


Abstract
Effects of prolonged nicotinic ligand exposure on the function of human α4β2- and α4β4-nicotinic acetylcholine receptor (nAChR) subtypes were studied using receptors heterologously expressed in SH-EP1 human epithelial cells. Magnitudes of acute, nAChR-mediated, specific 86Rb+ efflux responses to 1 mM carbamylcholine were reduced after pretreatment with specific nAChR ligands in effects that depended on pretreatment drug dose, duration of drug pretreatment, and duration of drug-free recovery. Fifty percent inhibition of α4β2-nAChR function following 5 min of recovery occurred after 1 min of pretreatment with 1 mM nicotine but also after 1-h pretreatment at 10 nM nicotine. Seventy-five percent loss in function persisted 1 h after drug removal following 15 min or more of exposure to 1 mM nicotine. However, functional recovery was nearly complete after 1 h in drug-free medium following 1 min to 24 h pretreatment with 0.1 to 1 μM nicotine, i.e., in the range of smoker plasma nicotine levels. α4β4-nAChR was similarly sensitive to persistent inactivation by prolonged nicotine exposure. Carbamylcholine exhibited slightly lower persistent inactivation potency than nicotine at both α4β2- and α4β4-nAChR. The nAChR antagonist, mecamylamine, exhibited persistent inactivation potency and efficacy similar to nicotine at α4β2-nAChR but had a reduced effect on α4β4-nAChR. These studies illustrate persistent inactivation of human α4β2- or α4β4-nAChR induced by prolonged exposure to nicotine and show that other ligands induce nAChR persistent inactivation in a subtype-specific manner.
Multiple nAChR subtypes are present in the brain, each having a unique distribution (reviewed in Lukas, 1998). nAChRs in the brain are suggested to play roles in attention and memory, in mood and emotion, and in sensation and perception (reviewed in Paterson and Nordberg, 2000). These nAChRs also have been implicated in nicotine dependence (Balfour, 1994) and in neurodegenerative and mood disorders (reviewed in Mihailescu and Drucker-Colin, 2000b).
Either through therapeutic use of nicotinic ligands to treat neuropsychiatric disorders or via habitual use of tobacco products, there will be or is sustained exposure of the brain and body to nicotinic ligands. Any effects of such exposure are likely mediated by effects on numbers and function of nAChRs. Historically, much attention has focused on the phenomenon of quantitative increases in nAChR-like radioligand binding sites (“up-regulation”) following chronic nicotine exposure (reviewed in Gentry and Lukas, 2002). However, up-regulation is predominantly attributable to changes in internal rather than cell surface pools of nAChR-like radioligand binding sites (Ke et al., 1998; Whiteaker et al., 1998), and its physiological relevance is unclear (Gentry and Lukas, 2002). By contrast, effects on nAChR function are of obvious, primary importance in determining physiological and behavioral effects of sustained exposure to either nicotine or nicotinic therapeutic agents (Gentry and Lukas, 2002).
The literature predominantly indicates that sustained exposure to nicotinic agonists induces losses in nAChR function. nAChR “desensitization”, which descriptively refers to a rapid-in-onset, quickly and fully reversible loss of function during or following brief (seconds) exposure to nicotinic agonists, was first discovered at the nerve-muscle junction by Katz and Thesleff (1957). Subsequently, other processes involving longer-lasting loss of nAChR function following longer-term (minutes to days) nicotine exposure became recognized for nAChR subtypes found in the periphery (Boyd, 1987; Lukas, 1991; Ke et al., 1998; Reitstetter et al., 1999). Most studies also found that sustained nicotinic agonist exposure induces more than one phase of functional loss for the predominant, high-affinity nicotine-binding nAChR in the brain, composed of α4 and β2 subunits (Flores et al., 1992), whether expressed naturally or heterologously in the oocyte system (Collins et al., 1990a; Marks et al., 1993; Hsu et al., 1996;Fenster et al., 1997; Gopalakrishnan et al., 1997; Olale et al., 1997;Gentry and Lukas, 2002). The longer-lasting losses of nAChR function have been called “specific chronic desensitization” (Ochoa et al., 1990; Collins et al., 1994), “stable desensitization” (Mihailescu and Drucker-Colin, 2000a), “functional down-regulation” (Marks et al., 1993), “long-lasting inactivation” (Kawai and Berg, 2001), or “persistent inactivation” (Lukas, 1991; Lukas et al., 1996; Ke et al., 1998). Nevertheless, further evolution in the understanding of nAChR functional state transitions is required to identify mechanisms and to specify processes involved in “desensitization” or any of the other, apparently distinct processes of longer-lasting nAChR functional loss. For example, there still is ambiguity about whether and to what extent “desensitization” due to conversion to a closed-channel form is “contaminated” with agonist-mediated open-channel block; it is not yet clear whether phosphorylation or other processes account for loss of nAChR function with agonist exposure (Gentry and Lukas, 2002). That is, before tangible definitions of desensitization or of other, longer-lasting phases of nAChR functional loss can be formulated beyond postulated conversions of agonist-receptor complexes to inactive forms, explanations for these phenomena require refinement.
Although most studies have shown functional loss for α4β2-nAChR on sustained exposure to nicotine, a recent electrophysiological study of human α4β2-nAChR heterologously expressed in a mammalian human embryonic kidney-derived cell line reported an enhancement of function under some experimental conditions following chronic exposure to nicotine (Buisson and Bertrand, 2001). This finding poses new questions about whether and how different expression systems and details of experimental design influence functional responsiveness of individual nAChR subtypes to chronic nicotinic ligand exposure.
This study examines functional responses of human α4β2- and α4β4-nAChR heterologously expressed in the SH-EP1 human epithelial line following pretreatment with nicotinic ligands. Whereas the α4β2-nAChR subtype is of obvious importance, nAChR β4 subunit messages as detected by in situ hybridization in nonhuman primate brain also are abundant and colocalize with α4 subunit messages (Quik et al., 2000). Moreover, α4 and β4 subunits combine to form a functional nAChR ion channel with high affinity for nicotine (Eaton et al., 2000). The current studies examined and contrasted effects of nicotine as a membrane-permeant agonist, carbamylcholine (carb) as an ionic nAChR agonist, and mecamylamine (mec) as a prototypical nAChR antagonist with potential as an aid to smoking cessation (Rose et al., 1998) and a therapeutic (Sanberg et al., 1997). A preliminary report of these findings has appeared (Gentry and Lukas, 2001).
Materials and Methods
Drug Dilutions.
All drugs were prepared fresh on the day of the assay as 10 mM stock solutions in cell culture medium (see cell culture) and diluted appropriately for each experiment. Efflux assay buffer consisted of 130 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 5 mM glucose, and 50 mM HEPES, pH 7.4. Ringer's buffer consisted of 115 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1.3 mM MgSO4, and 33 mM Tris supplemented with 1.5 mM NaN3. Buffer components as well as (−)-nicotine bitartrate, carb Cl, and mec HCl were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Model Cell Lines and Cell Culture.
The present study used low-passage (less than 50) SH-EP1 human epithelial cell clones stably and heterologously expressing human α4β2- or α4β4-nAChRs to examine functional responsiveness of receptors following pretreatment with nicotine or other nicotinic ligands. These clonal cell lines were generated using techniques that have been previously reported (Peng et al., 1999; Eaton et al., 2000; Lukas et al., 2002). Briefly, transfection with pcDNA3.1-zeo-human α4 and pcDNA3.1-hygro-human β2 constructs created in our laboratory [subcloned using human α4(S452) and β2 subunit cDNAs kindly provided by Dr. Ortrud Steinlein, Rheinishce-Friedrich-Wilhelms-Universitaet, Bonn, Germany] followed by isolation of a stable, high-expressing clonal line was accomplished to create SH-EP1-hα4β2 cells. Transfection with pCEP4-hygro-human α4 and pcDNA3.1-zeo-human β4 constructs created in our laboratory (the latter was subcloned using a human β4 subunit cDNA kindly provided by Dr. Jon Lindstrom, University of Pennsylvania, Philadelphia, PA) also, followed by isolation of a high-expressing clone, was used to generate SH-EP1-hα4β4 cells. Cells were maintained at 37°C, under 95% O2/5% CO2 in Dulbecco's modified Eagle's medium supplemented with 5% fetal bovine serum (Hyclone Laboratories, Logan, UT), 10% horse serum (Invitrogen, Carlsbad, CA), 1% sodium pyruvate (Cellgro AK; Mediatech Inc., Herndon, VA), 2% glutamine penicillin-streptomycin (Irvine Scientific, Santa Ana, CA), and 0.02% amphotericin B (Sigma). Cell culture medium was also supplemented with 0.01% hygromycin (Calbiochem, San Diego, CA) and 0.03% zeocin (Invitrogen) to maintain stable expression of α4 and β2 or β4 subunits.
Assays of nAChR Function.
86Rb+ efflux assays (Lukas and Cullen, 1988; Lukas et al., 2002) were performed using SH-EP1-hα4β2 or SH-EP1-hα4β4 cells with some assay modifications to account for the nicotinic ligand pretreatment protocol. Cells were cultured (∼2 × 105cells per 15.5-mm-diameter well; ∼150 μg of total cell protein per well) on Falcon 24-well culture plates (BD Biosciences, San Jose, CA) precoated with poly(d-lysine), mol. wt. 70,000 to 150,000 (Sigma). Cells were grown overnight to confluence, verified using light microscopy. 86RbCl (3.8 mCi/mg or 0.2 cpm/fmol at 40% counting efficiency) was obtained from PerkinElmer Life Sciences (Boston, MA).
In all cases, cells were loaded with 0.5 ml of serum- and antibiotic-supplemented cell culture medium also supplemented with86Rb+ (∼2 μCi per well) for 4 h or longer to ensure maximal86Rb+ uptake by the cells. Control samples were not subjected to ligand pretreatment but were otherwise processed identically. For 1-, 15-, or 60-min drug pretreatments, medium containing86Rb+ only was removed and replaced at specific times with medium containing86Rb+ plus the appropriate concentration of drug. For 24-h drug pretreatments, medium containing drug only was removed after 20 h and replaced for the final 4 h of pretreatment with medium containing86Rb+ plus the appropriate concentration of drug. Cells were kept in incubators in 5% CO2 at 37°C for drug pretreatments of more than 5 min and for 86Rb+ loading except during medium changes.
At the end of any drug pretreatment and86Rb+ loading period, medium was removed by aspiration to mark the beginning of the recovery period, and the “flip plate” (Lukas et al., 2002) method was employed to administer three rinses with 2 ml per well of drug- and86Rb+-free efflux assay buffer (22°C for recovery times of 5 min or less, 37°C for recovery times greater than 5 min) to remove extracellular86Rb+ and pretreatment drug. For rinse times of 3 min or more, the first rinse solution was applied for 1 min, the second rinse solution was applied for 1 min, and the third rinse solution was applied for the remainder of the experimentally defined length of time (1 min for 3-min recovery, 3 min for 5-min recovery, etc.). Each of the three rinses was for 20 s for 1-min recovery samples.
After the experimentally defined periods of recovery from ligand exposure, efflux assays were used to evaluate nAChR function. Some wells of cells on each 24-well cell culture plate were reserved for controls. Total 86Rb+efflux (or positive control condition) was defined by cells not exposed to ligand pretreatment and only exposed for 2 min to 1 ml of 1 mM carb alone in efflux buffer (“acute agonist challenge”). Nonspecific86Rb+ efflux was defined using other control samples exposed to efflux buffer alone or to efflux buffer containing both 1 mM carb and 100 μM d-tubocurarine (either negative control approach gave comparable results). Simultaneously, wells of cells that had received ligand pretreatment were exposed to 1 ml of 1 mM carb alone in efflux buffer for the 2-min acute agonist challenge period.
At the end of the efflux period, buffer samples were collected, and amounts of 86Rb+ released into extracellular fluids were quantified by Cerenkov counting using a Wallac Trilux system (40% efficiency) (PerkinElmer Life Sciences). One milliliter per well of 0.1% SDS/0.1 M NaOH was then added to lyse the cells and to prepare samples for determination of the amounts of remaining, intracellular86Rb+ (see Data Analysis below). Specific nAChR function was defined as total minus nonspecific 86Rb+efflux. Typical values for specific and nonspecific86Rb+ efflux in control cells not subjected to ligand pretreatment were 2500 cpm and 500 cpm, respectively, for SH-EP1-hα4β2 cells (loaded with 3000 cpm of 350,000 cpm of applied86Rb+) and 3500 cpm and 400 cpm, respectively, for SH-EP1-hα4β4 cells (loaded with 5000 cpm of 350,000 cpm of applied86Rb+).
Conditions for 86Rb+ Loading.
Prior to initiating experiments, a control study compared effects of three different methods for 15-min ligand pretreatment: 1) simply adding a concentrated aliquot of ligand in media to cells maintained in86Rb+ loading media, 2) using the flip plate method to replace86Rb+ loading media with86Rb+-free media containing ligand, and 3) using the flip plate method to apply ligand in86Rb+ loading medium. Regardless of the method used, double-normalized results obtained (seeData Analysis below) were nearly identical (data not shown). This indicated that final results were not influenced by differences in86Rb+ loading during ligand pretreatment.
Data Analysis.
To control for possible influences of ligand pretreatment and recovery conditions on86Rb+ loading, data were subjected to a double normalization process. Background radioactivity was subtracted from all samples. Amounts of86Rb+ loaded and present intracellularly at the start of the acute agonist challenge to initiate the functional efflux assay were determined as the sum for each sample of released (extracellular) and remaining (intracellular)86Rb+ present at the conclusion of the functional assay. Specific86Rb+ efflux was defined as total 86Rb+ efflux minus nonspecific 86Rb+ efflux (described above). Normalized specific86Rb+ efflux was then expressed as a percentage of specific86Rb+ efflux divided by86Rb+ loaded. Double normalized specific 86Rb+efflux was then expressed as a percentage of normalized specific86Rb+ efflux for experimental samples subjected to ligand pretreatment divided by normalized specific 86Rb+efflux for control samples not subjected to ligand pretreatment. Data were plotted as means ± S.E.M. using Prism 3.0 software (GraphPad Software Inc., San Diego, CA). Results from three or more independent experiments are shown. Appropriate to sample size and degrees of freedom, unpaired t tests (two comparisons) or one-way ANOVA followed by Bonferroni post-tests (three comparisons) or Tukey's post hoc tests (greater than three comparisons) were used to evaluate statistical significance of important data sets. Most of the statistically significant differences between and within panels of data are obvious, but statistically significant differences that are not as evident are specified in the narrative, whereas remarks about similarity between such sets of data refer to differences that are not statistically significant at the 95% confidence interval.
Radiolabeled Epibatidine Binding Studies.
Competition for [3H]epibatidine (EBDN) binding sites on cell membranes prepared from SH-EP1-hα4β2 cells (Lukas et al., 2002) was utilized as a sensitive method for quantifying residual nicotine in efflux buffer samples collected during successive rinses of cells pretreated for 48 h with nicotine. Assays were performed in a total volume of 200 μl of Ringer's buffer in a 96-well plate format. Each well used to define the concentration of nicotine in one rinse sample of efflux buffer by competition for EBDN binding sites contained 50 μl of a 400 pM concentration of EBDN (final concentration = 100 pM), 50 μl of efflux buffer from one rinse sample, and 100 μl of SH-EP1-α4β2 cell membrane suspension (diluted to ensure that binding site concentrations were no more than 10 pM). Total control binding was defined as the binding of EBDN in the absence of any rinse sample, which was substituted for by a 50-μl aliquot of Ringer's-NaN3 buffer. Following a 1-h incubation at room temperature (∼22°C), cell membrane-bound EBDN was separated from unbound EBDN using a cell harvester system (Inotech Biosystems International, Inc., Rockville, MD) by filtration through GF/C glass fiber filters (Inotech Biosystems International, Inc.) that had been presoaked in a 0.1% polyethyleneimine solution at 4°C for at least 30 min. Cell membranes embedded in the filters were subsequently rinsed twice with ice-cold Ringer's buffer before being transferred to Ready Safe Liquid Scintillation Cocktail (Beckman Coulter, Inc., Fullerton, CA) for determination of [3H] content by coincidence counting on a Wallac MicroBeta TriLux microplate liquid scintillation counter (PerkinElmer Life Sciences). Counts obtained were transformed for presentation to a percentage of control specific EBDN binding defined in each assay.
Results
Determination of Residual Drug Levels.
Because a recurring technical critique of some studies of effects of chronic drug treatment regards possible effects of residual pretreatment drug on the parameter being measured, experiments were conducted to ensure that the rinse method used in our nicotine pretreatment-acute challenge protocol was sufficient to remove pretreatment ligands, thereby allowing nAChR to recover from effects of drug pretreatment in a drug-free environment. Other studies showed that rinses of 30- to 60-s duration were adequate to allow equilibration in nicotine distribution between intra- and extracellular spaces (J. Fryer and R. D. Lukas, unpublished observations). Independent experiments were completed to measure residual nicotine levels present in buffers used to rinse SH-EP1-hα4β2 cells after nicotine pretreatment based on the ability of residual drug to activate nAChR-mediated86Rb+ efflux from fresh SH-EP1-hα4β2 cells (Fig. 1). These functional assays demonstrated that nicotine levels in first-rinse samples were >100-fold lower than the initial nicotine concentration (i.e., there was a more than 2 log unit, rightward shift in the dose-response profile for the first-rinse sample from that for acute nicotine effects). Residual nicotine present in second-rinse samples elicited ion flux only if initial nicotine concentrations were 1 mM, but with efficacy equivalent to 0.3 to 1 μM nicotine, indicating a 1000- to 3000-fold removal of drug. Levels of residual nicotine present in third-rinse samples had been reduced to below those capable of eliciting any 86Rb+ efflux; i.e., below 100 nM, a greater than 10,000-fold reduction. The efficiency of removal of nicotine in the second rinse appeared to be slightly lower following longer pretreatment times of 1 or 48 h relative to 20 s of nicotine pretreatment, but no pretreatment time-dependent difference in nicotine removal was observed after the third rinse. Thus, three rinses were routinely used to process samples.

Functional bioassay to assess effectiveness of removal of pretreatment nicotine. SH-EP1 cells heterologously expressing human α4β2-nAChR were pre-exposed to nicotine at the indicated concentration (abscissa, log M scale) for 20 s (top), 1 h (center), or 48 h (bottom). Pre-exposure medium was aspirated, and the pre-exposed cells were subjected to four successive rinses of 2 ml of drug-free assay buffer. The first rinse was for 1 min, and each subsequent rinse was for 2 min. Biologically active nicotine in the rinse buffers was assayed based on its ability to elicit function of human α4β2-nAChRs heterologously expressed in fresh SH-EP1 cells not previously exposed to nicotine (specific86Rb+ efflux; ordinate, percentage of control; see Materials and Methods). nAChR function was measured in the presence of fresh, acute nicotine (○) to generate a standard curve or in buffers collected from successive cell rinses (first rinse, ▴; 2nd rinse, ▿; third rinse, ♦; fourth rinse, ■). Smooth curves were drawn through data points, which are means ± S.E.M. from at least three separate experiments. Concentration-response profiles were used to determine nicotine levels in rinse buffers based on comparison with the standard curve and indicate progressively lower levels of residual nicotine with each rinse.
Additional and more sensitive bioassays using competition toward EBDN binding (see Materials and Methods) also were applied to quantify residual nicotine. Findings from these studies confirmed that third-rinse media contained nicotine at concentrations no higher than 10 nM (Fig. 2). Thus, three successive rinses with 2 ml per well of drug-free efflux buffer for a period of 1 min or greater reduced the concentration of drug in the media to a concentration below that capable of eliciting any nAChR channel opening detectable using ion flux assays.

Evaluation of residual nicotine levels after cell pretreatment based on radioligand binding competition assays. SH-EP1 cells heterologously expressing human α4β2-nAChRs were pre-exposed to nicotine at the indicated concentration (abscissa, log M scale) for 48 h. Samples were processed and rinse medium was collected as described in the legend to Fig. 1. Biologically active nicotine collected in rinse buffers was tested based on its ability to inhibit specific [3H]epibatidine binding (ordinate, percentage of control; see Materials and Methods) to cellular membrane fractions from fresh SH-EP1-hα4β2 cells. Assays done in the acute presence of fresh nicotine (○) were used to generate a standard nicotine dose-inhibition curve against which pretreatment nicotine dose-inhibition curves were compared for samples from successive cell rinses (first rinse, ▴; second rinse, ▿; third rinse, ♦; fourth rinse, ■). Smooth curves were drawn through data points, which were means ± S.E.M. from at least two separate experiments. Concentration-response profiles were used to determine nicotine levels in rinse buffers based on comparison with the standard curve and indicate progressively lower levels of residual nicotine with each rinse.
It is possible that residual nicotine at concentrations of 10 nM (achieved after ∼5 log unit reduction from an initial preincubation concentration of 1 mM) might be capable of stabilizing nonfunctioning nAChR conformers. However, an ∼5 log unit reduction from pretreatment concentrations on the order of 1 μM would yield residual nicotine concentration closer to 10 pM, much lower than the apparent binding affinity of nicotine for desensitized nAChR. Thus, demonstrated efficiency of the rinse protocol mitigates against, and at least establishes boundary conditions for, a role for residual pretreatment drug in long-lasting effects of drug pretreatment on nAChR function.
Effects of Nicotine Pretreatment on SH-EP1 Cell Human α4β2-nAChR Function.
Effects of prolonged nicotine exposure on heterologously expressed α4β2-nAChRs were examined. α4β2-nAChRs are the predominant form of high-affinity nicotine binding site in the brain, making them not only physiologically and pharmacologically important, but also the most likely effectors in nicotine dependence. Pre-exposure of SH-EP1-hα4β2 cells for 1 min to nicotine at concentrations as low as 1 μM produced an ∼20% loss of human α4β2-nAChR function when tested with an acute challenge dose of carb after 1 min of recovery from nicotine pre-exposure (Fig.3, top-left). Losses of function for 1-min pretreatment with 1 or 10 μM nicotine reversed (i.e., function elicited by an acute challenge dose of carb returned to control levels) after 1 h of recovery. However, after 1 h (or less) of recovery, losses of function were still evident in cells pre-exposed to 100 μM (∼20% loss for 1-h recovery) or 1 mM (∼30% loss for 1-h recovery) nicotine. In the absence of tangible definitions of desensitization and phases of longer-lasting losses of nAChR function induced by sustained nicotinic agonist exposure, explicit, operational definitions provided by Ke et al. (1998) distinguishing desensitization as a loss of nAChR function that is reversed during a 5-min period of recovery from agonist exposure from persistent inactivation, which was defined as a loss of nAChR function that is not reversed during a 5-min period of recovery from agonist exposure, were applied for their empirical utility. Thus, levels of persistent inactivation surviving the 5-min recovery period were ∼10% for pretreatment with 1 μM nicotine and ∼55% for pretreatment with 1 mM nicotine.

Time- and concentration-dependent effects of nicotine exposure on function of α4β2-nAChR measured using86Rb+ efflux assays. SH-EP1 cells heterologously expressing human α4β2-nAChRs were pre-exposed to nicotine at the indicated concentrations (abscissa, log M scale) for 1 min (top-left), 15 min (top-right), 1 h (bottom-left) or 24 h (bottom-right). Pre-exposure medium was aspirated and the pre-exposed cells were subjected to three successive rinses with 2 ml of drug-free assay buffer. The first and second rinses were for 1 min, and the third rinse consumed the remainder of the experimentally defined recovery time. Recovery times were 1 min (○), 3 min (▴), 5 min (▿), 15 min (♦), 30 min (■), or 60 min (▪). Specific86Rb+ efflux (ordinate, percentage of control), measured as described under Materials and Methods, was determined by eliciting nAChR function using acute (2 min) exposure to 1 mM carb. A 100% response was defined by untreated control cells' response to 1 mM carb. Linear curves are drawn through data points (means ± S.E.M. from at least three separate experiments). Over much of the dose profile, 1-min pretreatment results were significantly different from other results for either 15-min, or 1- or 24-h pretreatments. One- and 24-h pretreatment results were not statistically different from each other (ANOVA followed by Tukey's post hoc test, p < 0.05).
Extending the nicotine pretreatment period led to progressively longer-lasting and greater α4β2-nAChR functional losses, such that doses of nicotine as low as 10 nM (compare to smoker plasma/brain levels of 100 nM–1 μM) induced losses of nAChR function (Fig. 3).
Following nicotine exposure for 15 min, α4β2-nAChR exhibited a decline in functional responsiveness at pretreatment doses as low as 100 nM, 75% of α4β2-nAChR function was lost following exposure to 1 μM nicotine, and pretreatment with nicotine at 10 μM or greater caused nearly total loss of receptor function (Fig. 3, top-right). Whereas the functional loss resulting from pretreatment with 10 μM nicotine recovered almost completely within 1 h, nAChRs exposed to 1 mM nicotine regained only 20% of their function following 1 h in drug-free buffer. After 5 min of recovery, the extent of persistent inactivation varied between ∼15% for 100 nM nicotine pretreatment and ∼90% for pre-exposure to 1 mM nicotine.
Following a 1-h pretreatment (compared with 1- or 15-min pretreatment) with nicotine, additional functional decline was observed throughout the dose range examined (Fig. 3, bottom-left). The most remarkable effects were those at lower concentrations of nicotine. Pre-exposure to 10 nM nicotine caused ∼60% loss of α4β2-nAChR function at 1 min of recovery, ∼20% functional loss that persisted after 30 min of recovery, and ∼10% functional loss evident after 1 h of recovery. Losses in function at higher doses of nicotine pretreatment were even greater. Overall, for 1-h pretreatment, persistent inactivation remaining after 5 min of recovery ranged from ∼50% at 10 to 100 nM nicotine to no recovery of α4β2-nAChR response after 1 mM nicotine exposure.
Functional profiles for α4β2-nAChRs pretreated with nicotine for 24 h and allowed to recover for times ranging from 1 min to 1 h were similar to profiles for 1-h pretreatment (Fig. 3, bottom-right). One-hour recovery was sufficient to bring receptors exposed to 10 μM or less of nicotine back to levels of function matching that of untreated control cells. However, for the 5-min recovery period, losses of function ranged between ∼45% for 10 nM nicotine pretreatment and ∼100% for pre-exposure to 1 mM nicotine.
Effects of Nicotine Pretreatment on SH-EP1 Cell Human α4β4-nAChR Function.
Studies of effects of chronic nicotine exposure on function of human α4β4-nAChRs were also conducted because nAChR β4 subunit messages in nonhuman primate brain have been reported to be abundant and to colocalize with α4 subunit messages and because human α4β4-nAChRs also have high affinity for nicotine. Upon prolonged nicotine exposure, heterologously expressed, human α4β4-nAChRs in transfected SH-EP1 cells became less responsive to subsequent activation by 1 mM carb (Fig.4), and patterns of functional changes were similar to those of α4β2-nAChRs. Following 1 min of exposure to 1 mM nicotine, an ∼75% reduction in efflux from α4β4-nAChRs was observed (Fig. 4, top-left). Functional response remained at ∼50% of normal when cells were allowed to recover for 1 h in drug-free buffer. A dose of 100 μM nicotine led to ∼50% reduction in α4β4-nAChR function that did recover substantially (to ∼20% loss) following 1 h in drug-free buffer. One-minute pretreatment with nicotine at 1 μM or less did not induce persistent inactivation. Five-minute recovery allowed α4β4-nAChR function to return to ∼80%, 70%, or 40% of control, respectively, after 10 μM, 100 μM, or 1 mM nicotine pre-exposure.

Time- and concentration-dependent effects of nicotine exposure on function of α4β4-nAChR measured using86Rb+ efflux assays. SH-EP1 cells heterologously expressing human α4β4-nAChR were pre-exposed to nicotine at the indicated concentrations (abscissa, log M scale) for 1 min (top-left), 15 min (top-right), 1 h (bottom-left), or 24 h (bottom-right). Samples were processed as described in the legend to Fig. 3. Recovery times were 1 min (○), 3 min (▴), 5 min (▿), 15 min (♦), 30 min (■), or 60 min (▪). Specific86Rb+ efflux (ordinate, percentage of control) was measured as described in the legend to Fig. 3 (means ± S.E.M. from at least three separate experiments). Results were not significantly different for 1- or 24-h pretreatments at equivalent nicotine doses and recovery times, but these results and results from 1- and 15-min pretreatments were significantly different from each other over the entire dose profile (ANOVA followed by Tukey's post hoc test, p < 0.01).
α4β4-nAChRs exposed to nicotine for 15 min showed slightly greater functional decline than was observed following only 1 min of nicotine exposure and slightly less functional loss than was observed for α4β2-nAChRs following an equivalent nicotine exposure (Fig. 4, top-right). α4β4-nAChR function did not return to normal following 1 h of recovery in drug-free buffer except when pretreatment nicotine concentration was less than 100 nM. Persistent inactivation was observed at all doses tested (10 nM–1 mM).
Increasing the pretreatment time to 1 or 24 h led to greater loss of function for α4β4-nAChRs (Fig. 4, bottom, left and right). For example, 24-h nicotine pretreatment and 1-min recovery was significantly different from 1-min pretreatment and 1-min recovery (p < 0.05). Effects at 10 or 100 nM nicotine became more profound. Losses of function were ∼35 to 80% after 1-min recovery and remained at ∼10 to 25% after 1-h recovery. Pretreatment with 1 μM nicotine for 1 h followed by 1 min of recovery produced ∼75% loss of receptor function, and increasing the pretreatment time to 24 h completely eliminated responses to subsequent carb challenge. Function was entirely lost for 1- or 24-h pretreatment with 10 μM to 1 mM nicotine after 1 min of recovery, although function returned to between 30 and 89% of control after 1 h in drug-free medium. Using the persistent inactivation criteria, losses in function after 5-min recovery ranged between ∼30% at 10 nM nicotine and ∼90% at 1 mM nicotine.
To summarize the results of nicotine pre-exposure on α4β2- and α4β4-nAChRs, the extent of functional loss for each subtype depended on duration of nicotine pretreatment, duration of recovery in nicotine-free buffer, and concentration of nicotine during pretreatment. Loss of nAChR function occurred following pretreatment with low (10–100 nM) concentrations of nicotine, but only for pretreatment times of 15 min or longer.
Effects of Carbamylcholine Pretreatment on SH-EP1 Cell Human α4β2-nAChR Function.
carb was chosen for study as a representative ionic agonist to allow comparisons of its ability and that of nicotine as a membrane-permeant agonist to induce changes in nAChR function. One-minute pretreatments with 0.1 or 1 mM carb caused a reduction in heterologously expressed, human α4β2-nAChR function of 15% and 25%, respectively, when measured with a subsequent, acute carb challenge (Fig. 5, top-left). In both cases, receptor function fully recovered within 30 min. One-min pretreatment with carb concentrations of 10 μM or less had no effect. Only 100 μM or 1 mM pretreatments produced persistent inactivation.

Time- and concentration-dependent effects of carb exposure on function of α4β2-nAChR measured using86Rb+ efflux assays. SH-EP1 cells heterologously expressing human α4β2-nAChRs were pre-exposed to carb at the indicated concentrations (abscissa, log M scale) for 1 min (top-left), 15 min (top-right), 1 h (bottom-left) or 24 h (bottom-right). Otherwise, samples were processed as described in the legend to Fig. 3. Recovery times were 1 min (○), 3 min (▴), 5 min (▿), 15 min (♦), 30 min (■), or 60 min (▪). Specific86Rb+ efflux (ordinate, percentage of control) was measured as described in the legend to Fig. 3 (means ± S.E.M. from at least three separate experiments). For 1-min pretreatments at equivalent carb doses and recovery times, results were significantly different (less functional loss for 1-min pretreatment) for 1-min recovery at high carb doses, but 15-min, 60-min, and 24-h pretreatments were not significantly different from each other across the dose profile (ANOVA followed by Tukey's post hoc test,p < 0.01). Comparing results for studies of α4β2-nAChR, carb pretreatment produced significantly less functional inhibition across all dose profiles compared with effects of nicotine under equivalent dose and pretreatment and recovery times (ANOVA followed by Bonferroni post hoc test,p < 0.05) (see Fig. 3).
Fifteen-minute pretreatment with 1 mM carb reduced α4β2-nAChR function by more than 80% (Fig. 5, top-right). Following a 1-h recovery period, α4β2-nAChR function had returned to 75% of untreated control. With a 15-min pretreatment, functional effects were seen at concentrations as low as 10 μM. The 30% reduction in α4β2-nAChR function observed under these conditions recovered fully within 15 min. However, 15-min pre-exposure to carb at concentrations between 10 nM and 1 μM produced no functional loss, although α4β2-nAChR persistent inactivation of ∼25%, ∼50%, and ∼70% was observed for pre-exposure to 10 μM, 100 μM, and 1 mM carb, respectively.
An additional decline in receptor function was observed following 1-h pretreatment with carb (Fig. 5, bottom-left) when compared with 15 min of pretreatment. Most notable differences included some functional loss at lower concentrations (100 nM–1 μM carb) and an increase in the extent of functional loss at higher concentrations, e.g., to 100% for 1-h pretreatment with 1 mM carb followed by 1-min recovery. However, persistent inactivation was detectable at doses as low as 100 nM. Functional dose-response profiles for α4β2-nAChR inactivation following 24-h pretreatment with carb and 1-min recovery were similar to the profiles for 1-h pretreatment and equivalent times of recovery (Fig. 5, bottom-right).
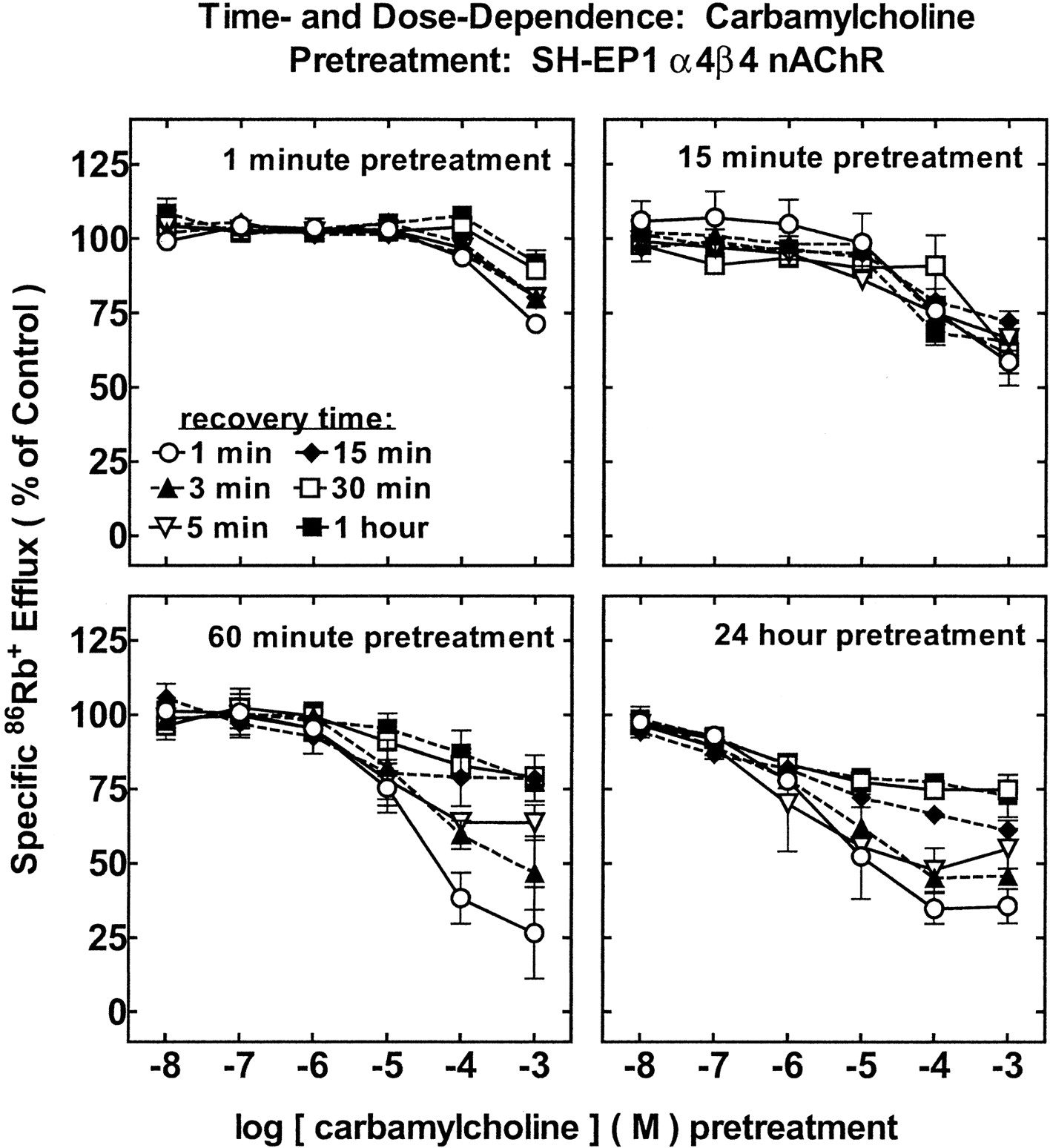
Effects of Carbamylcholine Pretreatment on SH-EP1 Cell Human α4β4-nAChR Function.
Following 1 min of carb exposure, heterologously expressed, human α4β4-nAChRs functioned normally except when pretreated with concentrations of carb greater than 100 μM (Fig. 6, top-left). One-minute exposure to 1 mM carb and 1-min recovery led to a reduction of efflux response (∼20% decline). Receptor function returned to normal after 30 min in drug-free buffer. Greater α4β4-nAChR functional effects were observed following 15 min of carb exposure (Fig. 6, top-right). After 1 h of incubation, pretreatment concentrations of carb as low as 10 μM began to have an effect on α4β4-nAChR function (Fig.6, bottom-left), although function recovered fully within 1 h of drug-free recovery. Functional losses of ∼60 to 70% occurred for 1-h pretreatment with 0.1 to 1 mM carb, and function recovered to only ∼80% of control after 1 h of recovery. Increasing carb exposure time from 1 to 24 h did not change α4β4-nAChR functional inactivation (Fig. 6, bottom-right), although the range for functional loss remaining at 1 h of recovery extended to 100 nM carb (∼10% loss; ∼25% at 0.01 to 1 mM carb). Persistent inactivation first became evident for 1-min pretreatment with 1 mM carb, for 15 min to 1 h of pretreatment with 10 μM carb, or for 24-h pretreatment with 100 nM carb.

Time- and concentration-dependent effects of carb exposure on function of α4β4-nAChR measured using86Rb+ efflux assays. SH-EP1 cells heterologously expressing human α4β4-nAChRs were pre-exposed to carb at the indicated concentrations (abscissa, log M scale) for 1 min (top-left), 15 min (top-right), 1 h (bottom-left), or 24 h (bottom-right). Otherwise, samples were processed as described in the legend to Fig. 3. Recovery times were 1 min (○), 3 min (▴), 5 min (▿), (♦) 15 min, (■) 30 min, or (▪) 60 min. Specific86Rb+ efflux (ordinate, percentage of control) was measured as described in the legend to Fig. 3 (means ± S.E.M. from at least three separate experiments). Linear curves are drawn through data points (means ± S.E.M. from at least three separate experiments). Results were not significantly different for 1- or 24-h pretreatments at equivalent carb doses and recovery times, but these profiles and those for 1- and 15-min pretreatments were significantly different from each other across the dose profile (ANOVA followed by Tukey's post hoc test, p < 0.05). carb pretreatment produced significantly less functional loss of α4β4-nAChR than did nicotine under all otherwise equivalent conditions tested (ANOVA followed by Bonferroni post hoc test,p < 0.05) (see Fig. 4). Moreover, 1-min or 15-min carb pretreatment produced significantly greater functional loss for α4β2-nAChR than for α4β4-nAChR (unpaired t test,p < 0.01) (see Fig. 5).
In summary, differences were observed in carb-treated α4β2- and α4β4-nAChRs when increasing pretreatment time and concentration. Differences were also observed between α4β2- and α4β4-nAChR responses to 1-min or 15-min carb treatment, with α4β2-nAChRs showing greater sensitivity to carb-induced functional loss (unpairedt test, p < 0.01).
Effects of Mecamylamine Pretreatment on SH-EP1 Cell Human α4β2-nAChR Function.
Studies were done to determine whether chronic exposure to mec could induce long-lasting nAChR functional changes, in part because of its potential utility as a smoking cessation aid and therapeutic, but also because its noncompetitive antagonism might occur through open-channel block, possibly mimicking effects of nicotine acting at high concentrations and/or chronically. Pretreatment with mec induced functional losses in heterologously expressed, human α4β2-nAChRs that persisted after removal of the drug from media (Fig. 7). It should be noted, however, that experiments evaluating effectiveness of rinses for removing mec after pretreatment (not shown) indicated that our protocol yielded an ∼1000-fold reduction in mec concentration, meaning that for samples pretreated with 1 mM mec, enough residual drug could remain to antagonize as much as 50% of receptor function. Nevertheless, these studies showed that residual mec levels would be less than those needed to acutely produce nAChR functional block if pretreatment with mec was done at concentrations of 100 μM or less. Therefore, although efficiency of mec removal was lower than for removal of nicotine, any functional loss observed following pretreatment with lower concentrations of mec can be attributed to desensitization and/or persistent inactivation rather than channel block. α4β2-nAChRs were functioning at 25% of normal following 100 μM mec pre-exposure for 1 min and 1 min of recovery, but 5 to 60 min of recovery allowed function to return to ∼85% of control levels (Fig. 7, top-left). Pretreatment for 1 min with mec at concentrations less than 10 μM had negligible effects on α4β2-nAChR function. Persistent inactivation evident after 5 min of recovery was about 15% for 1-min pretreatment with 10 μM mec.

Time- and concentration-dependent effects of mec exposure on function of α4β2-nAChR measured using86Rb+ efflux assays. SH-EP1 cells heterologously expressing human α4β2-nAChRs were pre-exposed to mec at the indicated concentrations (abscissa, log M scale) for 1 min (top-left), 15 min (top-right), 1 h (bottom-left) or 24 h (bottom-right). Otherwise, samples were processed as described in the legend to Fig. 3. Recovery times were 1 min (○), 3 min (▴), 5 min (▿), 15 min (♦), 30 min (■), or 60 min (▪). Specific86Rb+ efflux (ordinate, percentage of control) was measured as described in the legend to Fig. 3 (means ± S.E.M. from at least three separate experiments). Results for 1- or 24-h pretreatments at equivalent mec doses and recovery times were not significantly different, but these profiles and those for 1- and 15-min pretreatments were significantly different from each other across the dose profile (ANOVA followed by Tukey's post hoc test,p < 0.01). mec produced less functional loss of α4β2-nAChR than did nicotine, but more than carb, across virtually all conditions (ANOVA followed by Bonferroni post hoc test,p < 0.01; see Figs. 3 and 5).
After 1 min of recovery from 15-min pre-exposure to 10 μM mec, function was ∼75% of control, but 1 h after drug removal, function was at control levels (100%). Pretreatment with mec for 1 h led to receptor inactivation occurring at lower concentrations (Fig. 7, bottom-left). For example, pretreatment with mec at concentrations as low as 10 nM led to α4β2-nAChR functional loss of 25%, although this effect was fully reversed within 30 min of recovery in drug-free media. One hour was insufficient for complete recovery from functional loss after pretreatment with mec of concentrations 1 μM or higher. Defined as the loss of function after 5 min of drug removal, persistent inactivation of α4β2-nAChRs ranged from ∼10% at 10 nM mec to ∼90% at 100 μM mec. Concentration-response profiles following 24-h mec pretreatment were similar to those following 1 h of mec exposure (Fig. 7, bottom-right). Persistent inactivation ranged from 20% at 100 nM mec to ∼100% at 10 μM mec or higher.
Effects of Mecamylamine Pretreatment on SH-EP1 Cell Human α4β4-nAChR Function.
One minute of pretreatment with as much as 10 μM mec had no effect on subsequently assessed function of heterologously expressed, human α4β4-nAChRs (Fig.8, top-left). Following 1 min of 1 mM mec exposure and 1-min recovery, α4β4-nAChR functional response was 50% of untreated control. Functional assessment after 5 min of recovery found α4β4-nAChRs to function at 70% of normal, and recovery was to 95% of control after 0.5 to 1 h in drug-free medium. No significant differences were observed if mec pretreatment times were extended to 15 min (Fig. 8, top-right). One or 24 h of pretreatment with 1 mM mec yielded only modest increases in the extent of functional loss (Fig. 8, bottom, left and right). However, pre-exposure to 100 μM mec for 1 h induced an ∼30% loss in function, although recovery occurred within 5 to 60 min of drug-free incubation.

Time- and concentration-dependent effects of nicotine exposure on function of α4β4-nAChR measured using86Rb+ efflux assays. SH-EP1 cells heterologously expressing human α4β4-nAChRs were pre-exposed to mec at the indicated concentrations (abscissa, log M scale) for 1 min (top-left), 15 min (top-right), 1 h (bottom-left), or 24 h (bottom-right). Otherwise, samples were processed as described in the legend to Fig. 3. Recovery times were 1 min (○), 3 min (▴), 5 min (▿), 15 min (♦), 30 min (■), or 60 min (▪). Specific86Rb+ efflux (ordinate, percentage of control) was measured as described in the legend to Fig. 3 (means ± S.E.M. from at least three separate experiments). Linear curves are drawn through data points (means ± S.E.M. from at least three separate experiments). Results were not significantly different for 1- or 15-min pretreatments at equivalent mec doses and recovery times, but results for pretreament for 1 h, compared with 1 or 15 min, at 100 μM mec were significantly different for 1- and 3-min recovery times (ANOVA followed by Tukey's post hoc test, p < 0.05). mec pretreatment produced significantly less functional loss of α4β4-nAChRs than did nicotine under all otherwise equivalent conditions tested (see Fig. 4) and than did carb for 1- or 24-h pretreatments (ANOVA followed by Bonferroni post hoc test,p < 0.01; see Fig. 6). For nearly all conditions tested, mec pretreatment produced significantly greater functional loss for α4β2-nAChRs than for α4β4-nAChRs (unpaired ttests, p < 0.05; see Fig. 7).
In summary, α4β2-nAChRs exhibited a substantial reduction in functional responsiveness following mec pretreatments, whereas α4β4-nAChRs were less vulnerable to persistent inactivation caused by mec exposure.
Discussion
Major Findings.
One of the major findings of this study is that pre-exposure to nicotine induces losses in function of human α4β2- or α4β4-nAChRs heterologously expressed in the SH-EP1 cell line. Losses in function, measured at maximally efficacious challenge doses of acute agonist, are substantial after pretreatment with concentrations of nicotine (∼100 nM–1 μM) found in the plasma of human smokers (Russell et al., 1980). These losses in function are even more pronounced after pretreatment at higher doses of nicotine, i.e., at concentrations that have acute functional efficacy equivalent to acetylcholine when acting at active synapses. Using the operational definition of persistent inactivation as the loss of function after 5 min of drug-free incubation (Ke et al., 1998), the present study found that 1 h of nicotine pretreatment at “smoker doses” of 100 nM to 1 μM induces 60 to 80% loss of human α4*-nAChR function, and recovery to 90% of control levels requires ∼1 h after drug removal.
A second major finding is that carb is less effective than nicotine at inducing functional loss of either α4β2- or α4β4-nAChRs as assessed by functional inactivation potency (dose dependence), rate of functional inactivation (time dependence), and extent of functional inactivation (percentage of functional loss for a given dose and time of drug exposure). A third major finding is that human α4β2- and α4β4-nAChR subtypes differ in their sensitivity to functional inactivation after mec pretreatment. Pretreatment with mec powerfully induces functional inactivation of α4β2-nAChRs but has no lasting effect on α4β4-nAChR function except when mec concentrations during pretreatment are greater than 100 μM. These observations provide insight into molecular bases for functional inactivation.
Ligand and nAChR Subtype Selectivity of Functional Inactivation.
As an ionic species, carb does not freely traverse the membrane lipid bilayer and gain access to the intracellular space as readily as nicotine does. The finding that carb induces α4*-nAChR receptor desensitization and inactivation suggests that ligand access to cytoplasmic or other buried domains is not required for functional inactivation. Nevertheless, whether expressed in absolute terms or normalized to agonist acute functional efficacy and potency, nicotine has more persistent inactivation potency (1-h pretreatment, 5-min recovery) than carb at either α4β2- or α4β4-nAChRs (Table1). This suggests that the ability of nicotine to access the cell interior may indeed play a role in its strong induction of persistent inactivation. Whether expressed absolutely or after normalization to agonist acute potency, persistent inactivation potency of carb is higher (>6- to >20-fold) at α4β2- than at α4β4-nAChRs (Table 1), suggesting an influence of nAChR β subunit sequence on this effect.
Comparisons between nicotinic ligand functional inactivation and acute activation/inhibition potencies
Absolute or normalized persistent inactivation potency for mec is lower than that for nicotine but higher than that for carb at either α4β2- or α4β4-nAChRs (Table 1). The higher sensitivity of α4β2-nAChRs than α4β4-nAChRs to mec-induced persistent inactivation (Table 1) implicates β2-β4 subunit sequence differences in this effect. mec acts as an open-channel blocker of nAChRs (Varanda et al., 1985) and is not a steric inhibitor of nicotinic agonist binding to α4β2- or α4β4-nAChR (Peng et al., 1999; Eaton et al., 2000). Although it is possible that mec induces persistent inactivation of spontaneously opening α4*-nAChRs or activates transient channel openings not detected using contemporary ion flux or electrophysiological measurements, our data with mec suggest that channel opening is not required for conversion of α4*-nAChR to an inactivated state(s). Some noncompetitive antagonists as well as agonists can induce desensitization and/or persistent inactivation of other nAChR subtypes (Lukas, 1991). As previously discussed, open-channel blocking ability may be a common feature for agonists and antagonists that produce functional inactivation, although other possibilities include mutual interaction at novel regulatory sites (Lukas, 1991) or stabilization of distinct, closed-channel, nAChR conformations (Changeux et al., 1990). However, these conclusions are based on the assumption that mec is efficiently removed from association with α4*-nAChR after pretreatment, as was shown for nicotine.
Also of relevance is the finding that concentrations of nicotine below those needed to acutely activate α4*-nAChR induce nAChR persistent inactivation. This supports the hypothesis that channel activation is not required for α4*-nAChR persistent inactivation. It also is consistent with potential mechanisms involving open-channel block (but of infrequently opening channels), allosteric interactions at distinct ligand binding sites, or stabilization of α4*-nAChR in closed-channel states.
Comparisons to Other in Vitro Findings.
Findings in the present study using the SH-EP1 human epithelial cell expression system are consistent with numerous reports of functional inactivation of α4β2-nAChRs from different species in diverse experimental systems following chronic nicotine exposure (Peng et al., 1994; Hsu et al., 1996; Fenster et al., 1997; Olale et al., 1997). Other nAChR subtypes that undergo functional inactivation following prolonged nicotine exposure include human muscle-type α1*-nAChR naturally expressed in TE671/RD cells, α7-nAChR and α3*-nAChR expressed inXenopus oocytes, and ganglionic α3*-nAChR naturally expressed in rat PC12 or human SH-SY5Y cells (reviewed in Gentry and Lukas, 2002). Additionally, evidence from in vivo studies that allow evaluation of nicotine effects and recovery times on the order of days are consistent with the hypothesis that chronic nicotine inactivates brain nAChR (reviewed in Gentry and Lukas, 2002).
However, contrary to these and the current observations are reports that function of human α4β2-nAChR heterologously expressed in the K-177 cell variant of the HEK293 human embryonic kidney fibroblast cell line can be up-regulated in response to nicotine (0.1–1 μM for 8 h–7 days), although exposure to 10 μM or higher nicotine produced functional inactivation of α4β2-nAChR, whether measured using ion flux or whole-cell electrophysiological recording (Gopalakrishnan et al., 1997; Buisson and Bertrand, 2001). Reasons for these differences are not immediately evident, although the choice(s) of agonist(s), the concentration(s) of agonist(s) used for pretreatment and acute functional assessment, and the experimental model (host cell environment) used may be important factors. Use of oocyte as opposed to mammalian cell expression systems, studies of native or heterologously expressed nAChR, differences in temperature for cell maintenance, culture medium composition (presence of serum), and presence of kinases, phosphatases, or other means for post-translational modification also may influence effects of chronic ligand exposure on nAChR function (Buisson and Bertrand, 2001). Additionally, species differences, variations in pH, assay temperature, passage number (if cell lines are used) and confluence of cells (amount of cell-to-cell contact) (Boyd, 1987), and subunit sequence variations may play a role as well. Thus, more work is needed to reconcile whether there truly are discrepant observations and/or diverse responses of α4β2-nAChRs to chronic nicotine exposure and, if so, to elucidate the reasons for these differences.
Relationships Between Chronic Nicotine-Induced Functional Inactivation and nAChR Numerical Up-Regulation.
Several factors can account for changes in the level of measurable nAChR function. Among these are enhanced/reduced conductance of each channel, enhanced/reduced frequency of channel opening, or longer/shorter channel open time. Among these possibilities, changes brought about by nicotinic ligands may be influenced by membrane lipid composition effecting conformational equilibria of nAChR (Baenziger et al., 2000), electrostatic interactions (Song and Pedersen, 2000), enzymatic carboxymethylation (Kloog et al., 1980), conformation changes (Baenziger and Chew, 1997), phosphorylation (Hsu et al., 1997; Fenster et al., 1999a), endocytosis/exocytosis (Peng et al., 1994), or number of nAChRs on the cell surface (Ke et al., 1998).
Regarding numbers of nAChRs expressed at the cell surface, substantial evidence indicates that chronic nicotine exposure leads to up-regulation of nAChR binding sites via a post-transcriptional process(es) (reviewed in Gentry and Lukas, 2002). The proposition that nAChR desensitization initiates up-regulation (Marks et al., 1983;Schwartz and Kellar, 1985; Fenster et al., 1999b) seems untenable given that the processes occur with widely different time and concentration dependencies and because any up-regulated nAChR appearing on the cell surface would also quickly become inactivated in the presence of pretreatment ligand (Lapchak et al., 1989; Collins et al., 1990b;Lukas, 1991). Perhaps a different issue is the rate of recovery from functional inactivation. Although not a significant difference, human α4β2-nAChR responses on SH-EP1 cells exposed to nicotine for 24 h did recover somewhat more quickly than nAChR on cells exposed to nicotine for only 1 h. Perhaps this reflects the presence of more surface nAChR or nAChR precursors due to an increase in numbers of nAChR-like radioligand binding sites as would occur over the longer period of nicotine exposure.
Perspectives.
The current evaluation of effects of prolonged nicotinic ligand exposure on function of human α4β2- and α4β4-nAChR provides a comprehensive characterization of nAChR desensitization and persistent inactivation and adds to findings indicating that all nAChR subtypes experience functional loss in response to chronic ligand exposure. Knowledge of nAChR subtype selectivity with regard to rates, extents, and nicotinic ligand concentration dependence of nAChR activation, desensitization, and persistent inactivation is critical toward development of rational approaches for nicotinic drug therapies in treatment of neuropsychiatric disorders and successful approaches to smoking cessation recognizing that tobacco use is a form of nicotine self-medication (reviewed in Gentry and Lukas, 2002). This is because, as the current studies show, acute and more chronic effects of nicotinic drugs are integrated across nAChR subtypes, drug exposure time, and drug dose at sites of nAChR expression, inducing some balance between activation and inactivation not only of nAChR subtypes, but also of the excitatory or inhibitory effects they have on specific neuronal processes and functions.
Acknowledgments
We are very grateful for the kind gifts of human nicotinic acetylcholine receptor subunit DNA from Dr. Ortrud Steinlein of the Rheinische-Friedrich-Wilhelms-Universitaet and from Dr. Jon M. Lindstrom of the University of Pennsylvania. We also appreciate statistical analysis consultations with Drs. Kris Horn and Curtis Bay.
Footnotes
-
↵1 Current Address: Department of Behavioral Science, College of Medicine, University of Kentucky, Lexington, KY 40546-0236.
-
This work was supported by the Ford Foundation, by scholarship support from the International Chapter, P.E.O. Sisterhood, by a grant from the Arizona Disease Control Research Commission (10011), by endowment and/or capitalization funds from the Men's and Women's Boards of the Barrow Neurological Foundation, and by the Robert and Gloria Wallace Foundation, and was conducted in part in the Charlotte and Harold Simensky Neurochemistry of Alzheimer's Disease Laboratory. Work was previously presented in preliminary form (Gentry and Lukas, 2001). The contents of this report are solely the responsibility of the authors and do not necessarily represent the views of the aforementioned awarding agencies.
-
DOI: 10.1124/jpet.102.041756
- Abbreviations:
- nAChR
- nicotinic acetylcholine receptor
- carb
- carbamylcholine
- EBDN
- [3H]epibatidine
- mec
- mecamylamine
- ANOVA
- analysis of variance
- Received July 16, 2002.
- Accepted September 11, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}