


Abstract
The influence of secretory transporters on intestinal permeability characteristics of the H2 receptor antagonists ranitidine and cimetidine was studied in Caco-2 monolayers and rat intestinal mucosa mounted in Ussing chambers. Both drugs exhibited vectorial transport across rat ileum with significantly greater (2–4-fold) permeability in the serosal-to-mucosal than the mucosal-to-serosal direction, indicative of net mucosal secretion. Mucosal ranitidine secretion was also observed in rat distal colon, although to a lesser degree. Ileal ranitidine secretion was concentration dependent and significantly reduced by the P-glycoprotein (P-gp) substrates verapamil and cyclosporin. In contrast, probenicid, an inhibitor of the multidrug-related protein, had no effect on ranitidine permeability. The paracellular marker mannitol showed no evidence of asymmetric permeability or sensitivity to P-gp inhibitors. Significant expression of P-gp protein in rat intestinal epithelial cells was confirmed by immunoblotting. Caco-2 monolayers, which overexpress P-gp, also showed asymmetric permeability of ranitidine and cimetidine. In this model, ranitidine permeability in the mucosal-to-serosal direction decreased by ≈95% as monolayer resistance increased from 150 to 500 Ω/cm2, indicating a primarily paracellular route of transport. However, serosal-to-mucosal permeability was insensitive to resistance changes, consistent with a primarily transcellular route in this direction. These data indicate that ranitidine and cimetidine can act as substrates for intestinal P-gp and suggest that the balance between absorptive and secretory mechanisms as a factor in determining intestinal absorption needs to be a routine consideration even for compounds expected to have a predominantly paracellular route of absorption.
P-glycoprotein (P-gp) is a 170–180-kDa membrane glycoprotein that mediates the active, outward transport of a variety of mainly lipophilic compounds from cells (Gottesman and Pastan, 1993). Overexpression of P-gp by cancer cells poses a considerable problem in chemotherapy because several antitumor drugs, notably the vinca alkaloids (e.g., vinblastine), are substrates for the transporter, thus leading to the phenomenon of multidrug resistance (MDR) (Fardel et al., 1996; Ford and Hait, 1990). P-gp also is widely expressed in normal tissues, including epithelial cells in the gastrointestinal tract and kidney tubules and endothelial cells at the blood-brain barrier (Thiebaut et al., 1989;Cordon-Cardo et al., 1990). Although the function of P-gp in normal tissues is not fully understood, it may have a protective function in limiting the net permeability of xenobiotics across these barriers (Cordon-Cardo et al., 1990; Ford and Hait, 1990). A number of other transport proteins with broad substrate specificity are expressed in the intestine and may also be involved in drug excretion, including multidrug-related protein (MRP) and the polyspecific cation transporter, OCT1 (Gründemann et al., 1994; Barrand et al., 1997).
There is increasing evidence that P-gp may have a much broader substrate specificity than originally envisaged, including clinically important drugs such as dexamethasone and other steroids (Ueda et al., 1992) and the immunosuppressive agent cyclosporin A (Saeki et al., 1993). Recent studies using transgenic mice in which the mdrla P-gp gene is disrupted provide evidence that P-gp influences the tissue distribution and pharmacokinetics of a wide range of drugs, some not previously associated with MDR (Schinkel et al., 1995). Markedly increased drug levels in the brains of these animals suggest that P-gp acts to limit the permeability of the blood-brain barrier, which is likely to be important in defining the toxicity of many drugs, as has been demonstrated for ivermectin in mdr1a−/−mice (Schinkel et al., 1994).
The intestinal epithelium is an important site for the absorption of orally administered drugs via paracellular and transcellular routes and is exposed to a variety of factors present in the intestinal lumen. The significant expression of P-gp in normal intestine raises the questions of its functional role and of whether it can influence the efficiency of drug absorption. Studies using reconstituted epithelial monolayers derived from human colonic adenocarcinomas have shown that transepithelial permeabilities of classic P-gp substrates such as vinblastine, as well as other compounds not usually associated with multidrug resistance (Hunter et al., 1991, 1993; Hosoya et al., 1996), are modified by active transport via P-gp. The relevance of these findings to P-gp effects on drug permeability in normal intestine is uncertain, given that these cells are derived from a human colonic tumor and exhibit permeability characteristics different to normal epithelium (Tanaka et al., 1995). Although there are indications of functional P-gp in normal intestine (Hsing et al., 1992; Saitoh and Aungst, 1995; Terao et al., 1996), there remains little information on the influence of P-gp expression on the transport of common drugs in “normal” tissues, although there is evidence of the secretion of known P-gp substrates (Fricker et al., 1996; Terao et al., 1996). It has been shown that the inhibition of P-gp in the rat intestine leads to an increase in the bioavailability of digoxin (Su and Huang, 1996).
The H2 receptor antagonists ranitidine and cimetidine are small, relatively hydrophilic drugs believed to cross the intestinal epithelium passively via a predominantly paracellular route (Collett et al., 1996; Gan et al., 1993) and, as such, are not thought to be typical substrates for P-gp. Preliminary observations in the human colonic cell line Caco-2 (Cook and Hirst, 1994) suggest ranitidine may be a substrate for this transporter, but no detailed information is available on the role of P-gp in modulating permeability of these drugs in normal intestine. The present study provides evidence of polarized permeability of both ranitidine and cimetidine across the Caco-2 colonic tumor line consistent with active transport by P-gp and demonstrates a similar pattern of permeability in rat small and large intestine in vitro. These data suggest that P-gp may be a factor in the oral absorption and distribution of common drugs and that Caco-2 provides a useful paradigm of P-gp function in normal gut.
Experimental Procedures
Materials
Radiolabeled 14C-mannitol and3H-vinblastine were purchased from Amersham International (Buckinghamshire, UK), and unlabeled compounds were from Sigma or Aldrich Chemical Company Ltd. (UK).14C-Ranitidine and14C-cimetidine were prepared by GlaxoWellcome Research and Development Ltd. (Greenford, Middlesex, UK). Tissue culture reagents were purchased from Gibco Life Technologies Ltd. (Paisley, UK). P-gp antibody C219 was purchased from Signet Laboratories, Inc. (Dedham, MA).
Tissue Culture
Caco-2 cells (passage 90–110) were cultured as described previously (Collett et al., 1996). For drug transport studies, cells were seeded onto 12-mm polycarbonate filter cell culture inserts (Snapwell/Transwell, Costar, Ltd., Buckinghamshire, UK) at a density of 1 × 105 cells/cm2. Culture media was changed every 2 days, and cultures were used for permeability studies 22 to 27 days after seeding. The development of transepithelial electrical resistance (Rt) was monitored using an Evometer (World Precision Instruments, Sarasota, FL) fitted with “chopstick” electrodes.
Transport Studies
Rat Intestine.
Nonfasting male Sprague-Dawley rats were stunned and killed by cervical dislocation. The ileum and colon were immediately removed, washed, and stripped of muscle layers by blunt dissection. Segments of the mucosa were mounted in Ussing chambers (0.64-cm2 surface area) and bathed on mucosal and serosal aspects with 4 ml of serum-free Dulbecco’s modified Eagle’s medium, pH 7.4 (SFDM), at 37°C under continuous oxygenation. Spontaneous tissue potential difference (PD) and Rt, measured as the deflection in PD caused by a 100-μA current pulse, were monitored periodically throughout the experiment; at all other times, tissues were maintained under short circuit conditions (Is.c.). Only tissues in which these electrical parameters remained within 15% of the initial stabilized value during the course of the experiment were used.
After a 45-min equilibration period, labeled and unlabeled compound was added to either the mucosal (apical) or serosal (basolateral) chamber to give a final concentration routinely of 0.1 mM and approximately 0.3 μCi/ml. A 100-μl sample was removed from the donor compartment to determine the initial isotopic concentration. Samples (1 ml) from the receiving chamber were taken every 30 min and replaced with fresh SFDM.14C and 3H activities were determined by liquid scintillation counting. The background was always less than 1% and was not subtracted from the total. The use of short flux periods (15–30 min) maintained sink conditions and ensured a linear rate of drug transport.
Caco-2 Monolayers.
Drug transport across Caco-2 cell layers grown on Snapwell units was measured by a method similar to that described above for the rat intestine. Epithelial cell layers were removed from the growth medium, washed twice in SFDM, and placed in a modified Ussing chamber with a 1.0-cm2 diffusion window. Electrical parameters and permeability were measured in the same way as for intestinal mucosa.
The effect of modulating the paracellular pathway on ranitidine permeability in Caco-2 monolayers was measured by a method similar to that described previously (Collett et al., 1996). Briefly, epithelial cell layers were removed from the growth medium, washed twice in SFDM, and placed in a 12-well plate. Aliquots of SFDM were added to the apical (0.5 ml) and basolateral (1.5 ml) compartments. Cell layers were left for 60 min to equilibrate. To begin permeability measurements, a 100-μl aliquot was removed from the apical or basolateral reservoir and replaced with an equal volume of SFDM containing14C-labeled and unlabeled ranitidine to give a final concentration of 0.1 mM. At each time point, 100 μl was removed from the receiving chamber for analysis and replaced with fresh SFDM. A 20-μl sample was also taken from the donor compartment at the beginning and end of each experiment. For the pulsed reduction of extracellular Ca++, culture inserts were washed three times with SFDM solution containing 2.5 mM EGTA and incubated in this medium on both apical and basolateral sides for 15 min, after which extracellular Ca++ was replaced. This produced a rapid and reversible reduction inRt, as previously described (Collett et al., 1996).
Permeability was expressed as apparent permeability (Papp) in cm/s, obtained according to the equation:
Where indicated, net permeability values (Pappnet) are shown representing the difference between permeability in the serosal-to-mucosal (s-to-m) and mucosal-to-serosal (m-to-s) directions (i.e.,Pappnet =Papps-m −Pappm-s). A positive value ofPappnet represents net mucosal secretion, and negative values indicate net mucosal absorption. Pappvalues are presented as the mean of three or four 15-min periods.
None of the drugs whose permeabilities were studied in this work had a significant effect on tissue electrical parameters when used at the concentrations indicated.
Immunoblotting
Epithelial cell fractions were isolated from rat ileum and colon by Ca++ chelation using a modification of the method of Traber et al., (1991). For ileum, a 15-cm segment starting 5 cm proximal to the ileocecal junction was removed and flushed with ice-cold phosphate-buffered saline (PBS). The segment was filled with buffer A (96 mM NaCl, 27 mM sodium citrate, 1.5 mM KCl, 8 mM KH2PO4, 5.6 mM Na2HPO4, 40 μg/ml phenylmethylsulfonyl fluoride, pH 7.4) and incubated for 15 min at 37°C. At the end of this period, the luminal buffer was drained, and the segment was refilled with buffer B (109 mM NaCl, 2.4 mM KCl, 1.5 mM KH2PO4, 4.3 mM Na2HPO4, 1.5 mM EDTA, 10 mM glucose, 0.5 mM dithiothreitol, and 40 μg/ml phenylmethylsulfonyl fluoride, pH 7.4). This process was repeated for incubation periods of 4, 4, 7, 5, 7, 10, and 10 min. This procedure has been shown to produce a sequential isolation of epithelial cells from villus to crypt (Traber et al., 1991). For immunoblotting studies, fractions 1 to 3 were pooled to give a villus cell fraction, and fractions 7 and 8 were pooled to give a crypt fraction. Intervening fractions (4–6) were not used. For colon, an epithelial cell fraction containing primarily intact crypts was isolated from the distal 5 cm of rat colon as previously described (Warhurst et al., 1996). Cell fractions were pelleted by centrifugation at 100g for 5 min at 4°C and washed twice in ice-cold PBS. Confluent Caco-2 cultures grown in 75-cm2 flasks were harvested by scraping into PBS and pelleted by centrifugation.
Immunoblot analysis of P-gp was performed by a method similar to that previously described (Hosoya et al., 1996). Epithelial cells were suspended in lysis buffer (PBS containing 3% sodium dodecyl sulfate, 2 mM dithiothreitol, 0.2 mM pepstatin, 0.2 mM leupeptin, and 1 mM phenylmethylsulfonyl fluoride) incubated on ice for 45 min before a brief (30-s) homogenization in a Polytron homogenizer. The homogenate was centrifuged at 12,000g for 15 min at 4°C, and the supernatant was used for protein determination and immunoblotting. For each cell fraction, 50 μg of protein were separated on a 7.5% sodium dodecyl sulfate-polyacrylamide gel followed by electrophoretic transfer onto Hybond ECL nitrocellulose membrane (Amersham, UK). Membranes were blocked overnight in PBS containing 5% defatted milk and 0.2% Tween-20 at 4°C followed by incubation with anti-P-gp antibody (C219; 1:1000) in the same solution for 2 h at room temperature. After washing, antibody binding to the 170–180-kDa P-gp protein was detected using an enhanced chemiluminescence blotting system (Amersham, UK).
Statistical Analysis
Data are presented as mean ± S.E.M. Statistical comparisons were made using the Student’s t test for unpaired data or, where appropriate, the Mann-Whitney Utest. Values of p < .05 were considered significant, and n refers to the number of cell monolayers or, in case of animal tissues, the number of animals with replicate tissues from each.
Results
Influence of P-gp on Ranitidine and Cimetidine Permeability in Caco-2.
Initial studies examined the ability of P-gp to modulate ranitidine and cimetidine permeability in Caco-2 monolayers, a system that is commonly used to model intestinal drug transport and is known to express high levels of P-gp in the apical membrane (Hunter et al., 1993a, 1993b). Both ranitidine and cimetidine are relatively hydrophilic with a water-octanol partition coefficient (logP) of 0.27 and 0.4, respectively (Moriguchi et al., 1994; Coruzzi et al., 1996). Figure 1A shows the Pappof 0.1 mM ranitidine and cimetidine across Caco-2 monolayers in the m-to-s or s-to-m direction. Ranitidine permeability in the s-to-m direction is 3-fold greater than that observed for m-to-s indicative of polarized secretion of the drug (P < .01). The addition of 0.1 mM verapamil, a substrate of P-gp used to reverse the MDR phenotype, abolished this secretion, primarily by reducing s-to-m ranitidine permeability to a value not significantly different from m-to-s permeability. Verapamil also produced a minor, although not significant, increase in m-to-s permeability of ranitidine. Cimetidine exhibited a similar pattern with a net secretion that could be inhibited by verapamil (Fig. 1A). The greater permeability of ranitidine from the serosal side of Caco-2 monolayers was concentration dependent with an approximate EC50 of 0.4 mM (Fig. 1B). In contrast, mucosal permeability of the drug showed no significant concentration dependence. Similar studies were performed using vinblastine, a known P-gp substrate that is lipophilic (log P, 1.96;Margalit et al., 1991) and crosses the epithelia via a predominantly transcellular route, and the classic paracellular marker mannitol (logP, −3.10; Rubas et al., 1993) (Table1). The pattern of vinblastine permeability across Caco-2 was similar to that observed for ranitidine and cimetidine, with permeability being 5-fold greater in the s-to-m than the m-to-s direction. Verapamil (0.1 mM) again significantly reduced s-to-m and increased m-to-s vinblastine permeability (P < .05 in both cases). No attempt was made to examine higher concentrations of vinblastine because these caused a marked decrease in monolayer resistance (data not shown). In marked contrast, the permeability of mannitol in Caco-2 did not show asymmetry and was unaffected by the addition of verapamil (Table 1). These data are consistent with ranitidine and cimetidine being able to enter Caco-2 cells, at least from the serosal aspect, and act as a substrate for P-gp.
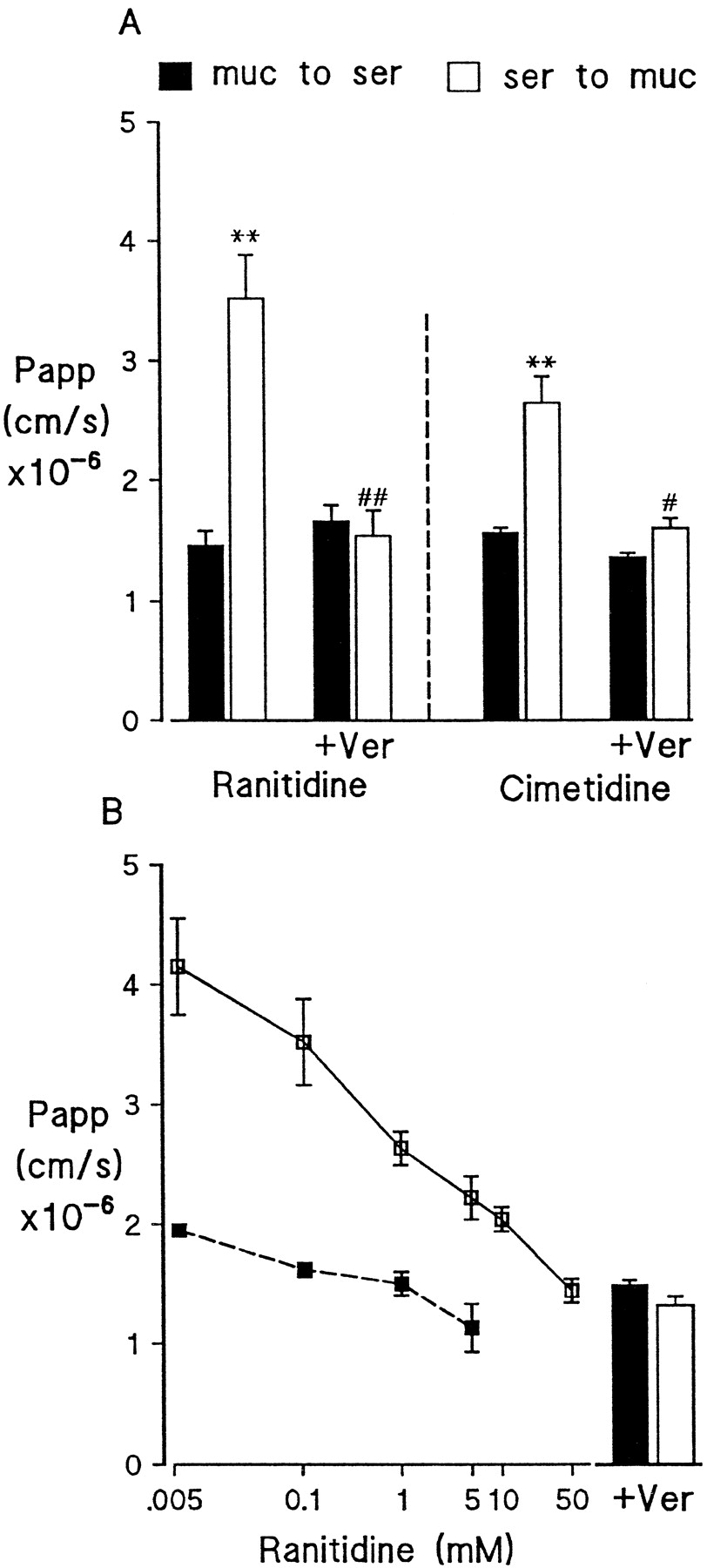
A, Papp of 0.1 mM ranitidine and cimetidine from the mucosal (▪) or serosal (■) side of Caco-2 monolayers in the presence or absence of 0.1 mM verapamil (Ver) (n = 4–7). B, concentration dependence of ranitidine permeability from mucosal (▪) or serosal (■) side of Caco-2 monolayers. Bar graph shows permeability on addition of 0.1 mM verapamil (n = 3–6). Permeability is expressed asPapp in cm/s as described under “Materials and Methods.” Verapamil was added to both mucosal and serosal bathing solution. Values are mean ± S.E.M. **P < .01 compared with control m-to-s permeability. #P < .05. ##P < .01 compared with control s-to-m permeability.
Permeability of mannitol and vinblastine across Caco-2 monolayers
Functional P-gp Expression in Rat Intestine.
Modulation of ranitidine and cimetidine permeability by P-gp in Caco-2 cells could be a function of their tumor origin and may not be relevant to their permeability in “normal” intestinal epithelium. We therefore went on to investigate whether ranitidine permeability could be similarly influenced in small and large bowel epithelium isolated from the rat. Immunoblotting studies with an anti-P-gp antibody showed significant expression of P-gp protein in Caco-2 and epithelial cells isolated from rat ileum and colon (Fig. 2). Caco-2 cells showed the highest levels of expression, followed by rat ileum and colon. In the case of the ileum, P-gp expression was unevenly distributed in the epithelium, being highest in villus cells with much lower levels in crypt epithelium.

Western blot analysis of P-gp expression in epithelial cells isolated from rat ileum and colon and Caco-2 monolayers. Two epithelial fractions are shown for ileum representing predominantly villus (V) or crypt (C) cells. Blots were probed with the anti-P-gp antibody C219. Arrow indicates the 170-kDa P-gp band.
In vitro permeability studies with isolated mucosal sheets of rat ileum and colon provide evidence for functional expression of P-gp. Serosal permeability of both ranitidine and cimetidine across rat ileum was 3- to 4-fold greater than mucosal permeability (Fig.3). Net mucosal secretion of both drugs was markedly inhibited by verapamil due primarily to a reduction in s-to-m permeability, although in the case of ranitidine there was also a small but significant (P < .05) increase in m-to-s permeability. Net mucosal secretion of ranitidine also exhibited concentration dependence with 10 mM ranitidine markedly reducing net secretion to a level similar to that observed in the presence of verapamil (Pappnet: ranitidine 0.1 mM, 15.0 ± 0.7 × 10−6 cm/s; ranitidine 0.1 mM + verapamil, 3.7 ± 0.38 × 10−6 cm/s; ranitidine 10 mM, 6.1 ± 1.7 × 10−6 cm/s). Rat distal colon also exhibited polarized efflux of ranitidine, although the magnitude of secretion was lower than that observed for small intestine with a 2-fold greater permeability in the s-to-m direction (m-to-s, 2.7 ± 0.3 × 10−6 cm/s; s-to-m, 6.2 ± 0.6 × 10−6 cm/s, P < .05). The addition of verapamil (0.1 mM) reduced s-to-m permeability to 4.0 ± 0.7 × 10−6 cm/s and slightly increased m-to-s permeability, resulting in marked inhibition of net mucosal ranitidine secretion from 3.5 ± 0.8 to 0.9 ± 0.5 × 10−6 cm/s (P < .05). The pattern of vinblastine and mannitol permeability in rat intestine closely mimicked that observed in Caco-2 with no evidence for asymmetric permeability of mannitol across rat ileum but a net mucosal secretion of vinblastine that was significantly reduced on the addition 0.1 mM verapamil (Fig. 4).

Asymmetrical permeability of ranitidine and cimetidine across mucosal sheets of rat ileum. Permeability of 0.1 mM ranitidine or cimetidine was measured in the m-to-s (▪) and s-to-m (■) directions in the presence or absence of 0.1 mM verapamil (Ver) as shown. Values shown represent mean Papp ± S.E.M. for n = 3–5 animals with two observations per animal. *P < .05, **P < .01 compared with control m-to-s permeability. #P< .05 compared with control s-to-m permeability.

Sidedness of permeability for mannitol (0.1 mM) and vinblastine (15 μM) across rat ileal mucosa. Values are shown for permeability (Papp) in the m-to-s (▪) or s-to-m (■) direction. The effect of addition of 0.1 mM verapamil (Ver) on permeability of mannitol and vinblastine is also shown. Results are mean ± S.E.M. for n = 3–5 animals with two observations per animal. **P < .01 compared with control m-to-s permeability. #P < .05 compared with control s-to-m permeability.
Role of MDR-Associated Protein in Mediating Mucosal Ranitidine Secretion.
MDR-associated protein (MRP) is a broad specificity drug efflux protein with a pattern of expression in the epithelium similar to that of P-gp. The role of MRP in mediating ranitidine efflux was investigated using probenicid, a known inhibitor of the transporter (Versantvoort et al., 1995). Under the same conditions in which verapamil markedly inhibited ranitidine secretion, probenicid (100 μM) had no effect on ranitidine permeability in either direction across rat ileum, resulting in net ranitidine secretion being unchanged [Papp: control, 14.06 ± 1.88 × 10−6 cm/s; + probenicid (100 μM), 15.25 ± 1.3 × 10−6 cm/s].
Funtional Characteristics of P-gp in Caco-2 and Rat Intestine.
Table 2 summarizes the permeability data for ranitidine and vinblastine in Caco-2 and rat ileum and compares the functional characteristics of the P-gp activity in terms of sensitivity to inhibition by other compounds thought to interact with the transporter. As described above, these epithelia exhibit net mucosal secretion of ranitidine and vinblastine. In both systems, secretion is markedly inhibited by the addition of either verapamil or cyclosporin, which are established substrates for P-gp. In contrast, dideoxyforskolin inhibits net secretion of ranitidine across Caco-2 monolayers but has no detectable effect in rat ileum preparations. A similar lack of effect of dideoxyforskolin was observed in rat distal colon (data not shown).
Influence of P-gp inhibitors on permeability of ranitidine and vinblastine in Caco-2 and rat ileum
Route of Transepithelial Ranitidine Transport.
The finding that ranitidine can be transported by intestinal P-gp implies an intracellular presence for this compound, which is surprising given the evidence that it crosses the intestinal epithelium via a passive paracellular route (Gan et al., 1993). To address this, we investigated the relationship between mucosal and serosal permeability of ranitidine and the transepithelial electrical resistance (Rt) in Caco-2 monolayers, an indicator of the “leakiness” of the paracellular pathway (Fig. 5). Ranitidine permeability in the m-to-s direction decreases by ≈95% as Rt increases from 150 to 500 Ω/cm2, indicative of a primarily paracellular route of transport from the mucosal compartment. In marked contrast, s-to-m permeability of ranitidine shows little or no change over the same Rtrange, suggesting that it readily crosses the basolateral membrane and is moving predominantly transcellularly in this direction. In contrast, mean mannitol permeability across Caco-2 decreased from 3.3 × 10−6 cm/s at 150 Ω/cm2 to 0.3 × 10−6 cm/s at 500 Ω/cm2 in both m-to-s and s-to-m directions.

Influence of Rt on ranitidine (0.1 mM) permeability across Caco-2 monolayers measured in the m-to-s (■) or s-to-m (▪) direction. The Rt values shown include monolayers with resistances lying 25 Ω/cm2 either side of that value (i.e., 400 Ω/cm2 includes values for 375–425). Rt levels below 200 Ω/cm2 were generated by exposing monolayers to 2.5 mM EGTA for 15 min. Results plotted are mean values for four monolayers in each group.
Discussion
The gastrointestinal tract is a major site for the absorption of orally administered drugs, and an understanding of the factors that influence the efficiency of absorption is crucial to the optimization of drug design and delivery. This study demonstrates that P-gp expressed in normal rat intestine has a functional activity broadly similar to that observed in the human colon cancer line Caco-2, stimulating a net mucosal secretion of common drugs with varying lipophilicity that could have an influence on their overall permeability in normal intestine. The H2 receptor antagonists ranitidine and cimetidine are relatively hydrophilic drugs that have been shown to undergo predominantly passive paracellular absorption (Gan et al., 1993; Collett et al., 1996). These drugs exhibited asymmetric permeability across both Caco-2 monolayers and rat intestinal sheets in vitro with s-to-m fluxes ≈4-fold greater than m-to-s fluxes, indicative of a net mucosal secretion of the drugs. In both systems, secretion was shown to be concentration dependent and markedly inhibited by verapamil. This pattern of permeability was similar to that of the lipophilic drug vinblastine, which is transported by P-gp in Caco-2 cells (Hunter et al., 1993b) and shown here to be a substrate for P-gp in rat intestine. Vinblastine also inhibits verapamil transport in the rat gut (Saitoh and Aungst, 1995). Taken together, these data indicate the presence of a transporter located on the mucosal membrane of rat small and large bowel epithelium, which is functionally similar to that expressed by colon cancer cells. This is likely to be the 170-kDa P-gp protein known to confer MDR in cancerous cells (Ford and Hait, 1990; Fardel et al., 1996). Significant expression of this protein was confirmed in all three epithelia by immunoblotting, supporting previous studies (Cordon-Cardo et al., 1990; Hsing et al., 1992), with the highest levels found in Caco-2 monolayers consistent with P-gp overexpression in cancer cells. In addition, the inability of probenicid to inhibit mucosal secretion of ranitidine argues against a role for MRP, the other broad-specificity drug efflux system known to be present in the intestinal epithelium.
Our data for vinblastine support a recent study showing that P-gp-induced efflux can alter the permeability of lipophilic compounds, such as verapamil and organic cations, in rat intestine (Saitoh and Aungst, 1995). Finding that permeabilities of relatively hydrophilic compounds like ranitidine and cimetidine are similarly affected is particularly interesting, given the suggestion that lipophilicity is a determining property of substrates of P-gp (Gottesman and Pastan, 1993). However, P-gp may have wider substrate specificity than previously thought (Schinkel et al., 1995), including data showing that cimetidine is transported by P-gp in renal epithelia (Dutt et al., 1994; Pan et al., 1994). Cimetidine and ranitidine are secreted into rat milk and cross the placenta from maternal to fetal blood by unknown active processes (McNamara et al., 1996van der Aa et al., 1996), in both cases possibly via the P-gp pump. The hypothesis that hydrophilic compounds can access intestinal efflux pathways is supported by a recent study showing active secretion of a hydrophilic cyclic peptide fibrinogen antagonist in rat intestine in vitro (Aungst and Saitoh, 1996).
How is active efflux of ranitidine via a P-gp-like transporter on intestinal enterocytes consistent with the accepted view of a paracellular route of absorption for this compound? The verapamil-sensitive component of ranitidine permeability observed in this study is assumed to be transcellular and modified by P-gp, whereas the verapamil-insensitive component represents either paracellular or passive transcellular transport, which is not modified by P-gp. We have confirmed that paracellular transport is insensitive to verapamil by showing a null effect on the classic paracellular marker mannitol. In Caco-2 monolayers, the lack of effect of verapamil on apical-to-basolateral flux of ranitidine is most easily explained by the inherent limited ability of ranitidine to enter the cell across the apical cell membrane such that passage via the paracellular route is dominant in this direction. In contrast, marked inhibition of basolateral-to-apical flux by verapamil suggests that ranitidine is able to cross the basolateral membrane much more effectively and, hence, the transcellular route and the influence of P-gp become more important. The relationship between Rt and polarized ranitidine flux provides further support in that although apical ranitidine permeability is highly dependent on monolayer Rt, absorption of the drug from the basolateral aspect is largely independent of this parameter. Data from rat ileum are similar except that verapamil significantly increases ranitidine permeability from the mucosal side as well as lowering serosal permeability. This suggests an enhanced apical transcellular component in rat ileum, possibly due to the greater surface area provided by well-developed microvilli. Based on these observations, selective blocking of P-gp activity could increase the efficiency of oral drug absorption, although further studies clearly are necessary, particularly to determine whether similar effects are observed in human tissues.
The relatively small increase in m-to-s ranitidine permeability caused by verapamil treatment in rat intestine suggests that P-gp may have only a minor effect on the efficiency of intestinal ranitidine absorption in vivo. However, it is conceivable that a significant “back-flux” of ranitidine could occur. The intestine can act to clear compounds from the blood (Wacher et al., 1996), and the relatively high serosal permeability to compounds like ranitidine and cimetidine observed in rat intestine could allow a clearance route for these and other drugs that are substrates for P-gp. In addition, it has recently been suggested that there is an overlap in the substrates for intestinal cytochrome P450 and P-gp, which would allow the intestine to both metabolize and secrete xenobiotics from the gut (Gan et al., 1996;Wacher et al., 1996).
How compounds like ranitidine and cimetidine enter epithelial cells has not been specifically addressed in this study. Passive diffusion across the cell membrane may well be a factor, although whether this can account for the apparently high level of P-gp-dependent ranitidine transport from the serosa is questionable, given that both drugs are relatively hydrophilic. An intriguing possibility is that these drugs are actively transported across the serosal membrane. Ranitidine and cimetidine are secreted into the renal tubules by an organic cation transporter that is distinct from P-gp and inhibited by tetraethylammonium (TEA) (Gründemann et al., 1994), and a similar transporter may be involved in drug secretion into the intestine (Saitoh et al., 1996). However, in preliminary studies, TEA at concentrations up to 500 μM applied either mucosally or serosally had no effect on the asymmetric permeability of ranitidine across rat ileum, which, at this stage, argues against a role for a TEA-sensitive cation transporter in either the uptake or efflux of these drugs in intestinal epithelial cells (results not shown). However, it is possible that gut epithelial cells express a novel transporter mediating the uptake of ranitidine and cimetidine. Certainly, a proton-coupled, 4,4′-diisothiocyanato-stilbene−2,2′−disulfonic acid (DIDS)-sensitive cimetidine transporter has recently been reported on the apical membrane of renal cells (Dudley and Brown, 1996).
In general, the functional properties of P-gp activity in Caco-2 and rat intestine appear similar. The relative mucosal and serosal permeabilities of ranitidine and vinblastine in both systems were similar even though P-gp content was greater in Caco-2 than in rat intestine. Interestingly, a difference in substrate specificity between P-gp activity in Caco-2 and rat intestine exists as judged by the ability of different compounds to inhibit the verapamil-sensitive transport. Cyclosporin A, a well established substrate of P-gp, inhibits the polarized transport of P-gp substrates (Wang et al., 1995). In the present study, cyclosporin A inhibited the mucosal secretion of ranitidine and vinblastine across both Caco-2 monolayers and rat ileum to a level similar to that produced by verapamil, providing further evidence for the involvement of P-gp in these systems. Dideoxyforskolin, which antagonizes vinblastine secretion in monolayers of Caco-2 (Hunter et al., 1993a) and the canine kidney cell line MDCK (Hunter et al., 1991), essentially abolished ranitidine secretion in Caco-2. In contrast, however, we found that dideoxyforskolin elicited no inhibitory effect on ranitidine secretion in either rat ileum or colon. This may be due to the different substrate specificity of variants of P-gp known to be expressed by human and rat. The human intestine expresses the mdr1 gene product, whereas a different gene, mdr3 (also referred to as mdr1a), is responsible for intestinal P-gp activity in rat (Lee et al., 1993). Our data suggest that the products of these two genes have different sensitivities to dideoxyforskolin. Nevertheless, overall, the Caco-2 line provides a useful model in which to test the potential effects of P-gp on intestinal permeability of drug compounds.
In conclusion, evidence is provided that P-gp expressed in the epithelium of normal intestine may influence the permeability of not only lipophilic compounds but also relatively hydrophilic drugs such as ranitidine and cimetidine. This would suggest that the balance between absorptive and secretory mechanisms as a factor in determining intestinal absorption needs to be a routine consideration even for compounds that are expected to have a predominantly paracellular route of absorption. The implication that this could reduce the efficiency of absorption of common, orally absorbed drugs will need to be tested in an in vivo model. However, there already is emerging evidence that intestinal efflux processes do act to limit oral bioavailability of some compounds. The present study also points to Caco-2 as a useful semiquantitative model for assessing the possible effects of P-gp on intestinal absorption. Recognition of the much broader specificity of P-gp and its functional effects on intestinal drug transport could lead to strategies for improving absorption, either by incorporating structural features in drug design that reduce interaction with P-gp or by the use of specific P-gp inhibitors such as those currently undergoing clinical trials as adjuncts for cancer chemotherapy (Ferry et al., 1996).
Footnotes
-
Send reprint requests to: Dr. Geoffrey Warhurst, Department of Medicine, Clinical Sciences Building, Hope Hospital, Salford M6 8HD, UK.
-
↵1 This work was supported by GlaxoWellcome Research and Development.
- Abbreviations:
- P-gp
- P-glycoprotein
- MDR
- multidrug resistance
- MRP
- multidrug-related protein
- Rt
- transepithelial electrical resistance, PD, potential difference
- TEA
- tetraethylammonium
- Received February 17, 1998.
- Accepted July 10, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}