


Abstract
Opioid receptors often couple to multiple effectors within the same cell. To examine potential mechanisms that contribute to the specificity by which δ-receptors couple to distinct intracellular effectors, we stably transfected rat pituitary GH3 cells with cDNAs encoding for δ-opioid receptors. In cells transfected with a relatively low δ-receptor density of 0.55 pmol/mg of protein (GH3DOR), activation of δ-receptors produced inhibition of adenylyl cyclase activity but was unable to alter L-type Ca2+ current. In contrast, activation of δ-receptors in a clone that contained a higher density of δ-receptors (2.45 pmol/mg of protein) and was also coexpressed with μ-opioid receptors (GH3MORDOR), resulted in not only the expected inhibition of adenylyl cyclase activity but also produced inhibition of L-type Ca2+ current. The purpose of the present study was to determine whether these observations resulted from differences in δ-opioid receptor density between clones or interaction between δ- and μ-opioid receptors to allow the activation of different G proteins and signaling to Ca2+ channels. Using the δ-opioid receptor alkylating agent SUPERFIT, reduction of available δ-opioid receptors in GH3MORDOR cells to a density similar to that of δ-opioid receptors in the GH3DOR clone resulted in abolishment of coupling to Ca2+ channels, but not to adenylyl cyclase. Furthermore, although significantly greater amounts of all G proteins were activated by δ-opioid receptors in GH3MORDOR cells, δ-opioid receptor activation in GH3DOR cells resulted in coupling to the identical pattern of G proteins seen in GH3MORDOR cells. These findings suggest that different threshold densities of δ-opioid receptors are required to activate critical amounts of G proteins needed to produce coupling to specific effectors and that δ-opioid receptors couple more efficiently to adenylyl cyclase than to L-type Ca2+channels.
Opioids are members of the large superfamily of G protein-coupled receptors that traverse the plasma membrane seven times and activate intracellular G proteins (Raynor et al., 1994). δ-Opioid receptors often couple to multiple effectors within the same cell and/or tissue. In neuroblastoma cells δ-opioid receptors regulate adenylyl cyclase activity (Prather et al., 1994c), Ca2+ channels (Hescheler et al., 1987), and phospholipase C (Jin et al., 1992). However, in other tissues the coupling of δ-opioid receptors to intracellular effectors appears to be more restricted. For example, in rat dorsal root ganglion neurons, δ-opioid receptors are able to inhibit adenylyl cyclase activity (Makman et al., 1988), but fail to couple to Ca2+channels (Moises et al., 1994).
We have studied the binding and functional properties of cloned μ- and δ-opioid receptors expressed either alone or in combination in GH3 cells (Piros et al., 1995, 1996a). Consistent with the previous studies in dorsal root ganglion neurons, we found that δ-opioid receptors expressed alone in GH3DOR cells bound opioid ligands and that their activation led to the inhibition of adenylyl cyclase without having an effect on Ca2+ channel activity (Piros et al., 1996a). In contrast, μ-opioid receptors in GH3MOR cells coupled to both adenylyl cyclase and L-type Ca2+ channels (Piros et al., 1995). Interestingly, activation of either δ- or μ-opioid receptors in GH3 cells that contained both receptor subtypes (GH3MORDOR cells) also resulted in inhibition of both adenylyl cyclase and L-type Ca2+ channel activity.
The dissimilar signaling of δ-opioid receptors expressed in these clones could be explained by the observation that GH3DOR cells contained a relatively low density of δ-opioid receptors (0.55 pmol/mg of protein) compared with the greater number of δ-opioid receptors expressed in GH3MORDOR cells (2.45 pmol/mg of protein). If confirmed, this would indicate that different thresholds of δ-opioid receptor density might be required for coupling to different effectors in the same cell. This is supported by studies in which increasing the density of transfected G protein-coupled receptors enhances both agonist potency and efficacy for regulation of several intracellular effectors. This has been investigated most extensively for β2-adrenergic stimulation of adenylyl cyclase (Whaley et al., 1994), but also has been observed for opioid inhibition of adenylyl cyclase (Hirst et al., 1997). Additionally, in CHO cells transfected with low densities of M2-muscarinic receptors, carbachol produces only inhibition of adenylyl cyclase. However, when M2-receptor density is increased, activation of phospholipase C is also observed (Ashkenazi et al., 1987). Lastly, overexpression of α2-adrenergic receptors in CHO cells results in the ability of the receptor to couple not only to Giα but also to Gsα, a G protein with which it does not normally couple (Eason et al., 1992). Consequently, it is possible that δ-opioid receptors couple to a unique blend of intracellular effectors in neurons, depending upon the stoichiometric ratio of receptors, G proteins, and effectors present within specific neurons.
Because δ-opioid receptors are able to couple to Ca2+ channels only in GH3cells that contain both μ- and δ-opioid receptors, it is also possible that these receptors interact to allow the activation of different G proteins to allow signaling to Ca2+channels. Indeed, recent studies have demonstrated that the binding and signaling of some G protein-coupled receptors, including opioid receptors can be influenced by the formation of heterodimers (Hebert et al., 1996; Jordan and Devi, 1999). In the case of GABAB receptors, coupling to K+ channels is not observed unless the GABABR1 and GABABR2 subunits are expressed together (White et al., 1998). Furthermore, the coexpression of κ- and δ-opioid receptors, both of which function individually, produces a heterodimer combination with different binding and functional properties seen when either of the individual receptors is expressed alone (Jordan and Devi, 1999). The changes in receptor signaling that are seen upon expression of receptors either alone, or in combination with another receptor subtype, are presumably caused by the differential activation of G proteins. Hence, the formation of μ/δ-opioid receptor dimers may enable coupling to Ca2+ channels through the activation of specific G proteins not activated when δ-opioid receptors are expressed in GH3 cells alone.
Therefore, the purpose of the present study was to determine whether the differential coupling of δ-opioid receptors to these two effectors results from differences in receptor density and/or the activation of specific G proteins. Using the δ-opioid receptor alkylating agent SUPERFIT, reduction of δ-opioid receptors in GH3MORDOR cells to a density similar to that of δ-opioid receptors in the GH3DOR cells resulted in abolishment of coupling to Ca2+ channels, but not to adenylyl cyclase. Furthermore, although significantly greater amounts of all G proteins were activated by δ-opioid receptors in GH3MORDOR cells, δ-opioid receptor activation in GH3DOR cells resulted in coupling to the identical pattern of G proteins as that in GH3MORDOR cells. These findings suggest that a threshold density of δ-opioid receptors is required to activate critical amounts of individual G proteins needed to produce coupling to certain effectors and that δ-opioid receptors couple more efficiently to adenylyl cyclase than to L-type Ca2+ channels.
Experimental Procedures
Materials.
[α-32P]GTP (3000 Ci/mmol) and antisera (EC2 and GC2) were purchased from NEN Life Science Products (Boston, MA). SUPERFIT was a generous gift from Dr. Kenner C. Rice (National Institutes of Health, Bethesda, MD). [3H]Adenine (27 Ci/mmol), [3H]diprenorphine (36 Ci/mmol), [α-32P]ATP (17 Ci/mmol), and ECL reagents were purchased from Amersham Life Science Products (Arlington Heights, IL). [3H]DPDPE (18 Ci/mmol) was provided by the National Institute on Drug Abuse (Bethesda, MD). DPDPE, CTOP, and somatostatin were obtained from Peninsula Laboratories (Belmont, CA). All tissue culture reagents, including geneticin (G418) and hygromycin-B, were purchased from Life Technologies (Gaithersburg, MD). All other reagents were purchased from Sigma (St. Louis, MO).
Cell Culture.
GH3 cells (CCL 82.1), obtained from the American Type Culture Collection (Rockville, MD), were maintained in DMEM with 10% (v/v) fetal calf serum, 0.05 I.U./ml penicillin and were incubated in a humidified atmosphere of 10% CO2, 90% O2 at 37°C. To prevent cells from adhering to one another, 30% conditioned media were included in all incubation media. Cells were harvested once each week by detachment with 0.1% phosphate-buffered saline supplemented with 0.4% EDTA (PBS/EDTA) and reseeded at 20% of their original density. The incubation medium was changed every 2 to 3 days. All experiments were conducted with cells maintained between passages 4 and 10.
Transfection.
Stable cell lines that expressed only δ- (GH3DOR) or both μ- and δ- (GH3MORDOR) opioid receptors were generated for this study. The transfection and subsequent characterization of GH3 cells to obtain the GH3MORDOR cell lines (Piros et al., 1996a) has been reported previously. To produce the GH3DOR cell line, GH3 cells (5 × 106 in 0.5 ml of phosphate-buffered saline, pH 7.4) were stably transfected by electroporation (500 μF, 250 V) in the presence of 10 μg of pCDNA1neo plasmids, which contained the cDNA encoding for the δ-opioid receptor (DOR-1). The DOR-1 construct was subcloned into the XhoI site of the pCDNA1neo plasmid (Invitrogen, San Diego, CA) and consisted of a 1.8-kb cDNA representing the coding region and a 700-base pair 3′-noncoding region of the mouse δ-opioid receptor. GH3 cells that stably incorporated these plasmids were selected by picking colonies that survived culturing in the presence of 1 mg/ml Geneticin (G418). Confirmation of δ-opioid receptor expression was determined by performing competition for [3H]diprenorphine (2 nM) binding by DPDPE (1 μM) as described below. The clone that expressed the highest level of δ-opioid receptor binding was selected for future studies and was designated as the GH3DOR clone.
Receptor Binding.
Membranes used in binding assays were prepared from GH3 cells by centrifugation of homogenates minus nuclei at 100,000g for 60 min as described previously (Prather et al., 1994a). All opioid receptor binding was performed using 250 μg of membrane protein. Binding incubation was performed in 50 mM Tris-HCl, pH 7.6, with 10 mM MgCl2, at room temperature for 90 min, as described previously (Prather et al., 1994b). For saturation binding studies using GH3DOR membranes, concentrations of [3H]diprenorphine from 0.1 to 10 nM were used. For saturation binding studies using GH3MORDOR membranes, concentrations of 0.05 to 20 nM [3H]DPDPE were used. In all experiments, nonspecific binding was determined in the presence of nonradioactive DPDPE (1 μM). Data obtained were subjected to Scatchard analysis, and estimates of affinity (Kd) and receptor density (Bmax) were obtained using the LIGAND computer program. In competition binding experiments, the ability of increasing concentrations (0.01 nM–100 μM) of DPDPE to compete for the binding of [3H]DPDPE (10 nM) or [3H]diprenorphine (2 nM) was assessed. The concentration of opioid receptor ligands to produce a 50% reduction in radioligand binding (IC50) was determined by the computer program Sigmaplot (Jandel Scientific, San Rafael, CA) and then converted to a measure of receptor affinity (Ki).
For determination of specific binding after SUPERFIT pretreatment, GH3MORDOR cells were grown to 80% confluence in T175 tissue culture flasks. The cells were washed once with warmed DMEM (serum and antibiotic free) and subsequently harvested in 10 ml of PBS/EDTA. Cells from individual T175 flasks were then resuspended in 20 ml of DMEM containing various concentrations of SUPERFIT (0.1–100 nM) and incubated at 37°C for 45 min in an orbital shaker (150 rpm). Cells were then centrifuged at 1500 rpm for 10 min and washed three times with 50 ml of PBS to remove residual SUPERFIT before binding. Final pellets were resuspended in binding buffer and aliquots taken for protein determination. The percentage of specific binding remaining relative to control cells (i.e., those not treated with SUPERFIT) was calculated for each SUPERFIT concentration and the best sigmoidal curve fit of these data was calculated using the computer program Sigmaplot.
Measurement of Opioid-Mediated Inhibition of Adenylyl Cyclase Activity.
The conversion of the [3H]adenine-labeled ATP pools to cyclic AMP was used as a measure of opioid ligand effect on cyclic AMP levels as described previously (Prather et al., 1994a). Briefly, measurements were made with GH3 cells seeded into 17-mm (24-well) plates (4 × 106 cells/plate), which resulted in 100% confluence when cultured for 4 days. The incubation medium was changed 24 h before the assay. On the day of the assay, media were removed and replaced with incubation mixture (warmed to 37°C) (DMEM containing 0.09% NaCl, 500 μM 3-isobutyl-1-methylxanthine, and 2 μCi/well [3H]adenine) for 1 h. At the time of the assay, plates were placed in an ice-water bath for 5 min. The incubation mixture was then removed and replaced with ice-cold assay mixture [Krebs-Ringer HEPES buffer (110 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 25 mM glucose, 55 mM sucrose, and 10 mM HEPES, at pH 7.4) containing 500 μM 3-isobutyl-1-methylxanthine, 10 μM forskolin] and the opioid ligand to be tested. The plates were then incubated at 37°C for 15 min and placed back in the ice-water bath for 5 min. After termination of incubations with 50 μl of 3.3 N perchloric acid and subsequent addition of [32P]cyclic AMP as an internal standard, radioactive cyclic AMP was separated from other3H-labeled nucleotides by a double-column chromatographic method. Seven milliliters of scintillation fluid was then added and samples were immediately counted in a Beckman LS2800 scintillation counter.
For determination of maximal opioid inhibition of adenylyl cyclase activity after SUPERFIT or β-FNA pretreatment,GH3MORDOR cells were cultured in 24-well plates as described above. On the day of the assay, media were removed and replaced with warmed incubation mixture (see above) containing various concentrations of SUPERFIT (0.1–600 nM) or β-FNA (10 or 20 nM) for 45 min. At the time of the assay, the incubation mixture was removed and cells were washed three times with 0.5 ml of warmed DMEM (serum and antibiotic free) to remove residual SUPERFIT. The remainder of the assay was conducted as detailed above. The percentage of maximal inhibition was calculated by dividing the amount of inhibition produced by DPDPE (10 or 100 nM) or DAMGO (1 μM) in cells pretreated with SUPERFIT or β-FNA, by the inhibition produced by agonists in control cells (i.e., those not treated with SUPERFIT or β-FNA). To prevent any action of DPDPE on μ-opioid receptors, 300 nM CTOP was included in all assay mixes. To prevent any action of DAMGO on δ-opioid receptors, 300 nM Tyr-Tic Phe-Phe was included in all assay mixes. The best sigmoidal curve fit of these data was calculated using the computer program Sigmaplot.
Electrophysiological Recordings.
Single cells were voltage clamped, and voltage-activated Ca2+ channel activity was recorded from whole GH3 cell clones using a List EPC-7 patch-clamp amplifier as described previously (Piros et al., 1995, 1996a). Before recording, culture dishes containing cells were superfused (flow rate, 2 ml/min) with a solution containing 140 mM NaCl, 2.8 mM KCl, 2 mM MgCl2, 1 mM CaCl2, and 10 mM HEPES (adjusted to pH 7.2 with NaOH). After a high-resistance seal was established and access to the interior of the cell was obtained, the external solution was replaced by a solution containing 125 mM NaCl, 5.4 mM CsCl, 10.8 mM BaCl2, 1 mM MgCl2, 10 mM HEPES, and 0.5 μM tetrodotoxin (adjusted to pH 7.2 with NaOH). The recording electrode contained 120 mM CsCl, 10 mM EGTA, 1 mM MgCl2, 3 mM magnesium-ATP, and 10 mM HEPES (adjusted to pH 7.2 with CsOH). Ba2+ currents were activated by step depolarizations of membrane potential from a holding potential of −80 mV for 100 ms. Capacitance and series resistance compensations were achieved using the patch-clamp amplifier. Residual artifacts and leakage currents were nulled using a P/4 subtraction. Patch electrodes were manufactured from thin-walled, borosilicate glass pipettes (World Precision Instruments, Sarasota, FL) using a Narishige (PP-83) electrode puller. Whole-cell currents, monitored using the EPC-7 amplifier, were low-pass filtered with an eight-pole Bessel filter (Frequency Devices, Haverhill, MA) at 1 kHz, digitized (Labmaster DMA interface; Axon Instruments, Foster City, CA) at a frequency of 5 kHz, and stored on an IBM PC hard disk. Data were acquired and analyzed using pCLAMP software (Axon Instruments). Drugs (prepared daily from frozen stock solutions) were applied to the bath, and all recordings were performed at room temperature (20–22°C).
For determination of maximal opioid inhibition of Ba2+ currents after SUPERFIT or β-FNA pretreatment, GH3MORDOR cells were cultured in 35-mm dishes as described above. On the day of the assay, the medium was removed and replaced with warmed DMEM (serum and antibiotic free) containing various concentrations of SUPERFIT (0.6–600 nM) or β-FNA (10 nM) for 45 min. At the time of the assay, the incubation mixture was removed and cells were washed three times with 5 ml of warmed DMEM to remove residual SUPERFIT or β-FNA. The percentage of maximal inhibition produced by opioid agonists DPDPE (10 or 100 nM) or DAMGO (100 nM) in cells pretreated with SUPERFIT or β-FNA, was calculated as described above for the adenylyl cyclase experiments. The best sigmoidal curve fit of these data was calculated using the computer program Sigmaplot.
Ba2+ current inhibition was measured by two different approaches. Our previous study demonstrated that approximately 25% of GH3MORDOR cells fail to respond to DPDPE (Piros et al., 1996a). Therefore, in the present study the effect of DPDPE was averaged over all cells tested when analyzing data from SUPERFIT experiments in which complete abolition of current inhibition by DPDPE was observed. Under these circumstances it is impossible to determine whether a lack of agonist effect is due to an absence of original response or irreversible alkylation of receptors. This approach was not necessary for analysis of either DAMGO or DPDPE effects after β-FNA treatment in which inhibitions induced by the opioid agonists remained discernible and therefore nonresponding cells were left out of the analysis.
Photoaffinity Labeling of Gα Subunits with [α-32P]AA-GTP.
The method for synthesis and purification of [α-32P]AA-GTP can be found inPrather et al. (1994a). The photoaffinity labeling of Gα subunits with [α-32P]AA-GTP has also been recently reported (Prather et al., 1994a,b, 1995; Chakrabarti et al., 1995). Plasma membranes (50 μg/assay) were incubated in the presence or absence of agonist for 6 min at 30°C in 100 μl of buffer I (50 mM HEPES, pH 7.4, 0.1 mM EDTA, 10 mM MgCl2, 30 mM NaCl, 50 μM GDP). After agonist incubation, [α-32P]AA-GTP (1 μCi/assay) was added, and samples were incubated for an additional 6 min at 30°C. The reaction was terminated by placing samples on ice. Membranes were then collected by centrifugation at 12,000g for 10 min and resuspended in 100 μl of buffer II (50 mM HEPES, pH 7.4, 0.1 mM EDTA, 10 mM MgCl2, 30 mM NaCl, 2 mM dithiothreitol). Resuspended pellets (droplets) were then irradiated at 4°C with 240 mJ from an ultraviolet lamp (254 nM, 150 W) at a distance of 15 cm. Samples were centrifuged as before, resuspended in sample buffer, and separated by SDS-PAGE (see below). After electrophoresis, [α-32P]AA-GTP-labeled Gα subunits were visualized autoradiographically by a Molecular Dynamics Inc. PhosphorImager 445 SI (Sunnyvale, CA) and quantified by densitometry using the NIH Image software program (version 1.56). To determine the amount of radioactivity incorporated by individual G proteins, the area of each band was traced, multiplied by its mean density, and the femtomoles of radioactivity determined by comparison with the density produced by a range of 32P standards using linear regression.
SDS-PAGE and Immunoblotting.
To identify Gα subunits, membranes were separated on 20-cm separating gels containing 10% acrylamide and 6 M urea (Prather et al., 1994a,b, 1995; Chakrabarti et al., 1995). Before separation, samples were resuspended in 80 μl of electrophoresis loading buffer (0.065 M Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 5% 2-mercaptoethanol), and heated at 90°C for 2 min. The ECL method of immunoblotting was used (Amersham Life Science Products). Gels were transferred to Hybond-ECL nitrocellulose membranes and incubated overnight at 4°C with 10% milk in blotting buffer (TBS-0.1%) (25 mM Tris-HCl, pH 7.6, 0.09% NaCl, 0.1% Tween 20). Blots were then washed three times (5 min each) with TBS-0.1% and incubated with primary antibodies (1:1000–2000) for 1 h at room temperature while shaking. The primary antibodies were then removed and blots were washed as described previously. Secondary antibody (donkey anti-rabbit or anti-mouse immunoglobin horseradish peroxidase, 1:5000) was then added and incubated for 30 min, with shaking. The secondary antibody was removed and blots were washed with 3 × 5-min washes with TBS-0.3%, followed by 3 × 5-min washes with TBS-0.1%. Blots were then incubated for 1 min with equal volumes of ECL detection reagents 1 and 2, wrapped in Saran plastic wrap, and exposed to Hybond-ECL X-ray film for periods varying between 30 s and 10 min.
The Gα-antisera used were EC2 selective for Giα3 (Simonds et al., 1989), GC2 for Goα (Spiegel, 1990), LEP4 for Giα2, and antisera 978 for Giα1 (Fargin et al., 1991). LEP4 was developed in the laboratory of Dr. P. Y. Law (University of Minnesota, Minneapolis) by immunizing rabbits with a Giα2 C-terminal peptide. Antiserum 978 is identical with AS190 used by Laugwitz et al. (1993) to identify Giα1.
Data Analysis.
Unless otherwise stated, data reported represent the mean ± standard error of at least three separate experiments that were each performed in triplicate. Data obtained from full dose-response curves using selective opioid agonists were subjected to sigmoidal curve fitting. The minimum and maximum plateau values for the amount of Gα subunits activated (expressed in fmol/mg of protein) and the amount of agonist required to produce 50% of maximal activation (i.e., ED50) were determined from the best-fit curves. The maximum amount of Gα subunits activated was defined as the difference between the minimum and maximum plateau values. Percentage increase in G protein activation was defined as the maximal femtomoles per milligram of protein of [α-32P]AA-GTP incorporated in the presence of agonist, divided by basal incorporation, times 100%. Statistical significance of the data was determined by ANOVA followed by comparison using either the nonpaired two-tailed Student's t test or Tukey's method.
Results
δ-Opioid Receptor Binding and Regulation of Adenylyl Cyclase and Calcium Channels in Transfected GH3DOR and GH3MORDOR Cells.
GH3 cells were stably transfected by electroporation with plasmids containing cDNAs encoding for opioid receptors to obtain clones that expressed only δ- (GH3DOR) or both δ- and μ-opioid receptors (GH3MORDOR) (Piros et al., 1996a). The density of each receptor (Bmax) and the affinity of the δ-opioid receptor-selective ligand DPDPE for the expressed receptors were determined in both clones by saturation and competition binding, respectively. The results of these characterizations are presented in Table 1. The density of δ-opioid receptors in the GH3MORDOR clone was almost 5-fold greater than that observed in the GH3DOR clone (P < .01). Additionally, the affinity of DPDPE for δ-opioid receptors in both clones reported here is similar to those reported for mammalian brain membranes (Goldstein and Naidu, 1989).
δ-Opioid receptor binding and regulation of adenylyl cyclase and calcium channel activity in transfected GH3DOR and GH3MORDOR cells
We examined the ability of the expressed opioid receptors to regulate adenylyl cyclase activity in both GH3 clones (Table 1). In GH3DOR cells, the δ-opioid agonist DPDPE produced a potent (IC50 = 2.2 nM) and efficacious (Imax = 37%) inhibition of adenylyl cyclase activity, whereas the μ-opioid agonist DAMGO only produced a slight reduction in cAMP levels at a concentration of 1 μM (data not shown). Overnight incubation of GH3DOR cells with pertussis toxin (100 ng/ml) completely blocked the ability of DPDPE to reduce cAMP accumulation (data not shown). In GH3MORDOR cells, DPDPE also potently (IC50 = 1.1 nM) and efficaciously (Imax = 78%) reduced the production of cAMP. Although this peptide is a relatively selective agonist, our previous studies demonstrated in this clone that DPDPE can displace the μ-opioid receptor-selective agonist [3H]DAMGO with a Ki value of 49 nM (Piros et al., 1996a). Therefore, to eliminate any inhibition of adenylyl cyclase activity produced at higher concentrations of DPDPE due to activation of μ-opioid receptors, we conducted all dose-response curves in the presence of a maximally effective concentration (300 nM) of a selective antagonist for μ-opioid receptors, CTOP.
In GH3DOR cells, neither DPDPE (1 and 10 μM) nor the less selective δ-/μ-opioid agonist [d-Ala2,d-Leu5]enkephalin (100 nM) was able to produce a significant decrease in Ba2+ currents (Table 1). We observed a similar lack of coupling of δ-opioid receptors to Ca2+channels when several other GH3DOR clones were initially screened (data not shown). Endogenous somatostatin (SRIF) receptors in GH3 cells are also coupled to L-type Ca2+ channels (Kleuss et al., 1991). Therefore, to confirm that GH3DOR cells expressed functional G protein-sensitive Ca2+ channels, SRIF (100 nM) was applied and produced a 13.0 ± 4% inhibition of Ba2+ currents. In marked contrast to our results obtained using GH3DOR cells, activation of δ-opioid receptors in GH3MORDOR cells inhibited Ca2+ channel activity (Table 1) (Piros et al., 1996a). DPDPE produced a dose-dependent inhibition of Ba2+ currents of more than 20%, with an IC50 in the low nanomolar range.
Effect of the δ-Opioid Receptor Alkylating Agent SUPERFIT on Opioid Receptor Binding, Inhibition of Adenylyl Cyclase Activity, and Ba2+ Currents in GH3MORDOR Cells.
Because GH3MORDOR cells expressed 5-fold more δ receptors than GH3DOR cells, we examined whether the differential coupling of δ-opioid receptors to Ca2+ channels in these clones was the result of differences in receptor density. To accomplish this, the alkylating agent SUPERFIT (Zhu et al., 1996) was used to selectively and irreversibly block δ-opioid receptors in GH3MORDOR cells and subsequently examine opioid receptor binding and the ability of the δ-opioid agonist DPDPE to inhibit adenylyl cyclase activity and the amplitude of Ba2+ currents (Fig.1). Pretreatment of GH3MORDOR cells with increasing concentrations of SUPERFIT (37°C for 45 min) followed by extensive washing reduced binding of the δ-opioid agonist [3H]DPDPE in a concentration-dependent manner with an IC50value of 6.1 nM (Fig. 1, top). This was similar to the IC50 of 7.1 nM observed in CHO cells expressing δ-opioid receptors alone and suggests that the presence of the μ-opioid receptors does not influence the affinity of SUPERFIT (Zhu et al., 1996). Furthermore, the binding of the μ-opioid agonist [3H]DAMGO was not significantly altered when cells were pretreated with a 100 nM SUPERFIT, suggesting that the δ-opioid selective antagonist does not interact with μ-opioid binding sites (data not shown). These data argue against a confounding influence of a significant level of heterodimers (underDiscussion). To confirm that the reduction in δ-opioid binding resulted from a decrease in receptor number and not receptor affinity, Scatchard analysis of [3H]DPDPE saturation binding was performed (Table2). Pretreatment of GH3MORDOR cells with SUPERFIT resulted in a dose-dependent (IC50 = 4.9 nM) reduction in δ-opioid receptor Bmax, without a significant reduction in Kd. These results are consistent with the previous observation of Zhu et al. (1996) that SUPERFIT was a highly selective irreversible δ-opioid receptor ligand and further demonstrated that we could selectively reduce the density of available δ-opioid receptors in GH3MORDOR cells.

Effect of SUPERFIT on δ-opioid receptor binding, inhibition of adenylyl cyclase activity, and Ba2+currents in GH3MORDOR cells. A, GH3MORDOR cells were pretreated with increasing concentrations of SUPERFIT (0.1–100 nM) for 45 min at 37°C, followed by extensive washing. The percentage of specific [3H]DPDPE binding remaining relative to control cells (i.e., those not treated with SUPERFIT) was calculated for each SUPERFIT concentration as described under Experimental Procedures. Each point represents the mean ± S.E. of three separate experiments. B, GH3MORDOR cells were cultured in 24-well plates (adenylyl cyclase assays; ○) or in 35-mm dishes (electrophysiological experiments; ▪). Cells were incubated with SUPERFIT (0.6–600 nM) for 45 min at 37°C and subsequently washed extensively with warmed DMEM. The remainder of the assay was conducted as described under Experimental Procedures. For both adenylyl cyclase assays and electrophysiological experiments, the percentage of maximal inhibition was calculated by dividing the amount of inhibition produced by DPDPE (10 nM) in cells pretreated with each SUPERFIT concentration, by the inhibition produced by DPDPE in control cells (i.e., those not treated with SUPERFIT). For the adenylyl cyclase assays, each point represents the mean ± S.E. of three separate experiments. For the electrophysiological experiments, each point represents the mean ± S.E. for measurements obtained from a minimum of four cells. The best sigmoidal curve fit for all of these data was calculated using the computer program Sigmaplot. a, statistically different from the inhibition obtained after 600 nM SUPERFIT pretreatment. b, statistically different from the inhibition of adenylyl cyclase activity obtained after pretreatment with a corresponding concentration of SUPERFIT.
δ-Opioid receptor density in membranes prepared from GH3MORDOR cells pretreated with increasing concentrations of the δ-alkylating agent SUPERFIT
When GH3MORDOR cells were pretreated with SUPERFIT (0.6–600 nM), a concentration-dependent decrease in the maximal inhibition produced by the δ-opioid agonist DPDPE (10 nM) of both adenylyl cyclase activity and Ba2+ currents was observed (Fig. 1, bottom). Nevertheless, DPDPE significantly inhibited adenylyl cyclase activity after application of SUPERFIT (12 nM) sufficient to block more than 80% of the δ-opioid receptors (Table 2). Even after pretreatment with 100 nM SUPERFIT, a concentration sufficient to reduce specific binding by 99%, more than 60% of the maximal inhibition of adenylyl cyclase activity by DPDPE, was retained. It is important to note that 1% of the δ-opioid receptor population in GH3MORDOR cells would be predicted to be represented by approximately 25 fmol/mg of protein of receptor. δ-Opioid agonists have been shown to produce efficacious inhibition of adenylyl cyclase activity in other cell lines expressing similar low densities of δ-opioid receptors (Prather et al., 1994b,c). In contrast to regulation of adenylyl cyclase activity, the inhibition of Ba2+ currents by DPDPE was almost abolished by 12 nM SUPERFIT. Additionally, SUPERFIT produced significantly greater reductions in δ-opioid receptor-induced maximal inhibition of Ba2+ currents relative to inhibition of adenylyl cyclase at all pretreatment concentrations. Inhibition of both adenylyl cyclase and Ba2+currents by δ-opioid receptors was totally abolished by pretreatment of cells with 600 nM SUPERFIT, a concentration predicted to alkylate all δ-opioid receptors. Interestingly, we observed a precipitous decline between the maximal inhibition of adenylyl cyclase activity by DPDPE from 64% when cells were pretreated with 100 nM SUPERFIT, to only 2% after 600 nM. This suggests that a certain “threshold” density of δ-opioid receptors (between 0 and 25 fmol/mg of protein) is required to produce significant inhibition of adenylyl cyclase activity.
Effect of δ- (SUPERFIT) and μ- (β-FNA) Opioid Receptor Alkylating Agents on μ- and δ-Opioid Receptor Inhibition of Adenylyl Cyclase Activity and Ba2+ Currents in Transfected GH3 Cells.
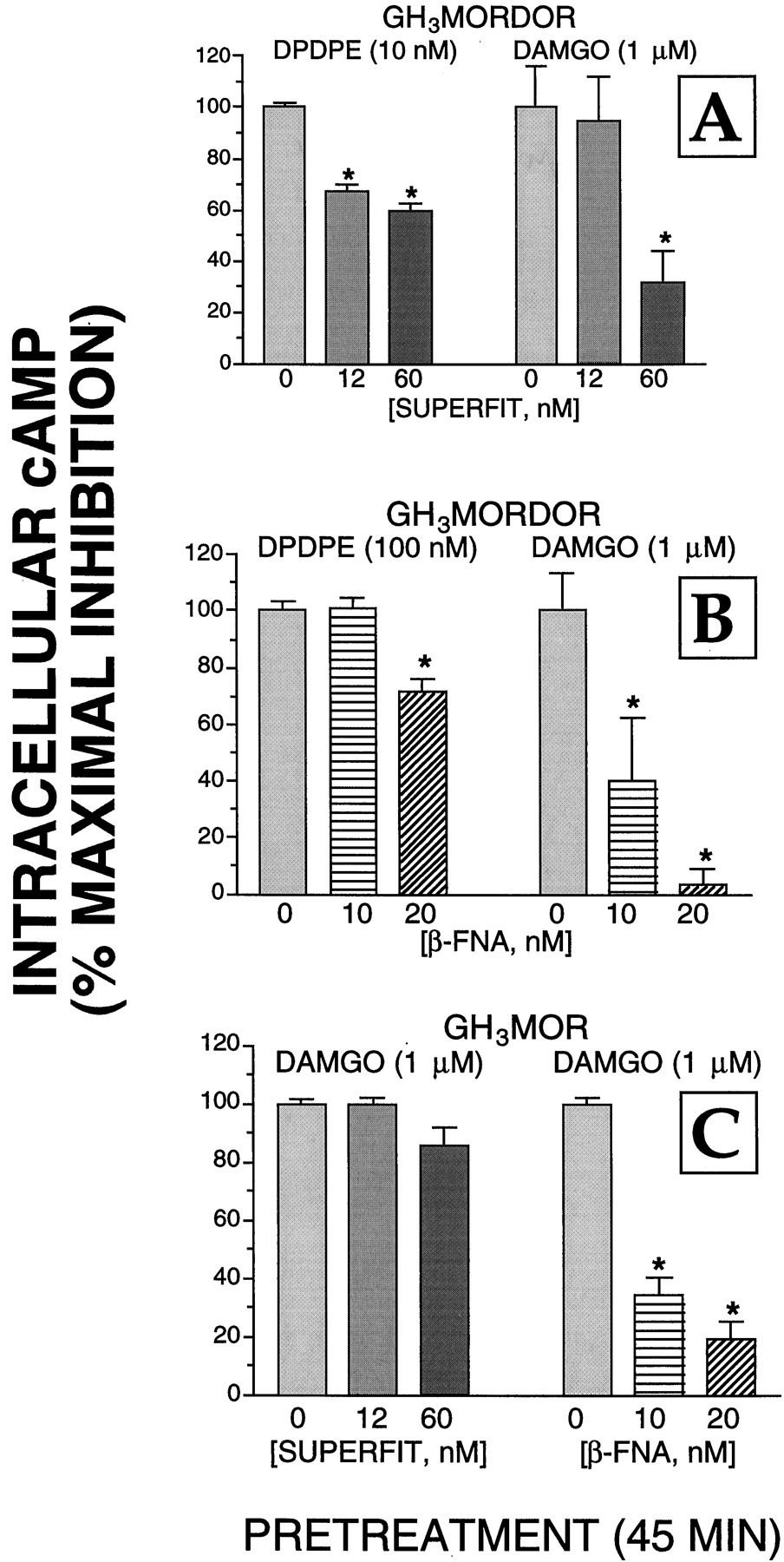
We performed additional experiments to determine whether the δ-opioid alkylating agent SUPERFIT influenced the ability of μ-opioid receptors to couple to adenylyl cyclase (Fig.2) and/or Ca2+channels (Fig. 3) in GH3MORDOR cells. For ease of comparison, the data presented in Fig. 1 for the effect of 12 and 60 nM SUPERFIT on inhibition of adenylyl cyclase activity and Ba2+currents produced by DPDPE (10 nM) are also presented in Figs. 2A and3A (left). Although both 12 and 60 nM SUPERFIT significantly reduced the maximal inhibition of adenylyl cyclase activity produced by DPDPE, SUPERFIT (12 nM) had no effect on the ability of the μ-opioid agonist DAMGO to reduce intracellular cAMP levels (Fig. 2A). However, the highest concentration of SUPERFIT tested (60 nM) did diminish the amplitude of the DAMGO (1 μM)-evoked inhibition of cAMP accumulation by 69%. Although 60 nM SUPERFIT also reduced the maximal inhibition of adenylyl cyclase activity by DAMGO in GH3MOR cells by 14%, this effect was not statistically significant. SUPERFIT (12 nM) also significantly reduced the ability of DPDPE to inhibit Ba2+ currents in GH3MORDOR cells; after pretreatment with the higher concentration of SUPERFIT (60 nM) the effect of DPDPE on Ba2+ currents was all but abolished (Fig. 3A). Both concentrations of SUPERFIT also reduced the ability of the μ-opioid agonist DAMGO to inhibit Ba2+ currents by 45 and 66%. The agent had no effect on Ca2+ channel function as evidenced by the unaltered amplitude of Ba2+ currents recorded from cells pretreated by 12 or 60 nM SUPERFIT (Fig. 3C). Therefore, these data suggest that at in cells that express both receptor subtypes SUPERFIT can decrease the function of μ- as well as δ-receptors.

Effects of SUPERFIT and β-FNA on the coupling of μ- and δ-receptors to adenylyl cyclase in GH3MORDOR and GH3MOR cells. A, GH3MORDOR cells were cultured in 24-well plates and incubated with SUPERFIT (0, 12, or 60 nM) for 45 min at 37°C. After extensive washing with warmed DMEM, cells were exposed to an assay mix containing either DPDPE (left) or DAMGO (right) for 15 min (described under Experimental Procedures). The percentage of maximal inhibition of adenylyl cyclase activity was calculated by dividing the amount of inhibition produced by agonists in cells pretreated with each SUPERFIT concentration by the inhibition produced by agonists in control cells (i.e., those not treated with SUPERFIT). B, same experimental design as presented in A was used except that GH3MORDOR cells were pretreated for 45 min with the μ-opioid receptor alkylating agent β-FNA (0, 10, or 20 nM). C, GH3MOR cells were incubated for 45 min with either SUPERFIT (0, 12, or 60 nM) or β-FNA (0, 10, or 20 nM), extensively washed, and the inhibition of adenylyl cyclase produced by DAMGO was measured. The values presented are the mean ± S.E. determined from four separate experiments. Statistical significance of the data was determined by a nonpaired two-tailed Student'st test. ∗, statistically different from cells receiving no pretreatment, P < .05.
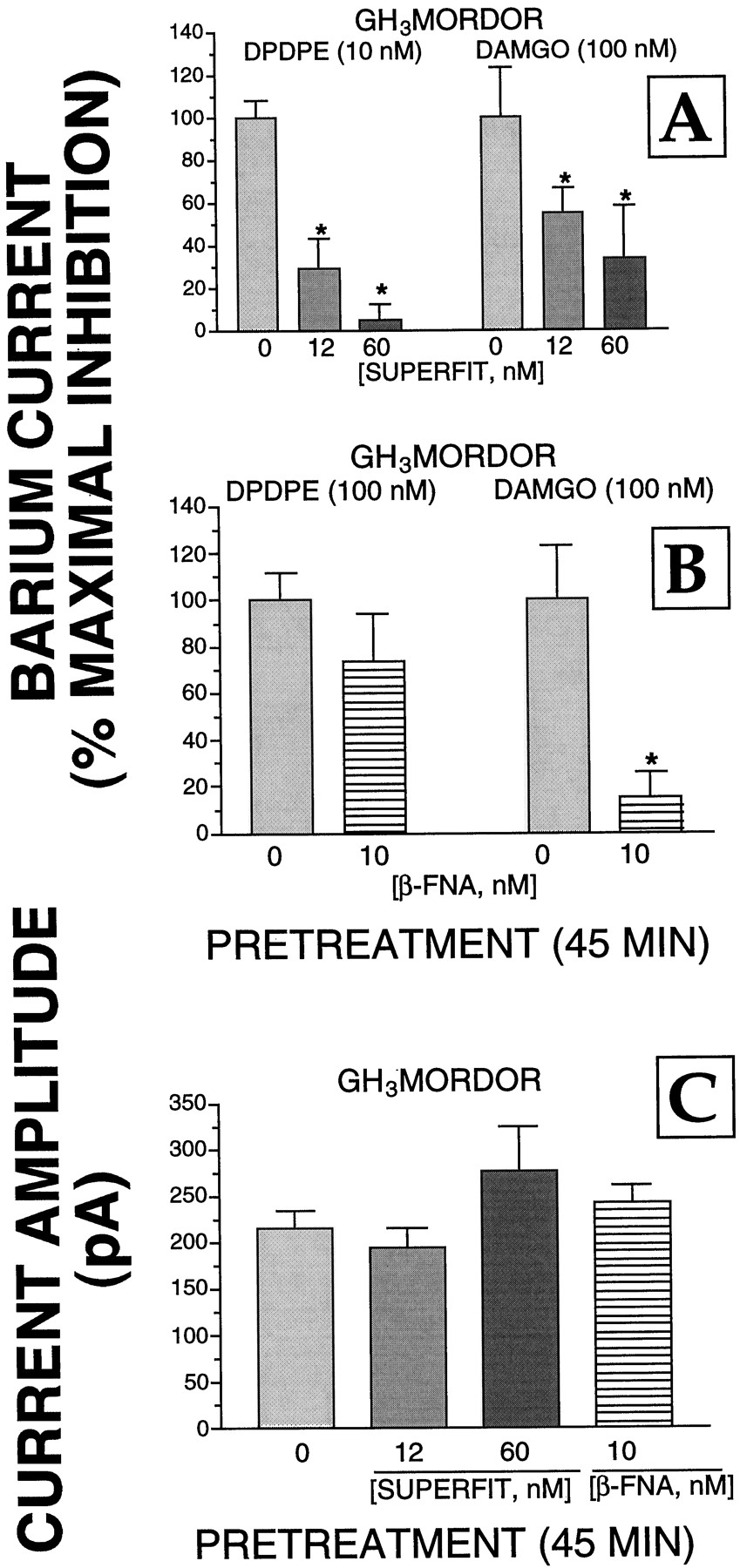
Effects of SUPERFIT and β-FNA on the coupling of μ- and δ-receptors to Ca2+ channels in GH3MORDOR cells. A, GH3MORDOR cells were cultured in 30-mm dishes and incubated with SUPERFIT (0, 12, or 60 nM) for 45 min at 37°C. After extensive washing with warmed DMEM, cells were exposed to either DPDPE (left) or DAMGO (right) for 15 min (described under Experimental Procedures). The patch-clamp technique was used to measure the percentage of maximal inhibition of Ba2+currents. This was calculated by dividing the amount of inhibition produced by agonists in cells pretreated with each SUPERFIT concentration by the inhibition produced by agonists in control cells (i.e., those not treated with SUPERFIT). The values presented are the mean ± S.E. determined from a minimum of four cells. B, same experimental design as presented in A was used except that GH3MORDOR cells were pretreated for 45 min with the μ-opioid receptor alkylating agent β-FNA (0 or 10 nM). C, neither SUPERFIT (12 and 60 nM) nor β-FNA (10 nM) had any effect on the amplitude of Ba2+ currents evoked by depolarizing GH3MORDOR cells from −80 to 0 mV. The values presented are the mean ± S.E. Statistical significance of the data was determined by a nonpaired two-tailed Student'st test. ∗, statistically different from cells receiving no pretreatment, P < .05.
In reciprocal experiments, we examined whether the μ-opioid-alkylating agent β-FNA (Chen et al., 1996) influenced the ability of δ-opioid receptors to couple to adenylyl cyclase and/or Ca2+ channels in GH3MORDOR cells (Figs. 2 and 3). Pretreatment of cells with 10 nM β-FNA significantly reduced the maximal inhibition produced by DAMGO of adenylyl cyclase activity and Ba2+ currents by 60 and 85%, respectively. In contrast, this concentration of β-FNA did not have a statistically significant effect on the maximal inhibition of either effector produced by DPDPE (Figs. 2B and 3B). However, the ability of DPDPE to reduce intracellular cAMP levels in GH3MORDOR cells pretreated with the highest concentration of β-FNA tested (20 nM) was significantly reduced by 28%.
GH3 Cells Express Four Pertussis Toxin-Sensitive Gα Subunits.
In addition to effects of receptor density, the differential coupling of opioid receptors to L-type Ca2+ channels in GH3 cells might also be explained by activation of a distinct pattern of G proteins by δ-opioid receptors in the two clones. Because opioid receptor-mediated inhibition of adenylyl cyclase activity was blocked by pretreatment of cells with pertussis toxin, which ADP-ribosylates only Gi/Go-type G proteins), we determined the identity of these Gα subunits in GH3 cells. Membranes (50 μg/sample) were incubated with the photoaffinity label [32P]AA-GTP, and labeled proteins were separated by urea/SDS-PAGE. After transfer to nitrocellulose membranes, blots were subjected to autoradiography, followed by immunoblotting with selective antibodies for individual Gαsubunits (Fig. 4). In the absence of opioid, [32P]AA-GTP was incorporated into three detectable bands (Fig. 4, A–D, lane 1). When membranes prepared from GH3MORDOR cells were incubated with [32P]AA-GTP in the presence of a δ-opioid agonist (Fig. 5), a fourth band incorporating the photoaffinity label was found that migrated above the first heavily labeled band. Consequently, these four bands were designated as proteins 1 to 4, from highest to lowest molecular weight.

Photoaffinity labeling and Western analysis of Gαs in GH3 membranes using specific antisera. Membranes (50 μg) from GH3 cells were incubated with the photoaffinity label [32P]AA-GTP and subsequently separated by urea/SDS-PAGE. Proteins were then transferred onto nitrocellulose membrane and subjected to autoradiography followed by Western analysis of the same immunoblot with antibodies selective for individual Gα subunits. A–D, depicted in lanes 1 and 2 are the autoradiogram (AR) and the subsequent immunoblot of that autoradiogram, respectively. Specific Gαsubunit antisera used were 978 (A, Giα1), EC2 (B, Giα3), GC2 (C, Goα1 and Goα2), or LEP4 (D, Giα1 and Giα2). Antibody-protein complexes were visualized using ECL and goat anti-rabbit conjugated with horseradish peroxidase as secondary antibodies.

Concentration-dependent [32P]AA-GTP labeling of individual pertussis toxin-sensitive G protein α-subunits by the δ-opioid agonist DPDPE in GH3 MORDOR membranes. Top, autoradiogram of Gα subunits photoaffinity labeled with [32P]AA-GTP (1 μCi) in the presence of increasing concentrations (0.3–1000 nM) of the δ-opioid agonist DPDPE in GH3 MORDOR membranes (50 μg), separated by urea/SDS-PAGE. Bottom, to determine the amount of radioactivity incorporated by individual Gαsubunits, the area of each band was traced, multiplied by its mean density, and the femtomoles of radioactivity determined by comparison with the density produced by a range of 32P standards using linear regression. The amount of each Gα subunit activated in femtomoles per milligram protein is plotted against the corresponding DPDPE concentrations. The values presented for each concentration represent the mean ± S.E. determined in five separate experiments.
The identity of the proteins that incorporated [32P]AA-GTP was determined by Western blot analysis immediately after autoradiography. Immunoblots were incubated with antisera 978 (Fargin et al., 1991), EC2 (Simonds et al., 1989), GC2 (Spiegel, 1990), and LEP4 (Prather et al., 1994b). No major immunopositive bands were observed when immunoblots were incubated with the Giα1-selective antisera 978 (Fig.4A, lane 2). However, 978 identified a very faint band that migrated with similar mobility as autoradiographic band 3 (subsequently identified as Giα2). This band was most likely due to cross-reactivity of 978 with Giα2 because other studies have also shown a lack of Giα1 in GH3 cells (Kleuss et al., 1991). Additionally, this band migrated between bands subsequently identified as Goα1 and Goα2. In other cell lines and tissues Giα1 migrates slower than both Goα subunits (Laugwitz et al., 1993). GC2, the antiserum selective for Goα1 and Goα2, recognized two bands that migrated with identical electrophoretic mobilities as bands 2 and 4 labeled by autoradiography (Fig. 4C, lane 2). These bands were concluded to be Goα1 and Goα2 from higher to lower molecular weight because they show similar relative mobilities in urea/SDS-PAGE to that observed for Goα1 and Goα2 in NG108-15 cells (Roerig et al., 1991). The antiserum selective for Giα1 and Giα2 (LEP4) recognized a single band that migrated with the identical electrophoretic mobility as band 3 (Fig. 4D, lane 2). Because this band was only very faintly labeled by antiserum 978 and Giα1 has been demonstrated previously to be absent from GH3cells (Kleuss et al., 1991), the band recognized by LEP4 was concluded to be Giα2. EC2 is an antiserum that is selective for Giα3, with cross-reactivity with Goα. From highest to lowest molecular weight, it recognized three major bands corresponding to autoradiographic bands 1 (radiolabeled only in the presence of agonist, Fig. 5), 2, and 4 (Fig. 4B, lane 2). Because autoradiographic bands 2 and 4 were previously identified as Goα1 and Goα2, band 1 was concluded to represent Giα3. In conclusion, comparison of the electrophoretic mobility of bands obtained by autoradiography with those detected by Western analysis of the same immunoblot indicated that the pertussis toxin-sensitive G proteins labeled by [32P]AA-GTP from higher to lower molecular weight were Giα3, Goα1, Giα2, and Goα2, respectively. Our findings of the Gα subunits present in GH3 cells is in agreement with those reported previously (Kleuss et al., 1991). Comparison of the abundance of Giα3, Goα1,Giα2, and Goα2 between GH3DOR and GH3MORDOR cells revealed no apparent variance in the relative abundance of any Gα subunit between the GH3 clones (data not shown).
Stimulation of a Higher Density of δ-Opioid Receptors in GH3MORDOR Cells by DPDPE Results in a Greater Amount, but Not a Different Pattern of G Protein Activation Than in GH3DOR Cells.
Pretreatment of cells with pertussis toxin blocked regulation of adenylyl cyclase activity in both cell lines. Therefore, the activation of pertussis toxin-sensitive Gi/oα subunits was examined in GH3 membranes. One approach to measure the activation of Gα subunits by receptors is to use agonist-stimulated incorporation of [32P]AA-GTP into Gαsubunits, followed by separation using urea/SDS-PAGE and subsequent autoradiography (Prather et al., 1994a,b, 1995; Chakrabarti et al., 1995). The results of a typical photoaffinity-labeling experiment using GH3MORDOR membranes are presented in Fig. 5. The δ-opioid receptor agonist DPDPE produced dose-related increases in the incorporation of [32P]AA-GTP into all four previously identified Gα subunits. As was described previously for inhibition of adenylyl cyclase activity in GH3MORDOR cells, to eliminate any nonselective stimulation of G protein labeling by higher DPDPE concentrations acting at μ-opioid receptors in GH3 MORDOR membranes, all photoaffinity experiments using GH3 MORDOR (but not GH3DOR) cells were conducted in the presence of 300 nM CTOP.
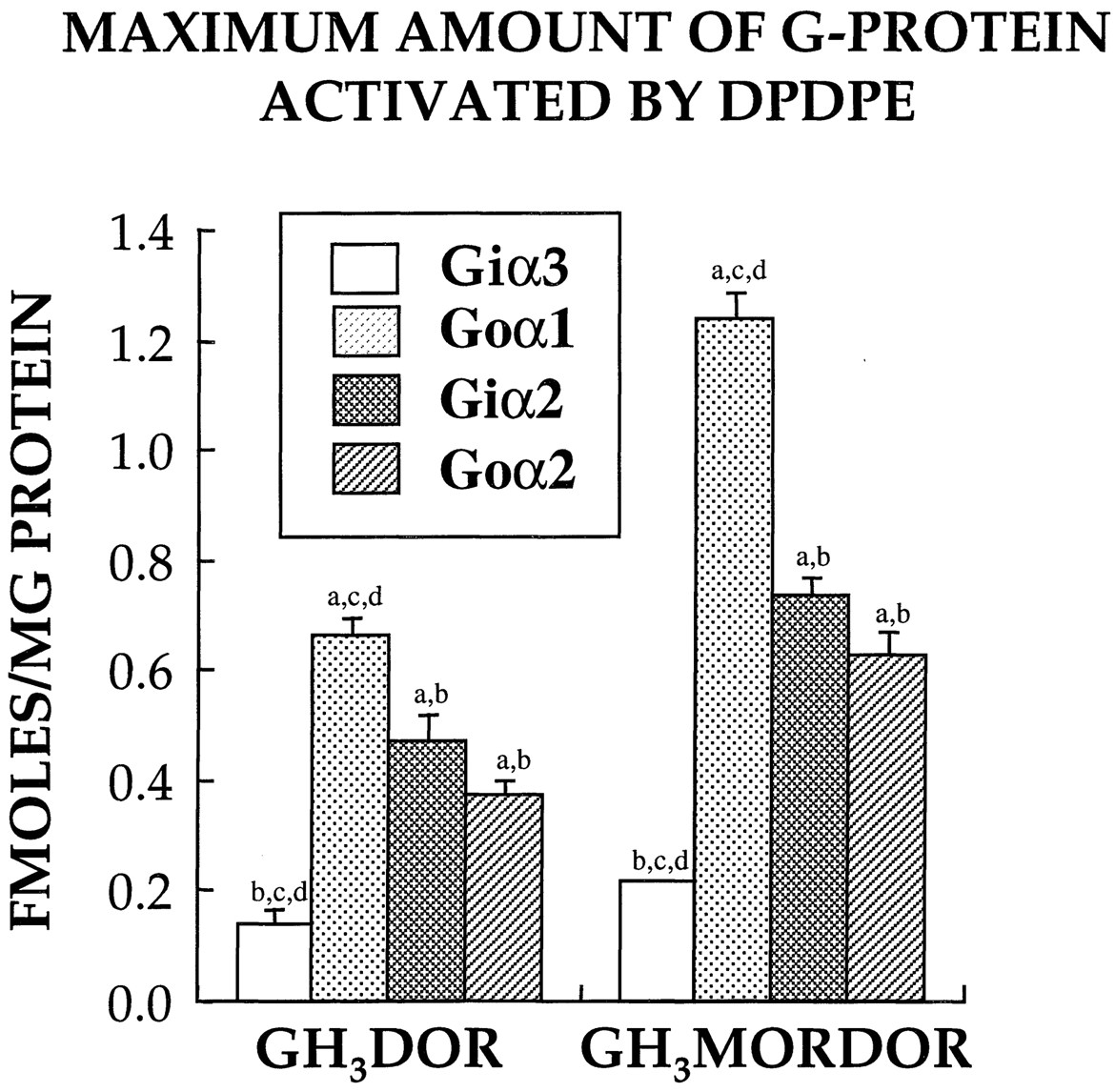
Using data obtained from full dose-response curves using the selective agonist DPDPE, we compared receptor/G protein interaction between δ-opioid receptors in the two GH3 clones (Fig.6). Stimulation of δ-opioid receptors in both GH3DOR and GH3MORDOR membranes by DPDPE produced a greater activation of Goα1 (0.67 and 1.24 fmol/mg of protein, respectively), followed by Giα2 (0.47 and 0.73 fmol/mg of protein) = Goα2 (0.37 and 0.62 fmol/mg of protein) > Giα3(0.14 and 0.21 fmol/mg of protein). As might have been expected based on a 5-fold greater receptor density (Table 1), activation of δ-opioid receptors in the GH3MORDOR clone produced a significantly (P < .01) greater activation of all G proteins than in GH3DOR membranes (1.65 versus 2.8 total fmol/mg of protein of G protein activated, respectively). When the data are presented as the maximum percentage increase in G protein activation relative to basal labeling, activation of δ-opioid receptors in GH3DOR and GH3MORDOR membranes by DPDPE resulted in a significantly (P < .01) greater percentage increase in activation of only Goα1 (146 and 304%, respectively) relative to all other G protein α subunits (data not shown). Finally, the amount of DPDPE required to produce half-maximal labeling of Gα subunits in all GH3 clones was also evaluated. Activation of δ-opioid receptors by DPDPE in both GH3DOR and GH3MORDOR membranes produced dose-dependent labeling of all four Gα subunits with similar potencies that were not statistically different [Giα3 (4.2 and 6.5 nM, respectively), Goα1 (11.1 and 14.6 nM), Giα2 (9.6 and 12.1 nM), or Goα2 (5.4 and 3.9 nM)].

Maximum amount of Gα subunits activated by δ-opioid receptors in GH3 clones. Full dose-response curves (0.01–1000 nM) using the δ-opioid agonist DPDPE to induce photoaffinity labeling of Gαsubunits with [32P]AA-GTP (1 μCi) were performed using 50 μg of membranes prepared from the indicated GH3 clones. Photolabeled Gα subunits were subsequently separated by urea/SDS-PAGE, exposed for autoradiography, and quantitated by densitometry. To determine the amount of radioactivity incorporated by individual G proteins, the area of each band was traced, multiplied by its mean density, and the femtomoles of radioactivity determined by comparison with the density produced by a range of32P standards using linear regression. Minimum and maximum plateau values (expressed in fmol/mg of protein) were determined by curve fitting of the sigmoidal dose-response curves. The maximum amount of Gα subunits activated was defined as the difference between the minimum and maximum plateau values. Data reported represent the mean ± S.E. of at least three to five separate experiments. Statistical significance of the data was determined by ANOVA followed by comparisons using Tukey's method. a, statistically different from Giα3. b, statistically different from Goα1. c, statistically different from Giα2. d, statistically different from Goα2.
Discussion
In some central nervous system neurons and several cellular models, δ-opioid receptors are able to regulate the activity of multiple intracellular effectors. For example, δ-opioids inhibit both Ca2+ currents (Stefani et al., 1994) and adenylyl cyclase activities (Childers et al., 1992) in striatal neurons. In addition, in neuroblastoma cells δ-opioid receptors regulate adenylyl cyclase activity (Prather et al., 1994c), Ca2+channels (Hescheler et al., 1987), and phospholipase C (Jin et al., 1992). Interestingly, in some other cells the coupling of δ-opioid receptors to intracellular effectors appears to be more restricted. In rat dorsal root ganglion neurons, μ- and κ- but not δ-opioid receptors are able to couple to Ca2+ channels (Moises et al., 1994) despite the fact that all three receptor subtypes are located on these cells (Ji et al., 1995). δ-Opioid receptors are also unable to regulate Ca2+ currents in several other central nervous system regions, including the nucleus tractus solitarius (Rhim and Miller, 1994), the basal forebrain (Soldo and Moises, 1997), and the neurohypophysis (Rusin et al., 1997). One possible mechanism that could underlie the coupling specificity of δ-opioid receptors to distinct effectors in the same cell might be the requirement for activation of different densities of receptors to regulate individual intracellular effectors.
In support of this hypothesis, we observed that in GH3 cells stably transfected with a relatively low δ-opioid receptor density of 0.55 pmol/mg of protein (GH3DOR), activation of δ-opioid receptors produced inhibition of adenylyl cyclase activity but was unable to alter current through Ca2+ channels. The density of δ-opioid receptors in GH3DOR cells is similar to that reported for the neuroblastoma cell line NG108-15 (0.52 pmol/mg of protein) and slightly higher than that demonstrated in the striatum (0.29 pmol/mg of protein) (Law et al., 1983; Sim et al., 1996). In contrast to our observation with GH3DOR cells, activation of δ-opioid receptors in a clone that contained a high density of δ-opioid receptors (2.45 pmol/mg of protein) coexpressed with μ-opioid receptors (GH3MORDOR), resulted in not only the expected inhibition of adenylyl cyclase activity but also produced inhibition of L-type Ca2+ currents. The coupling of opioid receptors to L-type Ca2+ channels is rarely seen in neuronal preparations and it is possible that the high levels of receptor expression achieved in our recombinant system enable this signal transduction pathway to function (Piros et al., 1996b). It is also possible that in those instances in which such coupling is seen in cells with native receptors that this could be due to elevated receptor expression. Alternatively, coupling of opioid receptors to L-type Ca2+ channels may be enabled by an interaction with a specific G protein not present in all neurons. Because GH3MORDOR cells expressed 5-fold more δ-opioid receptors than GH3DOR cells, in the present study we examined whether the differential coupling of δ-opioid receptors to Ca2+ channels in these clones was the result of differences in receptor density. Selective and irreversible blockade of δ-opioid receptors using the alkylating agent SUPERFIT (Zhu et al., 1996) resulted in a concentration-dependent reduction in the ability of DPDPE to maximally inhibit both adenylyl cyclase and Ba2+ currents. The inhibition of Ba2+ currents by DPDPE was barely detectable when cells were pretreated with a concentration of SUPERFIT (12 nM) that resulted in approximately an 80% reduction in available δ-opioid receptors (0.5 pmol/mg), a level similar to the density of δ-opioid receptors expressed in GH3DOR cells (0.55 pmol/mg). In contrast, more than 60% of the maximal inhibition of adenylyl cyclase activity by DPDPE was retained even when SUPERFIT pretreatment reduced specific binding by 99%. These results suggest that the inability of δ-opioid receptors to couple to L-type Ca2+ channels in GH3DOR cells results from an insufficient number of available receptors and that δ-opioid receptors couple more efficiently to adenylyl cyclase than they do to L-type Ca2+ channels. These results might also help explain a general observation that δ-opioid receptors, when present, are able to regulate adenylyl cyclase activity in most brain regions and cell lines studied, but in many of these same tissues fail to couple to Ca2+ channels.
Overexpression of several different G protein-coupled receptors results in coupling to G proteins and effectors that would not be regulated at lower receptor densities (Ashkenazi et al., 1987; Eason et al., 1992). Hence, the differential coupling of δ-opioid receptors to L-type Ca2+ channels in GH3 cells might be explained by higher densities of δ-opioid receptors producing activation of different (or additional) G protein(s) relative to those activated by lower densities of δ-opioid receptors. Therefore, we compared the pattern of maximal G protein activation produced by δ-opioid receptors in GH3DOR and GH3MORDOR cells. Dose-dependent activation of opioid receptors by agonists should produce a concomitant increase in the incorporation of [α-32P]AA-GTP into the G proteins with which they interact. Data obtained from full dose-response curves using selective agonists revealed that, stimulation of δ-opioid receptors in both GH3clones resulted in coupling to an identical, specific pattern of G proteins, the greatest activation of Goα1, followed by Giα2 = Goα2 > Giα3. Interestingly, the selective coupling of δ-opioid receptors to specific G proteins was different than observed previously when opioid receptors were transfected into CHO cells in which activation of μ-, δ-, or κ-opioid receptors resulted in nonselective, simultaneous coupling to multiple G proteins (Prather et al., 1994b, 1995; Chakrabarti et al., 1995). Although others have noted differential coupling of δ-opioid receptors to G proteins, the pattern of selectivity observed in the present study is different. Laugwitz et al. (1993) reported that δ- and μ-opioid receptors in SH-SY5Y cells preferentially activated Giα1 and Giα3, respectively, whereas both receptor subtypes interacted with Giα2 and Goα. The present findings suggest that increasing densities of δ-opioid receptors does not change the selectively of activation of different Gαsubunits.
Although the pattern of G protein activation produced by δ-opioid receptors was not different in the two GH3clones, the maximal amount of all G proteins activated was significantly greater in GH3MORDOR cells that expressed 5-fold more receptors. We have observed a similar direct relationship between δ- and μ-opioid receptor density and amount of G protein activated in other cells lines (Prather et al., 1994b;Chakrabarti et al., 1995). Based on these observations, it is tempting to speculate that stimulation of δ-opioid receptors in GH3MORDOR, but not GH3DOR cells resulted in activation of a critical threshold of specific G proteins required for Ca2+ channel inhibition. Several studies have attempted to identify the distinct G proteins responsible for coupling opioid receptors to Ca2+channels. For example, intracellular application of Goα subunits was more effective than application of Giα in reconstitution of the δ-opioid receptor-mediated inhibition of Ca2+ currents in NG108-15 cells treated with pertussis toxin (Hescheler et al., 1987). Furthermore, a mutant GoαA subunit resistant to pertussis toxin, was able to rescue δ-opioid receptor-mediated inhibition of Ca2+ channels in pertussis toxin-treated NG108-15 cells (Taussig et al., 1992). Intracellular dialysis of dorsal root ganglion sensory neurons with antibodies selective for Goα, but not Giα, blocked both μ- (Moises et al., 1994) and κ- (Wiley et al., 1997) opioid receptor inhibition of Ca2+ currents. Similarly, δ-opioid inhibition of Ca2+ channels in NG108-15 cells is attenuated by preinjection of antibodies selective for Goα, but not Giα (McFadzean et al., 1989). Collectively, this evidence suggests that opioid receptors couple to Ca2+ channels through interaction with G proteins associated with the Goα-class of subunits (either Gα and/or Gβγ subunits). In any case, the results from the present study suggest that activation of higher densities of δ-opioid receptors does not produce promiscuous coupling to additional G proteins, resulting in the inhibition of Ca2+ channels in GH3 cells. Alternatively, it seems more likely that a greater number of δ-opioid receptors are required to produce activation of increased amounts of specific G proteins that enable coupling to Ca2+channels.
Recent studies have demonstrated that the coexpression of fully functional κ- and δ-opioid receptors produces a heterodimer combination with different binding and functional properties seen when either of the individual receptors are expressed alone (Jordan and Devi, 1999). Therefore, it is also possible that the presence of the μ-opioid receptor and/or the formation of μ/δ-opioid receptor dimers may enable coupling to Ca2+ channels in GH3MORDOR cells. To examine this possibility, we conducted additional experiments with β-FNA at concentrations (10 and 20 nM) suitable to all but abolish μ-opioid receptor activation by DAMGO (Chen et al., 1996). This action had no significant effect on the ability of δ-opioid receptors to couple to either adenylyl cyclase or Ca2+ channels in GH3MORDOR cells. This suggests that the μ-opioid receptor is not influencing the function of δ-opioid receptors in this cell line.
We also performed additional experiments with SUPERFIT (12 and 60 nM) to determine whether this relatively selective δ-opioid receptor alkylating agent influenced the ability of μ-opioid receptors to couple to adenylyl cyclase and/or Ca2+ channels. Interestingly, we found that both concentrations of SUPERFIT diminished coupling of μ-opioid receptors to Ca2+ channels in GH3MORDOR cells. SUPERFIT had no direct effect on Ca2+ channel function as evidenced by the unaltered amplitude of Ba2+ currents recorded from cells pretreated with 12 and 60 nM. Additionally, although 12 nM SUPERFIT had no effect on the ability of μ-receptors to couple to adenylyl cyclase, the highest concentration of SUPERFIT (60 nM) tested did diminish the amplitude of the DAMGO (100 nM)-evoked inhibition of cAMP accumulation. Because the same concentrations of SUPERFIT produced a greater effect in GH3MORDOR cells than in GH3 MOR cells, it appears that SUPERFIT at high concentrations can decrease the function of μ- as well as δ-receptors in cells transfected with both receptor subtypes. This suggests that μ-opioid receptor function may be influenced by coexpression with δ-opioid receptors and that specific concentrations of SUPERFIT may preferentially block such heterodimers. This interesting possibility will be tested in a subsequent study. However, as mentioned above blockade of μ-receptors by β-FNA has no effect on the function of DPDPE in GH3MORDOR cells and δ-receptors couple to the same G proteins regardless of whether the μ-receptor is present. These findings lead us to conclude that differences in the coupling between δ-receptors and Ca2+ channels seen in GH3DOR and GH3MORDOR cells are best explained by the higher level of δ-receptor expression in the latter. This is supported by the fact that SUPERFIT reduces δ-receptor coupling to Ca2+ channels in the GH3 MORDOR cells at all concentrations tested.
Acknowledgments
We thank Dr. Patricia A. Claude for expert assistance in the transfection of the GH3 cells and Dr. Kenner C. Rice for the generous gift of SUPERFIT. We are also very grateful to Jamie Fornsaglio for skillfully performing some of the electrophysiological experiments.
Footnotes
-
Send reprint requests to: Paul L. Prather, Ph.D., Department of Pharmacology and Toxicology, Mail Slot 611, University of Arkansas for Medical Sciences, 4301 W. Markham St., Little Rock, AR 72205. E-mail: pratherpaull{at}exchange.uams.edu
-
↵1 This work was supported in part by National Institute on Drug Abuse Grants DA10936 (to P.L.P.), DA07234-07 (to P.L.P), DA05627-01 (to E.T.P.), DA05010 (to T.G.H.), DA07339 (to P.Y.L.), DA05695 (to P.Y.L.), and an intramural pilot study grant from the University of Arkansas for Medical Sciences (to P.L.P.).
- Abbreviations:
- CHO
- Chinese hamster ovary
- GABA
- γ-aminobutyric acid
- SUPERFIT
- cis-(+)-3-methylfentanyl isothiocyanate
- ECL
- enhanced chemiluminescence
- DPDPE
- [d-Pen2,5]-enkephalin
- CTOP
- d-Pen-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2
- DMEM
- Dulbecco's modified Eagle's medium
- β-FNA
- β-funaltrexamine
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- [α-32P]AA-GTP
- [α-32P]azidoanilido-GTP
- PAGE
- polyacrylamide gel electrophoresis
- TBS
- Tris-buffered saline
- Received April 3, 2000.
- Accepted August 3, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}