the presence of 1 mM KCN, nicotine metabolism was not complete until 16 min, whereas in the absence of KCN, nicotine metabolism was complete in <5 min. Interestingly, the primary nicotine metabolite in the presence of KCN was cotinine, not 5′-cyanonicotine. No 5′-cyanonicotine and little Δ1′(5′)iminium ion was detected at any time point. In contrast, when KCN was added at the end of the reaction, all of the Δ1′(5′)iminium ion was converted to 5′-cyanonicotine.
Discussion
We and others have previously reported that CYP2A13 and CYP2A6 are catalysts of nicotine 5′-oxidation (Nakajima et al., 1996; Messina et al., 1997; Yamazaki et al., 1999; Bao et al., 2005; Murphy et al., 2005). The unique observation reported here is that both CYP2A6 and CYP2A13 become inactivated during nicotine metabolism. CYP2A13 is both a better catalyst of nicotine metabolism and is more sensitive to nicotine-mediated inactivation than CYP2A6. The increased catalytic activity of CYP2A13 allowed a more complete characterization of nicotine-mediated inactivation by this enzyme. It seems likely that the mechanism of inactivation of both enzymes is similar but that it is modulated by the efficiency and specificity of metabolism of these two closely related enzymes. Investigation of nicotine-mediated inactivation of CYP2A6 is challenging because of the relatively long t1/2. Inactivation experiments with CYP2A6 need to be carried out for longer reaction times, during which a significant loss in enzyme activity occurs due to the futile cycle. However, characterizing the nicotine-mediated inactivation of CYP2A13 should shed some light on the mechanism by which CYP2A6-inactivation occurs and provide insight into the design of future experiments with CYP2A6.
In the case of CYP2A6, the loss in enzyme activity in the presence of nicotine required NADPH and was time- and concentration-dependent. It was not possible to establish linearity with concentration over a very wide range; however, the kinetic parameters of inactivation were estimated. The rate of inactivation reported here, 0.021 min-1, is three orders of magnitude less than the value, 71.1 min-1, reported previously (Denton et al., 2004). It is difficult to speculate on the significant difference in these two values, because no experimental detail was provided by Denton et al. (2004) on how the rate of inactivation was determined. It seems highly unlikely that the rate of 71.1 min-1 is correct, because according to our calculation, this would translate to a t1/2 of 0.01 min. This would mean that 50% of the enzyme is inactivated in <1 s. The authors of the earlier study refer to the observed time-dependent nicotine inhibition of coumarin 7-hydroxylation as mechanism-based inactivation but provide no data to support this designation.
In addition to characterizing the time and NADPH dependence of CYP2A6 inactivation by nicotine, here we report a third characteristic of mechanism-based inactivation, protection of the enzyme by an alternate substrate, S-NNN. We have not yet determined the reversibility of the observed CYP2A6 inactivation and therefore have not confirmed that the nicotine-mediated inactivation of CYP2A6 is mechanism-based.
The data presented here do support the characterization of CYP2A13 inactivation by nicotine as mechanism-based. The loss in enzyme activity in the presence of nicotine required NADPH, was time- and concentration-dependent, and was reduced by the presence of an alternate substrate. In addition, inactivation of the enzyme was not reversible after extensive dialysis or after washing on a spin column to remove any noncovalently bound small molecules. These data exclude the possibility that the observed inactivation is due to competitive inhibition by a nicotine metabolite with a high affinity for the enzyme.
Inactivation of CYP2A13 by nicotine was accompanied by a 30% loss in total native heme and a similar loss in the ability of the enzyme to form a reduced CO complex. These changes in the enzyme were irreversible and suggest that the heme molecule may be modified during inactivation. However, when CYP2A13 inactivation reactions were carried out with [5-3H]nicotine, no 3H-labeled heme was detected by radioflow HPLC. We were also unable to detect any tritiated modification of the apoprotein. A simple explanation for this would be that a tritiated adduct of either the heme or apoprotein was formed but that it was not stable to the denaturing acidic conditions of the analysis. A possible unstable adduct would be the Schiff base of the aldehyde tautomer of 5′-hydroxynicotine. Alternatively, a nonradioactive modification may be present.
The major product of CYP2A13-catalyzed nicotine metabolism is the Δ1′(5′)iminium ion; the next most abundant metabolite is nornicotine (Murphy et al., 2005). Nornicotine is formed by spontaneous demethylation of N-(hydroxymethyl)-nornicotine. The second product of this reaction is formaldehyde, which readily reacts with a number of amino acid side chains and may generate cross-links (Gubisne-Haberle et al., 2004; Metz et al., 2004). If formaldehyde modifications of CYP2A13 were generated during the metabolism of [5-3H]nicotine, the enzyme would not be radiolabeled. Inconsistent with formaldehyde mediating the inactivation of CYP2A13 by nicotine is the observation that the majority of enzyme inactivation occurs after nicotine is completely metabolized (Table 5). These data are more consistent with the role of a secondary metabolite of nicotine as the inactivating species.
Support for the role for a secondary or even tertiary nicotine metabolite as the agent of inactivation is provided by the detection of significant secondary metabolism of nicotine during the time when inactivation of the enzyme occurred (Fig. 5). Cotinine was routinely observed as a product of CYP2A13-catalyzed nicotine metabolism, even at short reaction times. Therefore, contrary to the prevailing dogma, the formation of cotinine from the Δ1′(5′)iminium ion (or 5′-hydroxynicotine; Fig. 1), does not require aldehyde oxidase. Both CYP2A6 and CYP2A13 catalyze this reaction. The suggestion that a P450 may catalyze this reaction was made some time ago by Shigenaga et al. (1988), who reported that cotinine was a product of nicotine metabolism by both rabbit liver microsomes and human liver microsomes. The detection of cotinine as a product of nicotine metabolism by both CYP2A6 and CYP2A13 has led us to hypothesize that the reactive species responsible for nicotine-mediated inactivation of these enzymes is a metabolite of the Δ1′(5′)iminium ion. Studies on the metabolism of the nicotine Δ1′(5′)iminium are now being carried out to test this hypothesis.
The Δ1′(5′)iminium ion has previously been reported to modify microsomal proteins during nicotine metabolism (Shigenaga et al., 1988). Subsequently, it was reported that the addition of either cytosol or GSH to the reaction mixture reduced the modification of microsomal proteins by greater than 85% (Williams et al., 1990). In contrast, neither GSH nor cytosol protected CYP2A13 from nicotine-mediated inactivation. Taken together, these data suggest that if metabolism of the Δ1′(5′)iminium ion is required to inactivate CYP2A13, then the Δ1′(5′)iminium ion does not leave the active site before its further metabolism.
In an attempt to trap the iminium ion, inactivation experiments were carried out in the presence of KCN. Cyanide does enter the active site of P450 enzymes and has been used by several investigators to trap nicotine iminium ion metabolites (Nguyen et al., 1979; Koop and Hollenberg, 1980; McCoy et al., 1986; Williams et al., 1990; Gorrod et al., 1991). KCN did seem to protect CYP2A13 from nicotine-mediated inactivation. However, the interpretation of these experiments is complicated by the fact that KCN inhibits P450 activity (Kitada et al., 1977; Koop and Hollenberg, 1980; Shigenaga et al., 1988). Enzyme inhibition by KCN seems to be the mechanism by which protection of nicotine-mediated CYP2A13 inactivation occurs. Inactivation still occurred in the presence of KCN; the rate was simply slowed as was the rate of nicotine metabolism. When the majority of the nicotine is metabolized, a similar loss in enzyme activity occurs as in the absence of KCN. Therefore, the metabolism of the Δ1′(5′)iminium ion was not inhibited and no conclusions on its role in enzyme inactivation can be drawn.
One surprising finding of our experiments with KCN was that when KCN was included in reactions with nicotine and CYP2A13, we did not detect any 5′-cyanonicotine. The major product detected was cotinine (Fig. 5C). At longer reaction times, the amount of cotinine formation increased but little Δ1′(5′)iminium was detected and no 5′-cyanonicotine was found (Fig. 5D). In contrast, when KCN was added at the end of the reaction, essentially all of the Δ1′(5′)iminium ion was converted to 5′-cyanonicotine. These data suggest that 5′-cyanonicotine is a substrate for CYP2A13. Precedent for a cyano adduct of an iminium ion as a P450 substrate exists for the metabolism of N-(benzyl)piperidine [described by Vickers and Polsky (2000)]. When KCN was present during the metabolism of N-(benzyl)piperidine by phenobarbital-induced rat liver microsomes, the formation of the α-lactam increased by 100% (Masumoto et al., 1991).
In summary, we have demonstrated that nicotine or, more accurately, a metabolite of nicotine is a mechanism-based inhibitor of CYP2A13. The reactive species responsible for enzyme inactivation has yet to be identified. Despite the fact that the primary pathway of nicotine metabolism by CYP2A13 is 5′-oxidation, methyl oxidation also occurs. Therefore, products of both of these pathways are being investigated as candidates for reaction with the enzyme.
Footnotes
-
This work was supported by the National Institutes of Health Grant CA-84529.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.105.091306.
-
ABBREVIATIONS: NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; GSH, glutathione; HPLC, high-performance liquid chromatography; NNN, N′-nitrosonornicotine; TFA, trifluoroacetic acid.
- Received June 21, 2005.
- Accepted September 26, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}