


Abstract
Ciproxifan, i.e., cyclopropyl-(4-(3-1H-imidazol-4-yl)propyloxy) phenyl) ketone, belongs to a novel chemical series of histamine H3-receptor antagonists. In vitro, it behaved as a competitive antagonist at the H3 autoreceptor controlling [3H]histamine release from synaptosomes and displayed similar Ki values (0.5–1.9 nM) at the H3 receptor controlling the electrically-induced contraction of guinea pig ileum or at the brain H3 receptor labeled with [125I]iodoproxyfan. Ciproxifan displayed at least 3-orders of magnitude lower potency at various aminergic receptors studied in functional or binding tests. In vivo, measurement of drug plasma levels, using a novel radioreceptor assay in mice receiving ciproxifan p.o. or i.v., led to an oral bioavailability ratio of 62%. Oral administration of ciproxifan to mice enhanced by ∼100% histamine turnover rate and steady state level oftele-methylhistamine with an ED50 of 0.14 mg/kg. Ciproxifan reversed the H3-receptor agonist induced enhancement of water consumption in rats with and ID50 of 0.09 ± 0.04 mg/kg, i.p. In cats, ciproxifan (0.15–2 mg/kg, p.o.) induced marked signs of neocortical electroencephalogram activation manifested by enhanced fast-rhythms density and an almost total waking state. In rats, ciproxifan enhanced attention as evaluated in the five-choice task performed using a short stimulus duration. Ciproxifan appears to be an orally bioavailable, extremely potent and selective H3-receptor antagonist whose vigilance- and attention-promoting effects are promising for therapeutic applications in aging disorders.
HA is a cerebral neurotransmitter exerting its actions on target cells via three classes of molecularly and/or pharmacologically well defined receptors designated H1, H2 and H3(reviewed by Hill et al., 1997; Schwartz et al., 1991, 1995). The H3 receptor is a presynaptic receptor regulating the synthesis and/or release of HA itself (Arrang et al., 1983) as well as a variety of other aminergic or peptidergic neurotransmitters (reviewed by Schlicker et al., 1994). It was initially defined by the design of two selective ligands: (R)α-MeHA, a full agonist, and thioperamide, an antagonist with nanomolar potency (Ki ≅ 4 nM). Thioperamide has become the prototypical H3-receptor antagonist, used in a large number of neurochemical, electrophysiological and behavioral studies because it is one of the few agents able to markedly enhance cerebral histaminergic transmissions in vivo via a selective mechanism. In agreement, few other actions of thioperamide were described, e.g., inhibition of P450 cytochromes (La Bellaet al., 1992) and 5-HT3-receptor blockade (Leurset al., 1995), which require higher drug concentrations than H3-receptor blockade and are therefore not relevant forin vivo studies.
Nevertheless thioperamide has several drawbacks: 1) its in vivo potency is rather low compared with its in vitropotency, suggesting that drug bioavailability, particularly its brain penetration, is restricted, 2) more importantly it displays a distinct liver toxicity on repeated administration which has prevented it being submitted to human clinical trials.
Because H3-receptor antagonists represent a novel class of agents with potentially interesting therapeutic applications, namely in psychiatry (Schwartz et al., 1995) sustained efforts have been devoted to the design of drugs more potent and safer than thioperamide (reviewed by Stark et al., 1996b).
As with thioperamide, all highly effective compounds obtained so far contain a monosubstituted imidazole ring, but the thiourea moiety of the latter, to which hepatotoxicity might be attributable, is replaced by numerous polar functionalities such as amine or carbamate, etc.
Recently, we have designed 3-(1H-imidazol-4-yl)propanol derivatives as a novel series of potent in vitroH3-receptor antagonists with a high capacity for oral absorption and brain penetration in some compounds (Stark et al., 1996a; Hüls et al., 1996). In these novel chemical classes of compounds, we have recently identified [125I]iodoproxyfan, i.e., [125I]3-(1H-imidazol-4-yl)propyl-(4-iodophenyl)methyl ether as a new probe for a sensitive assay and localization of the H3 receptor in brain (Ligneau et al., 1994).
We describe the biological properties of ciproxifan (fig.1), a highly potent and selective H3-receptor antagonist belonging to this novel chemical class of compounds which suggest its potential therapeutic interest as a waking and procognitive agent.

Chemical structure of ciproxifan, i.e., cyclopropyl-(4-(3-(1H-imidazol-4-yl)propyloxy)phenyl) ketone.
Materials and Methods
[3H]Histamine release from synaptosomes.
[3H]HA release experiments were performed according toGarbarg et al. (1992). Briefly, a crude synaptosomal preparation from rat cerebral cortex was preincubated for 30 min with [3H]l-histidine (0.4 μM) at 37°C. After extensive washing synaptosomes were resuspended in fresh 2 mM K+-Krebs-Ringer’s medium and in the presence of the appropriate drugs. After 5-min incubation synaptosomes were depolarized bringing the K+-concentration to 30 mM for 2 min. Incubations were ended by a rapid centrifugation and [3H]HA levels in the supernatant were determined after an ion-exchange chromatography purification. Release was expressed as the percent fraction of total [3H]HA initially present in the synaptosomal preparation. Typically total [3H]HA represented about 3,500 dpm/mg protein and total radioactivity about 100,000 dpm/mg protein in the test tube.
Assay of t-MeHA in brain.
Male Swiss mice (18–20 g) or male Wistar rats (140–160 g) (Iffa-Credo, L’Arbresle, France) were fasted for 24 hr before p.o. administration. After treatment animals were killed by decapitation, the brain was dissected out and homogenized in 10 volumes (w/v) of ice-cold perchloric acid (0.4 N). The clear supernatant obtained after centrifugation (2000 × g, 30 min, + 4°C) was stored at −20°C before measuring the t-MeHA level by radioimmunoassay as described (Garbarg et al., 1992). Changes were evaluated statistically by the Student’st test.
[125I]Iodoproxyfan binding assays.
The procedure for binding assays to rat striatal brain membranes was that described by Ligneau et al. (1994). Aliquots of membrane suspension [100 μl containing 15 to 20 μg of protein determined according to Lowry et al. (1951) using bovine serum albumin as standard] were incubated for 60 min at 25°C with 25 pM [125I]iodoproxyfan (Kd = 65 ± 4 pM) alone or together with competing drugs dissolved to give a final volume of 200 μl in a phosphate buffer medium (Na2HPO4/KH2PO4 50 mM, pH 6.8). Incubations performed in triplicate were stopped by four additions of 5 ml ice-cold medium followed by rapid filtration through glass microfiber filters (GF/B, Whatman, Maidstone, U.K.) presoaked in a 0.3% polyethylene imine ice-cold buffer. Radioactivity trapped on filters was measured on a gamma counter.
Histamine H1 receptor assay on guinea pig ileum.
The procedure used was that described by Pertz and Elz (1995).
Histamine H2 receptor assay on guinea pig right atrium.
The procedure used was that described by Pertz and Elz (1995).
Muscarinic M3 receptor assay on guinea pig ileum.
The procedure used was that described by Pertz and Elz (1995).
Adrenergic α1D receptor assay on rat aorta.
The procedure used was that described by Hirschfeld et al.(1992).
Adrenergic β1 receptor assay on guinea pig right atrium.
The procedure used was that described by Pertz and Elz (1995).
Serotoninergic 5-HT1B receptor assay on guinea pig iliac artery.
The procedure used was that described by Pertz (1993).
Serotoninergic 5-HT2A receptor assay on rat tail artery.
The procedure used was that described by Pertz and Elz (1995).
Serotoninergic 5-HT3 receptor assay on guinea pig ileum.
The procedure used was that described by Elz and Keller (1995).
Serotoninergic 5-HT4 receptor assay on rat esophagus.
The procedure used was that described by Elz and Keller (1995).
Histamine H3 receptor assay on guinea pig ileum.
The procedure used was that described by Ligneau et al.(1994). Briefly, longitudinal muscle strips from guinea pig small intestine were dissected out and incubated in a gassed O2/CO2 (95%/5%) modified Krebs-Ringer’s bicarbonate medium at +37°C in presence of 1 μM mepyramine to block the H1 receptor. After equilibration, contractile activity under stimulation (rectangular pulses of 15 V, 0.5 msec, 0.1 Hz) was recorded. Concentration-response curves of the effect of (R)α-MeHA alone or together with the antagonist were established.
Radioreceptor assay of H3-receptor ligands in serum.
Male Swiss mice (18–20 g, Iffa-Credo, L’Arbresle, France) were fasted for 24 hr before ciproxifan administration. At various times thereafter, mice were decapitated. Blood was collected at +4°C, serum collected after centrifugation (100 × g, 10 min, +4°C), and stored at −20°C. Ciproxifan levels were measured with the following radioreceptor assay derived from the [3H](R)α-MeHA binding assay (Garbarget al., 1992).
Male Wistar rats (160–180 g, Iffa-Credo, L’Arbresle, France) were decapitated and the brain was removed immediately. The cerebral cortex was dissected out and homogenized in 10 volumes (w/v) of ice-cold 50 mM Na2HPO4/KH2PO4 buffer, pH 7.5 using a Polytron. After a centrifugation (140 ×g, 10 min, +4°C), the supernatant was recentrifuged (23,000 × g, 10 min, +4°C). The last pellet was washed superficially with and then resuspended in fresh ice-cold phosphate buffer to constitute the membrane fraction used for the binding assay. One-ml aliquots of the membrane suspension containing 300 to 330 μg of protein [determined according to Lowry et al. (1951) using bovine serum albumin as standard] were incubated for 60 min at +25°C with 1 nM of [3H](R)α-MeHA alone or together with different concentrations of ciproxifan in diluted serum of ciproxifan-free mice (standardization curve) or with diluted serum samples of ciproxifan-treated mice. Specific binding was defined as that inhibited by 3 μM thioperamide. Incubations were performed in triplicate and stopped by four additions of 5 ml of ice-cold phosphate buffer followed by rapid filtration through glass microfiber filters (GF/C, Whatman, Maidstone, U.K.) presoaked in 0.3% polyethylene imine ice-cold phosphate buffer. Radioactivity trapped on filters was measured by liquid scintillation spectrometry (Wallac 1410, EG&G, Evry, France). The standardization curves were established using a one-site competition model (GraphPad Prism, San Diego, CA) and H3-receptor ligand concentrations in serum were calculated using these curves and expressed as ciproxifan concentrations. In the conditions retained, the detection limit of the radioreceptor assay corresponded to a concentration of 20 nM ciproxifan in serum. Changes in H3-receptor ligand concentrations in serum with time were analyzed using either a point to point nonlinear model or the two phase exponential decay analysis model (GraphPad Prism).
(R) α-MeHA-induced water consumption in rats.
The procedure used was that described by Clapham and Kilpatrick (1993) with slight modifications. Briefly, male Lister hooded rats (280–320 g, Charles River, St Aubin lès Elbeuf, France), housed in cages of 10, were allowed free access to food and water. Experiments were carried out between 10 a.m. and 1p.m. Drugs in 0.9% NaCl solution were administered i.p., each rat receiving two injections [vehicle or (R)α-MeHA 10 mg/kg/p.o., and vehicle or ciproxifan] of 250 μl each. After drug treatments, rats were returned to their home cage with food (standard pellet) and without water for 30 min. Then they were placed in individual cages with only a water supply and 10 min later, the amount of water consumed was recorded by weighing. Statistical evaluation of results was performed by Student’s t test.
Analysis of neocortical EEG power spectral density and sleep-wake in the cat.
Cats, a species in which the role of the H3 receptor in sleep-wake control has been demonstrated, were used in this experiment (Lin et al., 1990). Briefly, five adult cats of both sexes weighing 2.7 to 3.8 kg were chronically implanted, under pentobarbital anesthesia (25 mg/kg, i.v.), with electrodes for polygraphic recordings of neocortical and hippocampal EEG, ponto-geniculooccipital activity, electromyogram and electrooculogram. In addition, a thermistor (10 K3 MCD2, Betatherm, 10 KΩ at 25°C, outer diameter of 0.45 mm) was placed in the caudate nucleus to record brain temperature. After a recovery period of 7 days the cats were housed in a sound-attenuated and dimly illuminated cage set at 24 to 26°C and fed daily at 6 p.m. with a normal standard diet. Polygraphic recordings were performed for 4 days to obtain the basic qualitative and quantitative parameters of the sleep-wake cycle.
Ciproxifan at doses of 0 (placebo), 0.15, 0.3, 0.7 and 2 mg/kg was administrated orally at 11 a.m. Subsequent polygraphic recordings were made for at least 24 hr. They were then scored minute by minute according to previously described criteria (Lin et al., 1990) for wakefulness (W), light slow wave sleep (S1), deep slow wave sleep (S2) and paradoxical sleep (PS). In some animals, neocortical EEG signals from the first 6 hr after placebo or drug administration were digitized at a sample rate of 128 Hz and computed on a CED 1401 Plus (Cambridge Electronic Design, Cambridge, U.K.). The power spectral density was averaged over 30-sec epochs for the frequency range of 0.25 to 50 Hz by a Fast Fourier Transform routine using the CED program Spike2 and correlated with sleep-wake stages.
Five-choice task in rats.
Male Lister hooded rats (Olac, Bicester, U.K.) were housed in pairs in a temperature-controlled (21°C) room that was illuminated in accordance with an alternating 12-hr light/dark cycle. Rats were food deprived and maintained at 85% of their free-feeding weight (MRC Diet 41B laboratory food) throughout the experiment while water was available ad libitum.
The test apparatus and procedure were as described in detail by Muiret al. (1994). Rats were trained to discriminate a brief visual stimulus presented in one of five spatial location during 30-min sessions. Rats initiated a trial by opening the magazine panel. After a 5-sec inter-trial interval a light at the rear of one of the five apertures was illuminated for 0.5 sec. Correct responses to the stimulus location were rewarded with the delivery of food pellets. Having obtained stable performance on the task (>80% correct responses), attentional demand of the task was increased by reducing the stimulus duration to 0.25 sec in certain drug sessions as described previously (Muir et al., 1994, 1995). Drugs or the vehicle (saline) were administered i.p. 1 hr before the test session (0.5- or 0.25-sec stimulus duration) according to a Latin square design. A rest day followed by a baseline day separated each drug test day.
Analysis of data.
Maximal effects, ED50, EC50 and IC50 values were determined using an iterative computer least-squares method derived from that of Parker and Waud (1971) with the following nonlinear regression: effect of the drug = ([maximal effect of the drug].[drug dose-or-concentration])/([drug dose-or-concentration] + (ED50 or EC50 or IC50)).
Ki
values of H3-receptor antagonists are calculated from their IC50 values, assuming a competitive antagonism and by using the relationship (Cheng and Prussoff, 1973):
Radiochemicals and drugs.
[125I]Iodoproxyfan and [3H](R)α-MeHA (specific activities at reference date of 2000 and 38.0 Ci/mmol, respectively) were from Amersham (Amersham, U.K.). All drug doses are expressed as free base of compound. Administration to animals was performed with drug preparation in 1% methylcellulose for the oral route, in 0.9% NaCl for i.v. and i.p. routes. The drugs and their sources were as follows: ciproxifan [cyclopropyl-(4-(3-(1H-imidazol-4-yl)propyloxy)phenyl) ketone] synthesis was described in the patent application (Schwartzet al., 1996); thioperamide and (R)α-MeHA (Laboratoire Bioprojet, Paris, France); carboperamide [1-(heptanoyl)-4-(1H-imidazol-4-yl)piperidine] was from M. Robba (University of Caen, France); clobenpropit was provided by H. Timmerman (Vrije Universiteit, Amsterdam, The Netherlands); iodoproxyfan was synthesized at the Freie Universität Berlin; imetit was synthesized by S. Athmani (University College, London, U.K.). All other chemicals were obtained from commercial sources and were of the highest purity available.
Results
Effects of ciproxifan on H3-receptor functional models in vitro.
The H3-receptor agonist imetit inhibited the [3H]HA release with a maximal effect of 54 ± 2%. Pharmacological parameters were estimated using the H3-receptor controlled inhibition curve of the [3H]HA release. Imetit concentration required for half inhibitory effect of its own maximal effect was 1.6 ± 0.3 nM and the corresponding pseudo-Hill coefficient (nH) of this concentration response curve was close to unity (0.85 ± 0.15). Ciproxifan (15 nM) induced a parallel rightward shift of the concentration-response curve for imetit and tended by itself to increase (∼20%) the K+-induced [3H]HA release; the antagonism developed by ciproxifan was entirely surmounted in the presence of the highest imetit concentrations tested (fig.2). The half-maximal inhibitory concentration for imetit in the presence of ciproxifan (estimated considering the total H3-receptor controlled inhibition curve) was 12.8 ± 1.8 nM, leading to an apparentKi value of 1.9 ± 0.3 nM for the antagonist.

Effect of ciproxifan on the inhibition by imetit of the K+-induced [3H]HA release from synaptosomes of rat cerebral cortex. Synaptosomes preincubated with [3H]l-histidine were incubated for 5 min in the presence of imetit in increasing concentrations, alone or together with ciproxifan at a fixed (15 nM) concentration. They were subsequently depolarized for 2 min in the presence of 30 mM K+ (final concentration). The spontaneous efflux of [3H]HA (2 mM K+) represented 14 ± 2% of the total [3H]HA in synaptosomes. In the presence of added agents, [3H]HA release induced by 30 mM K+ (expressed in percent of total [3H]HA over spontaneous efflux) represented 17 ± 2%. Each point represents the mean ± S.E.M. of results from three different experiments with quadruplicate determinations each.
Histamine (1 μM) inhibited its own release by 55 ± 2%, and a series of H3-receptor antagonists progressively reversed this response with nH coefficients close to unity (fig. 3). IC50 values (nM) of the various compounds were 9.2 ± 1.8 (ciproxifan), 75 ± 19 (thioperamide), 377 ± 38 (carboperamide), 11 ± 3 (clobenpropit) and 99 ± 5 (iodoproxyfan) leading toKi values reported in table1, assuming a competitive inhibition.

Effects of H3-receptor antagonists on the inhibition by HA of the K+-induced [3H]HA release from rat cerebral cortex synaptosomes. Synaptosomes preincubated with 1 μM HA alone or together with one of the compounds at increasing concentrations. They were depolarized for 2 min in the presence of 30 mM K+. Each point represents the mean result from three different experiments with quadruplicate determinations each.
In vitro and in vivo potencies of H3-receptor antagonists
Ciproxifan (3–300 nM) competitively antagonized the (R)α-MeHA induced relaxation of the electrically stimulated guinea pig ileum longitudinal muscle (Ligneau et al., 1994) without significantly affecting the maximum response of the H3-receptor agonist in the absence of ciproxifan [91 ± 5 (3 nM ciproxifan, n = 2), 100 ± 4 (10 nM, n = 6), 110 ± 10 (30 nM,n = 6), 105 ± 9 (100 nM, n = 6) and 87 ± 7% (300 nM, n = 6) vs. 100% relaxation in the absence of ciproxifan]. Schild analysis revealed a slope of 0.92 ± 0.04, not significantly different from unity (n = 26). After imposing the unity constraint, a pA2 value of 8.38 ± 0.03 was calculated for ciproxifan.
Effect of ciproxifan and other H3-receptor antagonists on [125I]iodoproxyfan binding.
At 25 pM [125I]iodoproxyfan the specific binding to rat striatal membranes represented 8.4 ± 0.4 fmol/mg of protein,i.e., 46 ± 1% of the total, and was completely and monophasically displaced by all compounds (fig.4). From these displacement curves IC50 values for the compounds were deduced leading to theKi values presented in table 1.

Inhibition of [125I]iodoproxyfan binding to rat striatal membranes by various histaminergic agents. Membranes were incubated for 60 min at 25°C with 25 pM [125I]iodoproxyfan and unlabeled H3-receptor antagonists in increasing concentrations. Specific binding, defined as that inhibited by 1 μM (R)α-MeHA, represented 46 ± 1% of the total binding. Results are expressed as percentages of specific [125I]iodoproxyfan binding in the absence of unlabeled agents. Each point and vertical bars represent the mean ± S.E.M. of results from three different experiments with triplicate determinations each.
Receptor selectivity of ciproxifan.
The compound displayed low apparent affinity at other receptor subtypes as evaluated in functional tests on isolated organs (histamine H1 and H2, muscarinic M3, adrenergic α1D and β1, serotonin 5-HT1B, 5-HT2A, 5-HT3 and 5-HT4) (fig.5). In addition, ciproxifan displayed aKi value higher than 1 μM in a large variety of radioligand binding tests (Panlabs screen), except at [3H]pirenzepine binding to rat cerebral cortex membranes where its Ki was about 1 μM (data not shown).

Receptor selectivity profile of ciproxifan. The affinity of the compound at the H3 receptor (pKi from [3H]HA release assay) is compared to corresponding values obtained in functional tests in isolated organs. Other values represent mean ± S.E.M. pA2 (Arunlakshana and Schild, 1959) from n= 3 to 12 preparations except for the H2- and β1-receptor assay (pD’2 according to Van Rossum, 1963).
Changes in serum drug concentration in animals treated with ciproxifan.
After i.v. administration of 1 mg/kg ciproxifan to mice (fig. 6B), the H3-receptor ligand concentration in serum decreased progressively, fitting a typical biexponential decay model with half-times (t1/2) of 13 and 87 min for the distribution and elimination phases, respectively. The quality of this fit is given by an R2 value of 0.985. At 6 hr, the serum ligand concentration was still detectable with a value of 23 ± 6 nM. When ciproxifan was given orally, also at 1 mg/kg (fig. 5A), serum ligand level rose rapidly, being maximal at 30 min with a maximal concentration (Cmax) value of 420 ± 40 nM; then, the ligand concentration decreased but still remained measurable at 6 hr (27 ± 5 nM). The AUCs were 1425 and 890 nM.hr after i.v. and p.o. administrations, respectively, leading to an oral bioavailability coefficient (AUCp.o./AUCi.v..100%) of 62%.

Serum drug concentration in mice receiving ciproxifan. Mice were killed after p.o. (A) or i.v. (B) administration of ciproxifan (1 mg/kg), and drug concentrations were evaluated in serum by a radioreceptor assay. Means ± S.E.M. of values from six mice.
In rats (n = 3) receiving 1 mg/kg ciproxifan p.o., the time-course of changes in serum ligand concentration were comparable to those in mice with a mean AUC of 2,225 nM.h, a Cmax of 881 ± 322 nM, also observed at 30 min, and a level of 96 ± 4 nM at 6 hr (not shown).
In one cat receiving 3 mg/kg of ciproxifan orally serum levels (μM) were of 0.14 (0.5 hr), 4.0 (1 hr), 2.2 (1.5 hr), 0.27 (3 hr) and 0.12 (6 hr) leading to an AUC of 5.07 μM.hr at this dose.
Changes in brain t-MeHA level after administration of ciproxifan or other H3-receptor antagonists.
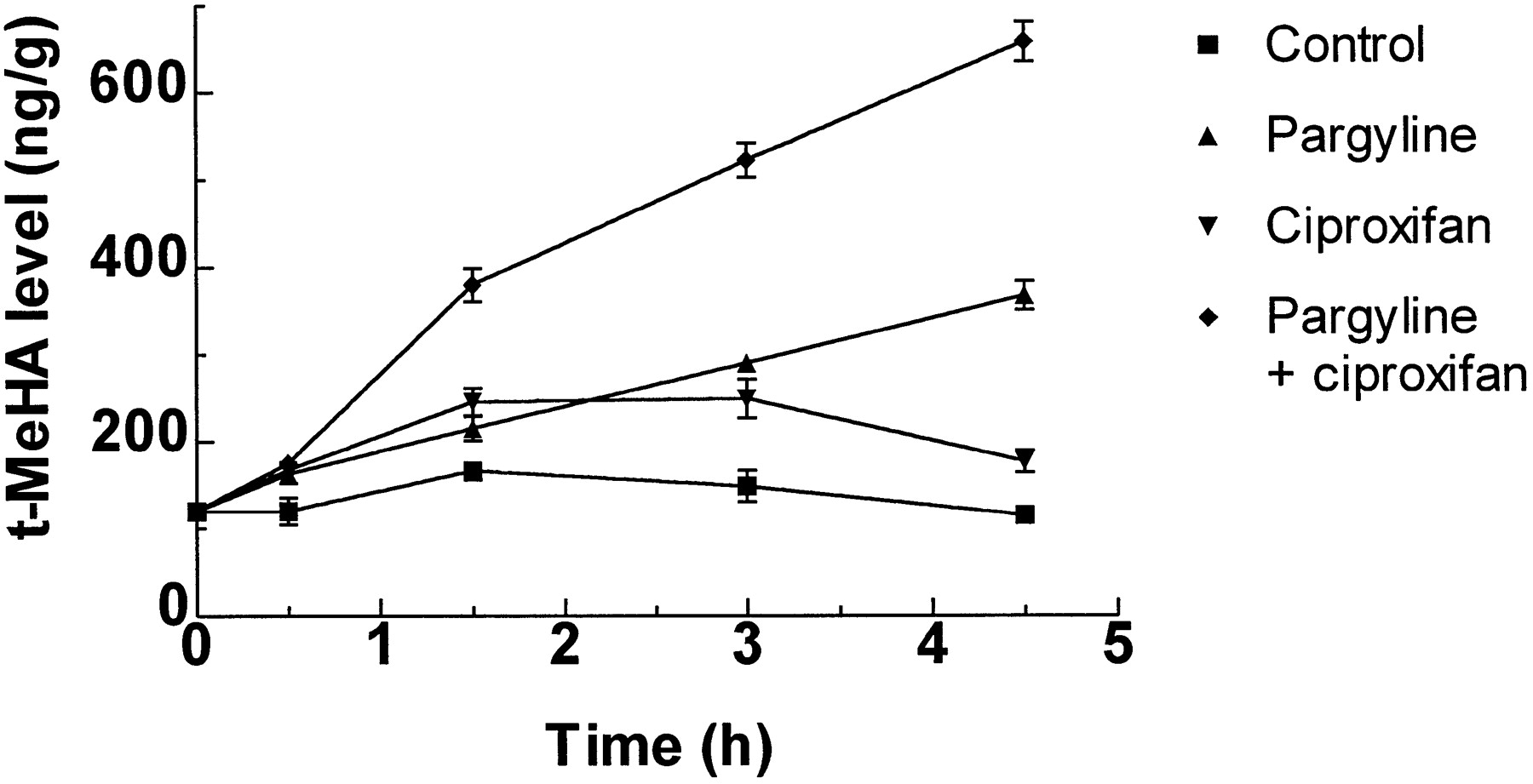
After the administration of ciproxifan (1 mg/kg, p.o.), brain t-MeHA level rose rapidly, being already significantly increased after 30 min, reaching a plateau between 90 and 180 min and still remaining enhanced after 270 min (fig. 7). In mice receiving pargyline, a monoamine oxidase inhibitor, t-MeHA level increased linearly with time at a rate of 55 ng/g/hr, whereas coadministration of pargyline and ciproxifan enhanced this rate to 120 ng/g/hr.

Changes in brain t-MeHA levels in control and pargyline-treated mice after administration of ciproxifan. Mice were killed at various times after simultaneous administration of vehicle or ciproxifan (1 mg/kg, p.o.) and vehicle or pargyline (65 mg/kg, i.p.). t-MeHA levels are expressed in ng/g of tissue. Means ± S.E.M. of values from 12 mice.
The dose-response curves of ciproxifan and a series of other H3-receptor antagonists were established by measuring t-MeHA levels 90 min after oral administration (fig.8). Ciproxifan, thioperamide and carboperamide, maximally increased t-MeHA level to an equivalent extents about 2-fold over basal values. However, clobenpropit, at the highest dosage tested (30 mg/kg, p.o.) maximally enhanced t-MeHA level by 45%, and iodoproxyfan (10 mg/kg, p.o.) did not significantly modify this level. The ED50 values of the compounds, derived from data of figure 8, are reported in table 1.

Changes in brain t-MeHA levels in mice receiving H3-receptor antagonists. Mice were killed 90 min after the p.o. administration of vehicle or drugs in increasing doses. t-MeHA levels in treated mice are expressed in percent increase as compared to levels in control mice (133 ± 7 ng/g). Means ± S.E.M. of values from 12 mice.
Similar experiments performed in rats receiving ciproxifan orally led to ED50 values (mg/kg) of 0.23 ± 0.04 in cerebral cortex, 0.28 ± 0.08 in striatum and 0.30 ± 0.08 in hypothalamus with similar maximal enhancements of about 100% (not shown).
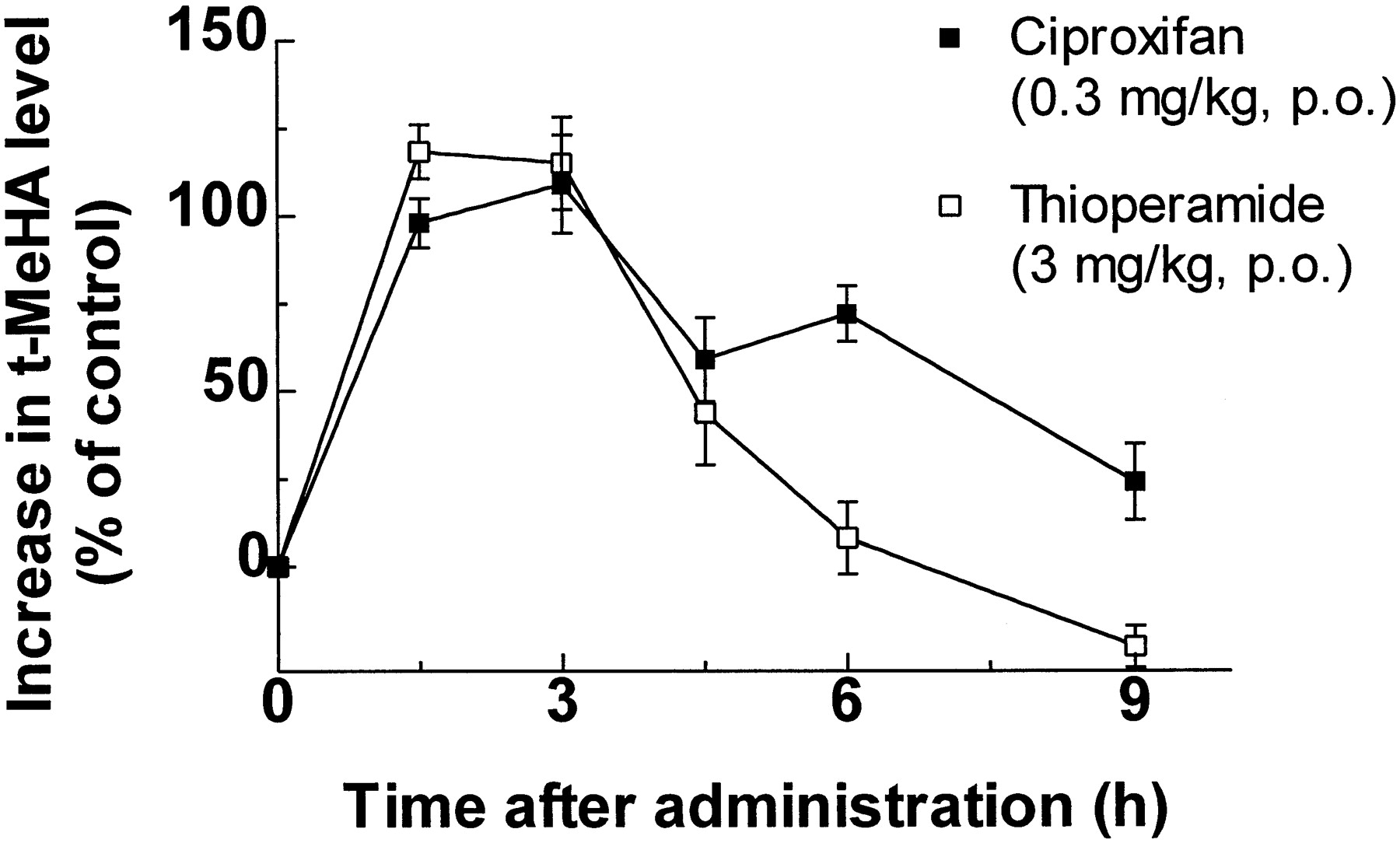
The time-course of the changes in t-MeHA levels in mouse brain elicited by oral administration of 0.3 mg/kg ciproxifan and 3 mg/kg thioperamide (fig. 9) were analyzed in terms of AUCs, leading to values (in percent increase.hr) of 597 and 425, respectively.

Changes in brain t-MeHA levels in mice receiving ciproxifan or thioperamide. Mice were killed at various times after the p.o. administration of vehicle, ciproxifan (0.3 mg/kg) or thioperamide (3 mg/kg). t-MeHA levels are expressed in percent increase as compared to levels in corresponding controls. Means ± S.E.M. of values from 18 mice.
Effect of ciproxifan on the water consumption induced by an H3-receptor agonist.
The H3-receptor agonist (R)α-MeHA (10 mg/kg, i.p.) markedly enhanced water consumption in rats, an effect that was progressively reversed by coadministration of ciproxifan in increasing dosage, the ID50 of the antagonist being 0.09 ± 0.04 mg/kg (fig.10). Ciproxifan alone (3 mg/kg) did not significantly modify water consumption (fig. 10).

Effect of ciproxifan on the (R)α-MeHA-induced water consumption in rats. Water consumption was measured over a 10-min period, 30 to 40 min after i.p. administrations of (R)α-MeHA and ciproxifan. Means ± S.E.M. of values from 5 to 10 rats per treatment group (data from two experiments). *P < .05 vs. vehicle.
Effect of ciproxifan in the five-choice task in rats.
Analysis of variance revealed a significant drug × stimulus duration interaction [F(1,9) = 12.19, P < .01]. As shown in figure 11, reducing the duration of the visual stimulus to 0.25 sec resulted in a significant reduction in the accuracy of performance compared to the baseline (0.5 sec) stimulus condition. Newman Keuls post hoc comparisons revealed that this reduction in performance was significant and that choice accuracy significantly increased after administration of 3.0 mg/kg of ciproxifan under the shorter stimulus condition compared to performance after administration of the vehicle (P < .05). There was no significant effect of this manipulation of the stimulus duration or of ciproxifan on any of the other measures recorded (i.e., speed, anticipatory or perseverative responses and errors of omission).
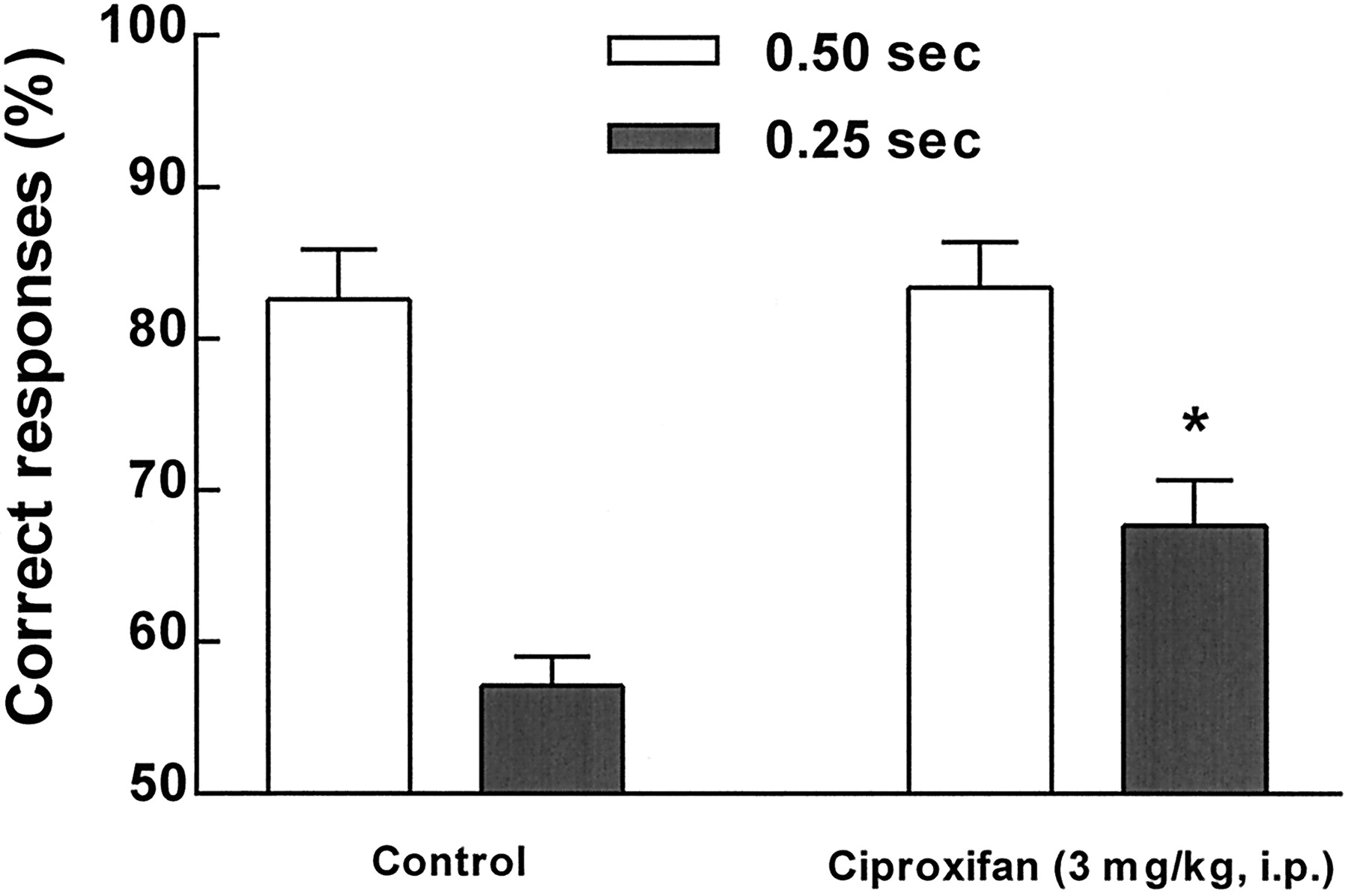
Effects of ciproxifan in the five-choice task in rats. The drug was administered (3 mg/kg, i.p.) 1 hr before the test session. The accuracy of responding, expressed as the percentage of correct responses, was significantly (*P < .05) improved by treatment only when the stimulus duration was 0.25 sec instead of 0.50 sec.
Effects of ciproxifan on neocortical EEG power spectral density and sleep-wake cycle in cat.
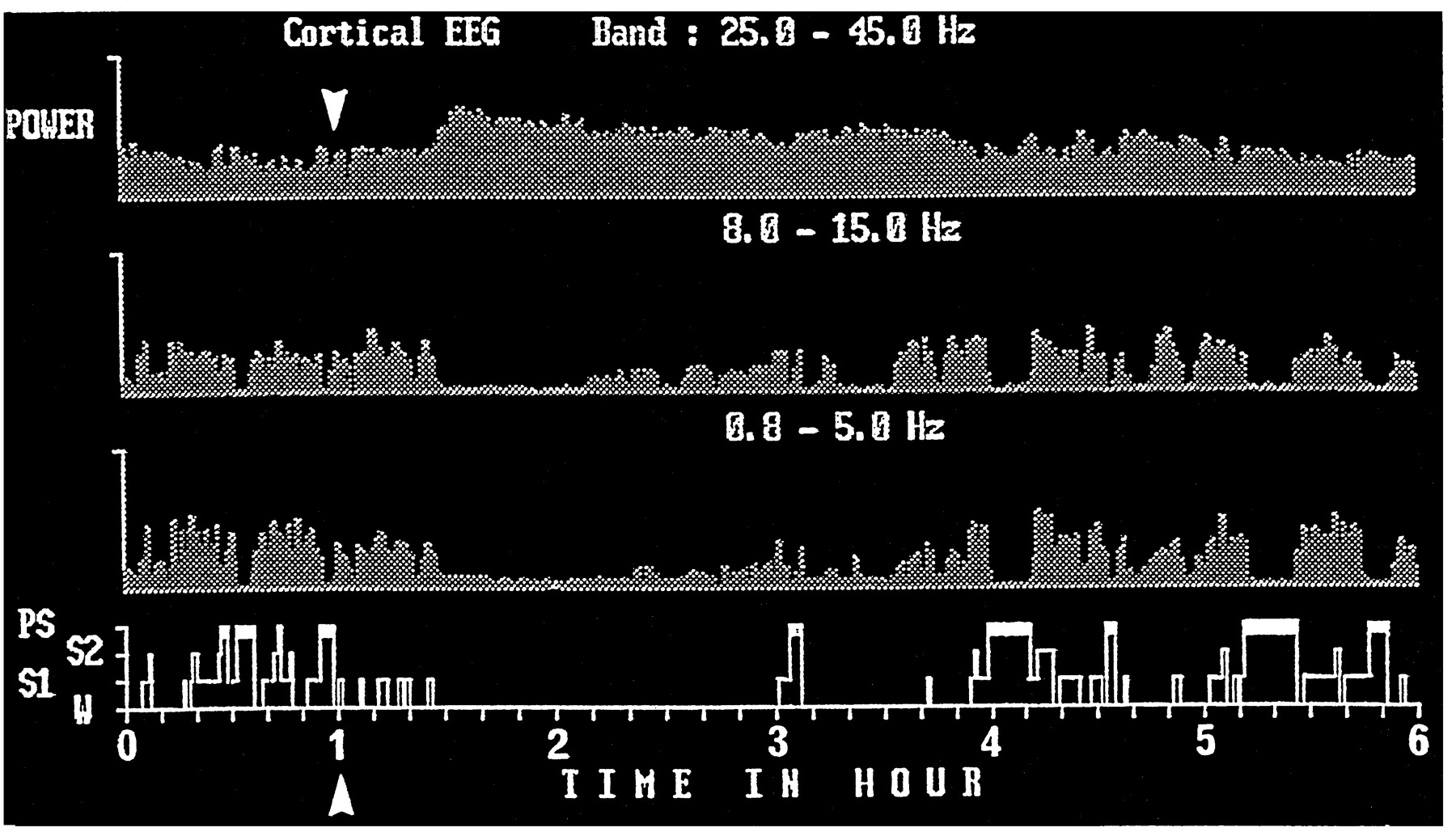
Administration of ciproxifan caused suppression or diminution (depending on the dose used) of neocortical slow activity (0.8–5 Hz) and spindles (8–15 Hz), resulting in a total cortical activation, i.e., low voltage electrical activity with dominant waves in the β and γ bands (mainly 25–45 Hz). Furthermore, ciproxifan increased the power density of these neocortical fast rhythms (fig. 12). These effects, occurring within 25 min after administration (fig. 12) were detectable at a dose of 0.15 mg/kg and became evident at a dose of 0.3 mg/kg or more (fig. 13).

An example of the effects of oral administration of ciproxifan on cortical EEG and sleep-wake cycle in cat. Neocortical EEG power density (mV2) in different frequency bands and sleep-wake cycle 1 hr before and up to 5 hr after administration of 2 mg/kg ciproxifan (arrow) are given. Note, from bottom to top, that ciproxifan elicited a total waking state accompanied by a suppression of cortical slow frequency activity (0.8–5 Hz) and spindle (8–15 Hz) and a marked increase in fast rhythms (25–45 Hz). Abscissa, Time in hours; ordinate, sleep-wake stages (PS, paradoxical sleep; S2, deep slow wave sleep; S1, light slow wave sleep; W, wakefulness).

Representative hypnograms (4 hr) obtained in cats after oral administration of ciproxifan at different doses. Note the dose-dependent waking effect. Abscissa, Time in hours; ordinate, sleep-wake stages (PS, paradoxical sleep; S2, deep slow wave sleep; S1, light slow wave sleep; W, wakefulness).
The effects of ciproxifan on neocortical EEG were manifested on polygraphic scoring as an almost total waking state, the duration of which was dose dependent. This waking effect was characterized by an increase in wake-episode duration and a delayed sleep latency. During the same period both slow wave sleep (especially S2) and PS were suppressed (fig. 13). After the period of induced total waking, cortical slow activity gradually reappeared, but an increase in waking could be seen during a period proportional to the doses used. No obvious sleep rebound was noted after the waking effect, and all sleep-wake parameters including power spectral density of neocortical EEG returned to control levels on the next day. The arousal effects of ciproxifan (0.3 mg/kg) were prevented or significantly reduced by prior (15 min) systemic injection of either mepyramine (1 mg/kg), an H1-receptor antagonist, or imetit (3 mg/kg), an H3-receptor agonist (not shown).
Discussion
From our present in vitro and in vivostudies, ciproxifan appears as a pure competitive antagonist at the histamine H3 receptor, one of the most potent so far available.
Our selection of the molecule was initially based on its ability to block the actions of histamine or imetit, a selective H3-receptor agonist, at the autoreceptor regulating the release of neosynthesized [3H]HA from K+-depolarized synaptosomes, according to a previously described model (Arrang et al., 1983; Garbarg et al., 1989, 1992). On this model, ciproxifan induced a parallel rightward shift of the concentration-response curve to imetit, indicative of a competitive antagonism with an apparent dissociation constant in the low nanomolar range. Ciproxifan in increasing concentration also progressively blocked the (R)α-MeHA-induced relaxation of the electrically stimulated longitudinal muscle of guinea pig ileum in a competitive fashion. At these various functional models as well as the [125I]iodoproxyfan binding tests, the drug displayed similarly low apparent dissociation constants (Ki = 0.5–4.2 nM), an observation that does not support the hypothesis of the existence of H3-receptor subtypes (Clapham and Kilpatrick, 1992). The hypothesis, mainly based on discrepancies in potency of some compounds in binding and functional models, is not supported either by the closely similar potencies of other antagonists in different models, examplified in table 1 (with the exception of iodoproxyfan for which the discrepancy can be fully explained by a slow equilibration rate).
The high degree of selectivity of ciproxifan toward the histamine H3 receptor was shown by the observation that the drug displayed lower affinity by about three orders of magnitude for any other receptor subtype on which it was tested (see “Results”).
Although some compounds in vitro were as potent as or even more potent than ciproxifan at the H3 receptor,e.g., clobenpropit and iodoproxyfan, ciproxifan given orally to mice enhanced brain t-MeHA levels at much lower dosage (ED50 = 0.14 mg/kg) than any of these compounds. A similar change, occurring with ED50 of 0.2–0.3 mg/kg p.o., was found in various areas of rat brain. t-MeHA is the product of the major metabolic pathway for endogenous HA in brain (Schwartz et al., 1971), and its steady-state level is a reliable index of histaminergic neuron activity (Oishi et al., 1983). The increase in t-MeHA level induced by the H3-receptor antagonists corresponds to an enhanced HA release, reflecting the tonic inhibition that the endogenous amine exerts on this process and on neuronal firing via stimulation of autoreceptors in the somatodendritic or terminal area of histaminergic neurons. In agreement, ciproxifan (1 mg/kg, p.o.) enhanced HA turnover rate in mouse brain, evaluated from the rate of t-MeHA accumulation after monoamine oxidase inhibition, from a value of 55 ng/g/hr, consistent with corresponding values obtained in the same species using either isotopic (Verdièreet al., 1977) or nonisotopic methods (Oishi et al., 1989), to a value of 120 ng/g/hr.
A similar maximal effect, corresponding to a nearly doubling of t-MeHA level, was obtained with thioperamide or carboperamide whereas, at the maximal dose tested of clobenpropit (30 mg/kg), this level was not reached and no significant change occurred after administration of iodoproxyfan (fig. 8) despite the high potency of these compoundsin vitro.
In the case of ciproxifan, a rather high oral bioavailability was evidenced by the ratio (>60%) of AUCs of H3 receptor binding activity in blood serum, measured by using a novel radioreceptor assay, following drug administration (1 mg/kg) by p.o. and i.v. routes, respectively. The rather slow kinetics of ciproxifan are indicated by a serum level still about 10 times above theKi value of the drug at the H3receptor 6 hr after oral administration. At this time, t-MeHA levels in brain are still enhanced by 24 ± 11% (fig. 9). Such comparison between drug levels in blood and a typical brain response in rodents might be useful to predict effective dosages in other species, particularly humans, in which only blood levels are available, assuming a similar ability of the drug to cross the blood-brain barrier. A similarly favorable bioavailability of ciproxifan on oral administration to rats and cats is suggested by measurements of t-MeHA and drug serum levels, respectively (see “Results”).
Characteristic behavioral responses were found in rats and cats receiving ciproxifan in low dosage. In water-deprived rats, ciproxifan blocked the enhancement of drinking elicited by (R)α-MeHA, an H3-receptor agonist (Clapham and Kilpatrick, 1993), with an ID50 value of ∼0.1 mg/kg. The exact site (central or peripheral) and mechanism of action of H3-receptor ligands in this test has not been clearly established. Thus, whereas the involvement of the renin-angiotensin system in HA-induced drinking was postulated by Kraly and Miller (1982), an AT1 antagonist did not affect the (R)α-MeHA-induced drinking (Clapham and Kilpatrick, 1993). The observation that (R)α-MeHA-induced drinking is blocked at doses of ciproxifan (this study), thioperamide and particularly clobenpropit (Barnes et al., 1993), close to those enhancing endogenous HA release in brain (table 1), suggests that H3 receptors in brain rather than in periphery are involved. The observation that ciproxifan or thioperamide given alone do not affect drinking suggests that the effect of (R)α-MeHA is mediated by H3 hetero- rather than autoreceptors. In agreement, H3 receptors on noradrenergic, serotoninergic, cholinergic, dopaminergic or peptidergic neurons do not appear to be, as with those on histaminergic neurons, tonically modulated by endogenous HA, because they respond to agonists but not to antagonists given alone (Schwartz et al., 1991,1995; Schlicker et al., 1994).
The marked dose-dependent waking effect of ciproxifan in cats is consistent with a large variety of experimental evidence showing that histaminergic neurons play a prominent role in cortical activation and arousal in cats and rats (reviewed by Lin et al., 1996;Schwartz et al., 1991, 1995). The arousing effects of H3-receptor antagonists, characterized by an enhancement of wakefulness at the expense of slow wave and paradoxical sleep, was previously shown in both animal species using thioperamide (Linet al., 1990; Monti et al., 1991) and carboperamide (Monti et al., 1996). The effect of ciproxifan was, as in the case of thioperamide (Lin et al., 1990), prevented by administration of mepyramine, an H1-receptor antagonist, suggesting that it resulted from a H3-receptor mediated enhancement of endogenous HA release. The brain site(s) at which endogenous HA promote(s) cortical EEG desynchronization via activation of the H1 receptor could be one of the brain areas to which ascending or descending histaminergic pathways project known to express the H1 receptor and to control sleep/wakefulness states. These potential targets comprise cortical neurons receiving direct histaminergic projections from the tuberomammillary nucleus, preoptic anterior hypothalamic neurons, thalamic relay neurons and basal forebrain or mesopontine tegmentum neurons (Lin et al., 1996).
Because the effect of ciproxifan in cats was to enhance fast cortical rhythms, known to occur during increased vigilance, and to cause a quiet waking state, a positive outcome in attentional tests could be anticipated. In confirmation the drug significantly enhanced choice accuracy in the five-choice serial reaction-time task when a visual stimulus of short duration (0.25 sec) was used. Such reduction of the stimulus duration increases the attentional load placed on the task, reduces choice accuracy and has been used to observe the effects of cholinergic agents on attentional function (Muir et al., 1994, 1995). “Pro-cognitive” effects of the H3-receptor antagonist thioperamide have been reported in other behavioural tasks,e.g., step-through passive avoidance response in senescence-accelerated mice (Meguro et al., 1995); elevated plus-maze performance in mice with scopolamine-induced learning deficits (Miyazaki et al., 1995) and in a test of social memory in rats (Prast et al., 1996). However, it has also been reported that thioperamide failed to improve scopolamine-induced attentional dysfunction in the same 5-choice task used in the present study (Kirkby et al., 1996).
Taken together these various observations suggest that ciproxifan is a potent, orally active H3-receptor antagonist and it seems of interest to assess its potential therapeutic applications, namely in aging or degenerative disorders in which vigilance, attention and memory are impaired.
Acknowledgments
The authors thank A. Galtier for processing this manuscript, P. Brugioti for technical help and A. Rouleau and M. Garbarg for helpful discussions.
Footnotes
-
Send reprint requests to: Dr. Jean-Charles Schwartz, Unité de Neurobiologie et Pharmacologie Moléculaire (U.109), Centre Paul Broca de I’INSERM, 2ter rue d’Alésia, 75014 Paris, France.
-
1 This work was supported by the Biomedical and Health Research Program EEC BMH4 CT96-0204 and the Direction des Recherches Etudes et Techniques (DRET 92/045).
- Abbreviations:
- HA
- histamine
- t-MeHA
- tele-methylhistamine
- (R)α-MeHA
- (R)α-methylhistamine
- AUC
- area under the curve
- Cmax
- maximal concentration
- W
- wakefulness
- S1
- light slow wave sleep
- S2
- deep slow wave sleep
- PS
- paradoxical sleep
- EEG
- electroencephalogram
- Received February 24, 1998.
- Accepted June 7, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}