


Abstract
Calcimimetics likeN-(3-[2-chlorophenyl]propyl)-(R)-α-methyl-3-methoxybenzylamine (NPS R-568) potentiate the effects of extracellular Ca2+ on parathyroid Ca2+ receptors and inhibit parathyroid hormone (PTH) secretion in vitro. When administered by gavage to normal rats in this study, NPS R-568 caused a rapid, dose-dependent (ED50, 1.1 ± 0.7 mg/kg) decrease in PTH levels that was paralleled by a subsequent decrease in plasma Ca2+ (ED50, 10.4 ± 3.7 mg/kg). At higher doses (≥3.3 mg/kg), PTH was reduced to a minimum level within 15 min, the duration of which was dose dependent. With doses of 10 to 100 mg/kg, the hypocalcemia was rapid in onset (<30 min) and, at 33 to 100 mg/kg, persisted for >24 h. Neither the magnitude nor the kinetics of the hypocalcemic response was affected by total nephrectomy, demonstrating that NPS R-568 does not induce hypocalcemia by acting on renal Ca2+ receptors to increase Ca2+ excretion. In contrast, parathyroidectomy (intact thyroid) abolished the hypocalcemic response to NPS R-568, regardless of whether the rats were hypocalcemic or rendered acutely normo- or hypercalcemic by calcium infusion before dosing. These data show that the parathyroid Ca2+ receptor can be selectively activated in vivo with a small organic compound to decrease plasma levels of PTH and Ca2+ and thus define the mechanism of action of this compound in vivo. Moreover, the data add pharmacological support to the view that the Ca2+ receptor is the primary molecular entity regulating systemic Ca2+ homeostasis.
Primary hyperparathyroidism (HPT) is characterized by chronically elevated plasma levels of parathyroid hormone (PTH) and ionized calcium (Ca2+). The treatment of this disease has been limited to surgical ablation of the affected gland(s), although many different approaches aimed at lowering plasma levels of PTH or Ca2+ or blocking the actions of PTH at target tissues have been attempted (Ljunghall et al., 1994; Silverberg and Bilezekian, 1996). Similarly, the management of secondary HPT with phosphate binders and calcitriol has been less than satisfactory. These treatments fail to lower plasma levels of PTH to target levels in many patients, and lowered levels of PTH often occur only at doses of calcitriol that cause hyperphosphatemia and/or hypercalcemia (Delmez and Slatopolsky, 1991; Coburn and Salusky, 1994). Moreover, because calcitriol therapy affects the synthesis rather than the secretion of PTH (Silver and Naveh-Many, 1994), its effects are slow in onset and often require months to become manifest.
A new approach for treating HPT is to target the mechanisms used by extracellular Ca2+ to regulate the moment-to-moment secretion of PTH. The Ca2+receptor is the first step in this process and enables parathyroid cells to detect and respond to changes in the concentration of extracellular Ca2+ (Nemeth and Scarpa, 1987;Brown, 1991; Brown et al., 1993; Garrett et al., 1995a). Activation of the Ca2+ receptor results in an immediate decrease in secretion of PTH. Unfortunately, the only known agonists of the Ca2+ receptor are either inorganic or organic polycations (Brown, 1991), all of which are unsuitable as pharmaceutical therapies. Recently, we discovered that certain small organic compounds, not polycations, are capable of activating the Ca2+ receptor. Structural modifications of these leads resulted in a class of compounds having potent and selective activity at the Ca2+ receptor, and they were shown to inhibit PTH secretion from bovine parathyroid cells in vitro (Nemeth et al., 1998). These compounds behave as positive allosteric modifiers of the Ca2+ receptor to increase the sensitivity of the receptor to activation by extracellular Ca2+. Such compounds are termed type II calcimimetics to distinguish them from Ca2+ and other polycations, which are true agonists and are termed type I calcimimetics (Nemeth et al., 1998).
One such type II calcimimetic isN-(3-[2-chlorophenyl]propyl)-(R)-α-methyl-3-methoxybenzylamine (NPS R-568). After oral administration, this compound caused a rapid decrease in plasma levels of PTH and Ca2+ in patients with either primary (Silverberg et al., 1997) or secondary (Antonsen et al., 1998) HPT. Although these findings seemingly provide proof of concept for this novel therapeutic approach, they do not provide any information regarding the mechanism by which NPS R-568 acts in vivo to lower the plasma levels of Ca2+. The same Ca2+ receptor is also expressed at high levels in C cells of the thyroid (Garrett et al., 1995b;Freichel et al., 1996) and the cortical thick ascending limb of the loop of Henle (Riccardi et al., 1995, 1996), tissues that play pivotal roles in the regulation of plasma Ca2+homeostasis. Thus, NPS R-568 could induce hypocalcemia in patients with primary HPT not only by inhibiting PTH secretion but also by activating C-cell Ca2+ receptors to stimulate the secretion of calcitonin and/or kidney Ca2+ receptors to inhibit the tubular reabsorption of Ca2+. Indeed, there is reason to suppose that all these sites would be affected by a calcimimetic compound in vivo and that they would all contribute to the observed hypocalcemia.
In this and the companion article (Fox et al., 1999), we use experimental manipulations in the rat to define the mechanism by which NPS R-568 decreases the plasma levels of Ca2+ in vivo. We show that the parathyroid cell Ca2+receptor can be targeted selectively by orally administered NPS R-568 and that the hypocalcemic response results largely, if not exclusively, from changes in the plasma levels of PTH and calcitonin. Ca2+ receptors in the kidney contribute little, if at all, to the hypocalcemic response to NPS R-568.
Materials and Methods
Animals, Diets, and Surgical Procedures.
Male Sprague-Dawley rats (Harlan Sprague-Dawley, Inc., Indianapolis, IN), weighing 250 to 300 g, were used in these studies. They were housed in hanging wire cages for at least 7 days before study and fed a commercial rodent chow (Purina 5001; Ralston Purina Co., St. Louis, MO) and tap water ad libitum. All surgical procedures were performed with a combination of ketamine (90 mg/kg i.m.) and xylazine (7 mg/kg i.m.) as anesthetic. A blood-sampling catheter was implanted in the abdominal aorta via the femoral artery, and, in some of the rats, a venous catheter for infusions was also implanted chronically in the inferior vena cava via the femoral vein (Fox, 1990). In one series of experiments, each rat was also parathyroidectomized (PTX). The parathyroid glands were exposed and removed by careful dissection leaving the thyroid gland intact. A plasma Ca2+ level of <1.0 mM (normal, ∼1.4 mM) 24 h after surgery was used to indicate successful removal of all parathyroid tissue. After catheterization, each rat was housed individually and studied no sooner than 2 days after surgery. All experimental procedures were approved by the Institutional Animal Care and Use Committee of NPS Pharmaceuticals, Inc.
Time Course and Dose Response to Orally Administered NPS R-568 in Normal Rats.
The first study tested the effects on plasma PTH and Ca2+ levels of NPS R-568 (as the hydrochloride salt) at doses of 3.3, 10, 33, and 100 mg/kg b.wt. The vehicle used to dissolve NPS R-568 was an aqueous solution of 2-hydroxypropyl-β-cyclodextrin (Research Biochemicals International, Natick, MA). The 100-mg/kg dose was dissolved (20 mg/ml) in 15% cyclodextrin. To keep the proportion of NPS R-568 to cyclodextrin constant at each dose, the lower doses of NPS R-568 were prepared by diluting the 20-mg/ml solution with water; i.e., the 33-mg/kg dose was administered in 5%, the 10-mg/kg dose in 1.5%, and the 3.3-mg/kg dose in 0.5% cyclodextrin. Vehicle-dosed rats received 15% cyclodextrin alone. Subsequent studies with an oral dose of 10 mg/kg showed no difference in the hypocalcemic response when NPS R-568 was administered in 1.5 or 15% cyclodextrin (data not shown). Blood samples (0.8 ml) were collected for assay of plasma Ca2+ and PTH levels, immediately before and at 0.25, 0.5, 1, 1.5, 2, 4, 6, 24, and 48 h after the administration of NPS R-568 or vehicle by oral gavage (1.0 ml/200 g b.wt.). To prevent excessive blood loss during the experiment, after removal of the plasma sample, the erythrocyte pellet was resuspended in an equal volume of normal rat plasma and reinjected.
Because the lowest dose used in the first study (3.3 mg/kg) maximally suppressed plasma PTH levels, a second, shorter experiment with lower doses of NPS R-568 (0.1, 0.33, 1.0, and 3.3 mg/kg) was performed in similar rats. As before, the 3.3-mg/kg dose was dissolved in 0.5% cyclodextrin in water, and lower doses were prepared by diluting that solution with water. Blood samples were collected immediately before and at 10, 20, 30, and 60 min after dosing.
Effects of Prevention of Hypocalcemia on Plasma PTH Response to NPS R-568.
Normal rats with chronic arterial and venous catheters received an oral dose of vehicle (1.5% cyclodextrin) or NPS R-568 (10 mg/kg). In one group of NPS R-568-dosed rats, calcium gluconate was infused i.v. at rates determined empirically to prevent the induced fall in plasma Ca2+ levels, i.e., to mimic the changes in vehicle-dosed rats. Blood samples were collected for 6 h after dosing.
Plasma Ca2+ Response to NPS R-568 in PTX Rats.
Four separate experiments were performed in PTX rats. Each rat received NPS R-568 (10 mg/kg) or vehicle (1.5% cyclodextrin) by gavage as described above. Study 1 investigated the plasma Ca2+ response to NPS R-568 in hypocalcemic PTX rats. Blood samples (0.1 ml) were collected for measurement of plasma Ca2+ levels before and 15, 30, 60, 90, 120, and 180 min after dosing. Studies 2, 3, and 4 tested whether the effect of NPS R-568 on plasma Ca2+ in PTX rats was dependent on the prevailing level of plasma Ca2+. In study 2, plasma Ca2+ was rapidly (<5 min) raised to normocalcemic levels (1.3–1.4 mM) and maintained for 60 min by the initially rapid and subsequently slower i.v. infusion of 10% calcium gluconate via the calcium-clamp technique (Fox, 1991). The calcium infusion rate was constant from 20 to 60 min, at which time NPS R-568 was administered and the infusion terminated. Blood samples (0.1 ml) for plasma Ca2+ assay were collected before; at 5, 15, 30, 45, and 59 min after the start of the calcium infusion; and at 15, 30, 60, 90, and 120 min after NPS R-568 administration. Study 3 was similar to study 2 except that the calcium infusion was continued at the same constant rate after the administration of NPS R-568 at 60 min. Study 4 tested the effects of NPS R-568 on hypercalcemic PTX rats. In these animals, plasma Ca2+ levels were rapidly raised to and maintained at ∼1.8 mM for 180 min. As in study 3, the calcium infusion rate was constant from 20 to 180 min. NPS R-568 was administered at 60 min and plasma Ca2+ levels monitored for another 120 min.
Plasma Ca2+ Response to NPS R-568 in Nephrectomized (NX) Rats.
This study assessed the role of the kidneys in the hypocalcemic response to NPS R-568. Each rat was anesthetized as described above, and anesthesia was maintained throughout the experiment by the periodic i.v. injection of anesthetic. After a catheter was implanted in the abdominal aorta, the renal vessels were ligated, and both kidneys were removed. A sham operation, which involved exposure of the kidneys and closure of the two flank incisions with wound clips, was performed in half the rats. Within 30 min of the completion of the nephrectomy or sham operation, NPS R-568 (1 mg/kg b.wt.) or vehicle (15% cyclodextrin) was injected via the arterial catheter (0.1 ml/100 g b.wt.). NPS R-568 was administered parenterally in this study because difficulties were encountered in instilling the solutions into the stomach of anesthetized rats. This dose is about 30 times higher than the ED50 for the acute reduction in plasma PTH levels by NPS R-568 administered i.v. in rats (our unpublished observations). Blood samples (0.1 ml) were collected for measurement of plasma pH and Ca2+levels immediately before NPS R-568 administration and at 15, 30, 60, 90, 120, 180, and 240 min after the injection.
Analyses.
Plasma pH and Ca2+ levels were measured immediately on duplicate 35-μl samples of heparinized whole blood with a model 634 Ca2+/pH analyzer (Ciba Corning, Medford, MA). PTH levels were determined with a two-site rat PTH-(1–34) immunoradiometric assay kit (Immutopics, San Clemente, CA). The detection limit averaged 1.0 ± 0.1 pg rat PTH-(1–34)/ml in six separate assays, and intra- and interassay coefficients of variation for an internal reference standard (49.5 pg/ml) averaged 5.5 and 4.5%, respectively. PTH levels in normal conscious rats were 18.7 ± 1.2 pg/ml (n = 60). PTH levels decreased from 15.7 ± 4.7 to 3.6 ± 0.5 pg/ml (n = 4) in normal rats 10 min after a calcium gluconate injection (100 μmol i.v.). PTH immunoreactivity was undetectable in the plasma of PTX rats. Plasma levels of phosphate were measured with a multichannel analyzer (Monarch 1000, Instrumentation Laboratory, Lexington, MA).
Statistical Analyses.
All data are presented as means ± S.E. Plasma pH and Ca2+ and PTH levels were initially subjected to two-factor ANOVA for repeated measures (SuperANOVA; Abacus Concepts, Berkeley, CA). Dunnett’s test was used to determine the significance of differences from control values. The Student-Newman-Keuls multiple-comparison test was used when comparisons other than to control were indicated. The ED50values for reduction in plasma PTH and Ca2+levels by NPS R-568 were determined via the Levenberg-Marquardt algorithm (Kaleidagraph; Abelbeck Software).
Results
Time Course and Dose Response to Orally Administered NPS R-568 in Normal Rats.
With the blood-sampling technique described inMaterials and Methods, plasma levels of PTH tended to increase in control rats for 2 to 3 h after vehicle administration but declined thereafter (Fig. 1). This change in plasma PTH probably resulted from a slight hypocalcemia, which was observed in vehicle-dosed animals. Because we were anticipating decreases rather than increases in plasma levels of PTH, these effects of blood sampling were not considered important.

Effect of oral administration of NPS R-568 on plasma PTH, Ca2+, and phosphate levels in normal rats. Values are means ± S.E.; n = 5 or 6/group. Some of these data have been reported previously (Nemeth et al., 1996).
In contrast to control animals, plasma PTH levels were reduced significantly to minimum levels within 15 min of NPS R-568 administration, and PTH levels remained significantly lower than control in all dose groups at 30 min. The duration of this minimum PTH level was dose dependent; PTH levels started to increase sooner and the rate of restoration to normal levels occurred more rapidly at the lower doses. Thus, whereas plasma PTH levels in rats receiving the two highest doses of NPS R-568 (33 and 100 mg/kg) remained depressed at 1 h after dosing, PTH levels in rats receiving the 3.3- or 10-mg/kg doses were no longer significantly lower. PTH levels in rats receiving the 33- and 100-mg/kg doses remained significantly lower than controls at 2 h after dosing, but PTH levels were no longer different from those in vehicle-dosed rats in any group from 4 to 48 h postdose.
Plasma Ca2+ levels decreased promptly after the administration of NPS R-568 (Fig. 1) and were significantly lower than control levels by 30 min postdose in rats receiving the three highest doses (10–100 mg/kg). At the highest doses, Ca2+levels continued to decrease to a nadir, although the time at which a minimum plasma Ca2+ level was achieved was dose dependent. As was observed with the changes in PTH levels, a nadir in plasma Ca2+ occurred sooner and the rate of restoration of normocalcemia was more rapid with the lower doses. Thus, plasma Ca2+ levels remained at the minimum levels until after 6 h in rats that received the 100-mg/kg dose, whereas plasma Ca2+ was not significantly different from control levels by 4 h postdose with 10 mg/kg. Plasma Ca2+ levels remained significantly below control in the rats receiving the 33- and 100-mg/kg doses at 24 h postdose, but all animals were normocalcemic by 48 h. In contrast, plasma Ca2+ levels in rats receiving the 3.3-mg/kg dose were not significantly different from those seen in vehicle-dosed rats at any time during this 48-h study (Fig. 1).
Changes in the plasma levels of phosphate after NPS R-568 administration were also dose dependent. At the 100-mg/kg dose, phosphate levels initially decreased and were significantly lower at 30 and 60 min (Fig. 1). However, this hypophosphatemic response then reversed, and phosphate levels were significantly elevated above levels in vehicle-dosed rats from 4 to 24 h after dosing. The 33-mg/kg dose produced a similar pattern of response, except that the initial decrease in phosphate levels was not significant, and a significant hyperphosphatemic response was only maintained from 4 to 6 h. Hypophosphatemia did not occur with the 3.3- and 10-mg/kg doses of NPS R-568, but both induced a significant increase in phosphate levels from 2 to 6 h.
Because plasma PTH levels were reduced to minimum levels by 15 min after NPS R-568 administration at all doses, a second experiment investigated the effects of lower doses of NPS R-568 on plasma PTH and Ca2+ levels. As was seen in the first study, plasma PTH levels increased in vehicle-dosed animals and decreased to a minimum at 10 to 20 min postdose in rats receiving the 3.3-mg/kg dose. The 0.1-mg/kg dose had no effect on plasma PTH levels, whereas the 0.33- and 1.0-mg/kg doses, although not decreasing PTH levels, tended to prevent the increase in PTH levels seen in control rats. However, PTH levels were significantly different from control at 10 to 30 min only in the rats receiving the 3.3-mg/kg dose (Fig.2). Plasma Ca2+levels tended to decrease in all animals after the oral administration of NPS R-568, but no significant differences between control and treated rats were observed (Fig. 2).

Effect of oral administration of low-dose NPS R-568 on plasma PTH and Ca2+ levels in normal rats. Values are means ± S.E.; n = 5 or 6/group.
Whereas plasma PTH levels were suppressed to similar levels by 15 min after the administration of NPS R-568 at doses ≥3.3 mg/kg, the rate of onset of hypocalcemia was different. When the data from experiments 1 and 2 were combined with those from other experiments that tested the same doses of NPS R-568, the results showed that the decrement in plasma Ca2+ levels at 30 min after the 3.3-mg/kg dose, although significantly greater than in vehicle-dosed animals, was significantly less than that seen with doses of 10 to 100 mg/kg (Fig.3).

Change in plasma Ca2+ levels at 30 min after the oral administration of NPS R-568 in normal rats. Values are means ± S.E.; n = 10 to 13/group.aP < .05 versus control rats;bP < .05 versus rats receiving the 3.3-mg/kg dose.
The results of experiments 1 and 2 were combined to determine the relationship between dose and the plasma PTH and Ca2+ responses to orally administered NPS R-568. Analysis of plasma PTH levels at 15 min (experiment 1) and the average PTH level in the 10- and 20-min samples (experiment 2) yielded an ED50 for reduction in plasma PTH levels of 1.1 ± 0.7 mg/kg of NPS R-568 (Fig.4). The calculated ED50 for reduction of plasma Ca2+ levels at 1 h postdose was 10.4 ± 3.7 mg/kg.

Relationship between oral dose of NPS R-568 and plasma PTH (●) and Ca2+ (○) levels in normal rats. PTH values are from 15-min samples of experiment 1 (Fig. 1) and from the mean of the 10- and 20-min samples of experiment 2 (Fig. 2). Plasma Ca2+ values are from 1 h postdose in both experiments. Values are means ± S.E.; n = 5 to 12/group.
Effects of Prevention of Hypocalcemia on Plasma PTH Response to NPS R-568.
Because hypocalcemia is the most important stimulus to PTH secretion, an experiment was performed to assess whether the induced fall in plasma Ca2+ levels played a role in the rapid restoration of normal PTH levels in animals given NPS R-568. The i.v. infusion of calcium into one group of rats given NPS R-568 resulted in a plasma Ca2+ profile similar to that observed in rats receiving vehicle. The minimum plasma PTH levels were maintained for >2 h in these rats, compared to <1 h in the rats in which hypocalcemia was allowed to develop (Fig.5). At 2 h after dosing, the difference in PTH levels between hypocalcemic and normocalcemic rats given NPS R-568 was statistically significant.
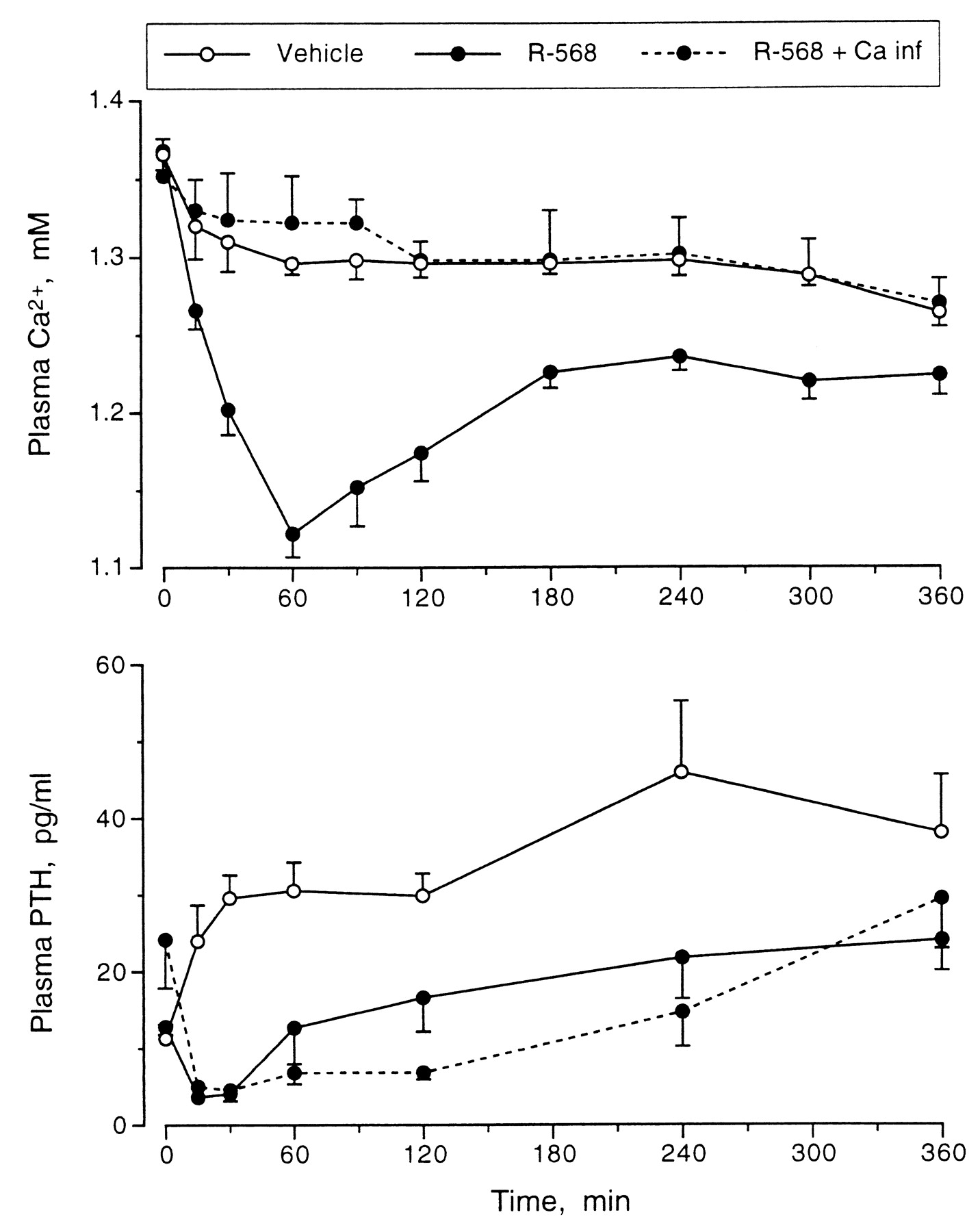
Effect of prevention of induced hypocalcemia by i.v. infusion of calcium gluconate on plasma PTH response to orally administered NPS R-568 (10 mg/kg) in normal rats. Values are means ± S.E.; n = 5/group.
Plasma Ca2+ Response to NPS R-568 in PTX Rats.
Four separate experiments tested the effects on plasma Ca2+ levels of NPS R-568 in PTX rats (Fig.6). Study 1 showed that NPS R-568 had no effect in hypocalcemic PTX rats. Plasma Ca2+levels decreased progressively, with no differences occurring between rats that received vehicle or NPS R-568. Studies 2 and 3 determined whether the acute restoration of normocalcemia by i.v. calcium infusion before NPS R-568 administration would reveal a hypocalcemic response. In study 2, the calcium infusion was terminated when NPS R-568 was administered; there was no effect of NPS R-568 on the rate of fall in plasma Ca2+ levels (Fig. 6). In study 3, the calcium infusion was continued throughout the experiment. Plasma Ca2+ levels tended to increase in control rats after dosing; NPS R-568 prevented this increase such that plasma Ca2+ levels were significantly lower from 60 to 120 min postdose. Study 4 determined the effect of NPS R-568 on plasma Ca2+ levels in acutely hypercalcemic PTX rats. The calcium infusion was also continued throughout this study. No differences in plasma Ca2+ levels were observed between vehicle and NPS R-568-dosed animals at any time (Fig. 6).

Effect of oral administration of vehicle (○) or NPS R-568 (●) (10 mg/kg) on plasma Ca2+ levels in PTX rats that were hypocalcemic (top) or rendered acutely normocalcemic (middle) or hypercalcemic (bottom) by i.v. calcium gluconate infusion before dosing. Values are means ± S.E.; n = 4 to 6/group.
Plasma Ca2+ Response to NPS R-568 in NX Rats.
This study determined the role of kidneys in the hypocalcemic response to NPS R-568. Although basal plasma Ca2+ levels tended to be lower in the NX rats, the differences were not significant. Plasma Ca2+ levels increased in both sham-operated and NX rats after vehicle administration, a probable result of the progressive decrease in blood pH (Fig.7). The administration of NPS R-568 resulted in a fall in plasma Ca2+ levels with similar kinetics and of similar magnitude in sham-operated and NX rats. The decrease in plasma Ca2+ levels at the nadir (2 h postdose) was 0.13 ± 0.01 and 0.15 ± 0.03 mM in the sham-operated and NX rats, respectively. There were no differences in blood pH between groups at any time.

Effects of i.v. injection of NPS R-568 (1 mg/kg) on plasma Ca2+ levels and blood pH in acutely NX or sham-operated, anesthetized normal rats. Values are means ± S.E.;n = 5 or 6/group.
Discussion
G protein-coupled receptors have been a classic site for pharmacological intervention in various diseases. As a relatively new member of this receptor family, the Ca2+ receptor would seem to be an ideal target for drugs useful in increasing or decreasing circulating levels of PTH. Our findings demonstrate selective pharmacological alteration of Ca2-receptor activity in vivo.
The hypocalcemic response to NPS R-568 clearly results from the ability of this compound to cause rapid, dose-dependent decreases in plasma levels of PTH. Thus, there is a temporal displacement of responses, and plasma levels of PTH always fall before those of Ca2+. Moreover, at low doses, NPS R-568 can decrease plasma levels of PTH without affecting those of Ca2+, whereas the converse is never observed in normal animals. NPS R-568 is more potent in depressing the plasma levels of PTH than those of Ca2+ as reflected in the dose-response curves for these two parameters. Finally, the dose-response curves for plasma levels of PTH and Ca2+ are parallel, suggesting a single site of action for NPS R-568.
This site of action is, in all likelihood, the parathyroid Ca2+ receptor. NPS R-568 acts selectively on the Ca2+ receptor in vitro and does not affect the activity of several other G protein-coupled receptors, including the structurally homologous metabotropic glutamate receptors (Nemeth et al., 1998). Type II calcimimetic compounds are potent inhibitors of PTH secretion in vitro [the IC50 of NPS R-568 for PTH secretion from isolated bovine parathyroid cells is around 20 nM (Nemeth et al., 1998)]. In dissociated bovine parathyroid cells, NPS R-568 inhibits only the regulated secretory pathway of PTH and, like extracellular Ca2+, does not affect constitutive secretion (Nemeth et al., 1998). Identical results are obtained in vivo; there is a residual level (∼15%) of plasma PTH that cannot be suppressed by a maximally effective dose of NPS R-568 or hypercalcemic stimulus. These results suggest that NPS R-568 and extracellular Ca2+ use the same mechanism in vivo to lower plasma PTH levels. Moreover, the hypocalcemic response to NPS R-568 is abolished in animals lacking the parathyroid Ca2+receptor (i.e., in PTX rats). In contrast, the hypocalcemic response to NPS R-568 persists in totally NX animals. The latter finding is especially important because it shows that NPS R-568 does not cause hypocalcemia by acting on Ca2+ receptors in the kidney (Riccardi et al., 1995; Brown and Hebert, 1997).
On the other hand, an action of NPS R-568 on Ca2+receptors expressed on C cells (Garrett et al., 1995b), thereby stimulating the secretion of calcitonin, may contribute to the hypocalcemic response. Indeed, at doses somewhat higher than those that depress PTH secretion, NPS R-568 does transiently increase the circulating levels of calcitonin (Fox et al., 1999). Such an effect might underlie several observations reported here. For example, the initial hypocalcemic response was less in rats given lower doses of NPS R-568, despite similar suppression of PTH levels. Moreover, PTX animals that were maintained normocalcemic by infusion of calcium gluconate and treated with NPS R-568 were slightly hypocalcemic compared with vehicle-dosed animals. This small effect was absent in similarly treated animals that were rendered hypercalcemic, presumably because calcitonin secretion was already maximally stimulated. Finally, the initial hypophosphatemic response observed with the higher doses of NPS R-568 may be attributable to calcitonin-mediated inhibition of bone resorption. Overall, however, the predominant effect of NPS R-568 that causes sustained hypocalcemia is a lowering of plasma levels of PTH. Our results show that the parathyroid Ca2+receptor can be selectively targeted in the whole animal with small organic compounds.
Inherited disorders of calcium homeostasis, such as familial benign hypocalciuric hypercalcemia and autosomal-dominant hypocalcemia, have been shown to result from inactivating and activating mutations, respectively, in the Ca2+ receptor (Pearce and Brown, 1996; Brown and Hebert, 1997). These molecular genetic studies have provided evidence for the pivotal role of the Ca2+ receptor in maintaining systemic Ca2+ homeostasis. Our findings show that selective activation of the normal Ca2+ receptor lowers plasma levels of PTH and Ca2+ and, as such, they provide the first pharmacological evidence supporting this essential role of the Ca2+ receptor.
Type II calcimimetic compounds, like NPS R-568, provide a novel means of lowering circulating levels of PTH without increasing plasma levels of Ca2+. The rate of onset and the magnitude of the effect on plasma PTH is the same when plasma levels of Ca2+ are clamped at normal levels or allowed to fall. In fact, plasma levels of PTH remain depressed even as the hypocalcemia induced by this effect becomes manifest. If hypocalcemia is prevented by infusion of calcium gluconate, the duration of the effect on plasma PTH levels is increased. Calcimimetic compounds like NPS R-568 can thus produce a reversible chemical parathyroidectomy, the duration of which can be controlled by altering the dosing regimen and/or pharmacokinetic properties of the compound. Calcimimetic compounds could provide a new adjunctive approach to managing secondary HPT and the first pharmaceutical treatment for primary HPT.
Footnotes
-
Send reprint requests to: John Fox, Ph.D., NPS Pharmaceuticals, Inc., 420 Chipeta Way, Salt Lake City, UT 84108. E-mail: jfox{at}npsp.com
- Abbreviations:
- HPT
- hyperparathyroidism
- NX
- nephrectomized
- PTH
- parathyroid hormone
- PTX
- parathyroidectomized
- Received November 10, 1998.
- Accepted March 31, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}