


Abstract
First-generation phosphodiesterase 4 (PDE4) inhibitors, such as rolipram, inhibit the activation of immune and inflammatory cells. The clinical use of these compounds is limited by gastrointestinal side effects, such as increased acid secretion and nausea. Consequently, the challenge has been to design novel PDE4 inhibitors that maintain the anti-inflammatory actions of rolipram while achieving an improved side effect profile. Among the first of this new class of PDE4 inhibitors specifically designed to have an improved therapeutic index relative to earlier compounds is SB 207499 (Ariflo) [c-4-cyano-4-(3-cyclopentyloxy-4-methoxy-phenyl)-r-1-cyclohexanecarboxylic acid]. In this study, we compared the anti-inflammatory and gastric secretogogue activities of SB 207499 with those of rolipram. The cellular models used were (1) histamine release from human basophils, (2) tumor necrosis factor-α generation in human monocytes, (3) degranulation of human neutrophils, (4) antigen-driven proliferation and cytokine synthesis from human T cells and (5) acid secretion from isolated rabbit gastric glands. SB 207499 inhibited the activation of a variety of immune and inflammatory cells in a concentration-dependent manner: (1) histamine release in basophils [−log IC25 = 6.6 ± 0.3 vs. 8.0 for (R)-rolipram], (2) lipopolysacchride-induced TNF-α formation in monocytes [−log IC50 = 7.0 ± 0.1vs. 7.2 ± 0.1 for (R)-rolipram], (3) fMLP-induced degranulation in neutrophils [−log IC15= 7.1 ± 0.2 vs. 6.4 ± 0.5 for (R)-rolipram], (4) house dust mite induced-proliferation of peripheral blood mononuclear cells [−log IC40 = 6.5 ± 0.3 vs. 6.4 ± 0.3 for (R)-rolipram] and (5) ragweed-induced production of interferon-γ [−log IC50 = 5.4] and interleukin-5 [−log IC50 = 5.0]. Although SB 207499 inhibits the activation of a variety of immune and inflammatory cells with a potency equal to that of rolipram, it is >100-fold less potent than the latter compound as an acid secretagogue [−log EC50 = 6.1 ± 0.1 vs. 8.3 ± 0.2 for (R)-rolipram]. Collectively, these data indicate that SB 207499 retains the anti-inflammatory activity of the prototypical PDE4 inhibitor rolipram but is substantially less likely to stimulate gastric acid secretion.
Interest in PDE4 as a molecular target for new antiasthmatic and anti-inflammatory drugs has increased greatly over the past few years (Giembycz, 1992; Torphy et al., 1994; Torphy and Undem, 1991). This heightened interest has been fueled by several factors: (1) the observation that PDE4 is a major if not dominant cAMP hydrolyzing activity in immune and inflammatory cells (Torphy et al., 1994; Torphy and Undem, 1991), (2) prototypical PDE4 inhibitors such as rolipram or Ro 20–1724 suppress the activation of these cells (Giembycz, 1992; Torphy and Undem, 1991) and (3) rolipram and other first-generation PDE4 inhibitors produce marked anti-inflammatory actions in animal models (Barnette et al., 1996a; Torphy and Undem, 1991). Unfortunately, the use of these earlier compounds was limited by gastrointestinal side effects, apparently as an extension of their pharmacological mechanism of action (i.e., inhibition of PDE4 in inappropriate tissues) (Torphy and Undem, 1991). The side effects observed include increased gastric acid secretion, nausea and vomiting (Horowski and Sastre-Y-Hernandez, 1985; Puurunen et al., 1978). Consequently, the challenge to contemporary drug discovery efforts has been the design of novel PDE4 inhibitors that maintain the anti-inflammatory actions of rolipram with a reduced potential to elicit side effects.
A mechanistic hypothesis for identifying compounds with improved therapeutic indices emerged from the observation that recombinant PDE4 enzymes exist in two distinct conformers, one of which is inhibited by rolipram with a Ki value of 1 nM and a second that is inhibited by rolipram with aKi value of ≈100 nM (Jacobitzet al., 1996; Torphy et al., 1993). These two conformers of PDE4 have been termed HPDE4 and LPDE4, respectively (Jacobitz et al., 1996)
Three critical observations regarding this two-conformation model of PDE4 behavior have led to the development of a mechanistic basis for improving the therapeutic index of PDE4 inhibitors. First, high-affinity rolipram binding sites (Kd = 1–2 nM), representing HPDE4 (Jacobitz et al., 1996; Torphy et al., 1993), are greatly enriched in certain tissues compared with others (Schneideret al., 1986). Second, the rank order potencies of various compounds for inhibiting HPDE4 differ from that for inhibition of LPDE4 (Torphy et al., 1992a). Finally, the functionally relevant conformer of the enzyme appears to differ among various tissues and cell types (Barnette et al., 1996a). For example, the ability of PDE4 inhibitors to reverse reserpine-induced hypothermia in mice (Schmiechen et al., 1990), to enhance acid secretion in isolated rabbit gastric glands (Barnette et al., 1995a) and to induce emesis in dogs (Barnette et al., 1996a) correlates with their ability to inhibit HPDE4. In contrast, suppression of LPS-induced TNF-α production in isolated human monocytes (Barnetteet al., 1996b; Semmler et al., 1993; Vergheseet al., 1995), suppression of guinea pig mast cell activation (Underwood et al., 1993) and guinea pig eosinophil superoxide production (Barnette et al., 1995b) are associated with the ability of compounds to inhibit LPDE4. Thus, one approach toward improving the therapeutic index of a new class of PDE4 inhibitors is by increasing the relative potency of compounds for LPDE4 vs. HPDE4.
SB 207499 (Ariflo) [c-4-cyano-4-(3-cyclopentyloxy-4-methoxy-phenyl)-r-1-cyclohexanecarboxylic acid] is among the first of a new generation of PDE4 inhibitors that was specifically designed to have decreased HPDE4 activity (Barnetteet al., 1994; Christensen et al., in press). SB 207499 inhibits HPDE4 and LPDE4 catalytic activity with equal potency (Ki ≈100 nM) (Barnette et al., 1994), whereas rolipram is ≈100-fold more potent against HPDE4 (Christensen et al., in press; Jacobitz et al., 1996). Thus, SB 207499 and rolipram are equipotent against LPDE4, but SB 207499 is 100-fold less potent against HPDE4. This profile suggests that SB 207499 should retain the anti-inflammatory activity of rolipram yet have a decreased tendency to produce side effects. To gather evidence in support of this proposal, the present study was conducted to compare the anti-inflammatory activities of SB 207499 in vitro with those of rolipram and to determine whether the reduction in affinity for HPDE4 seen with SB 207499 was associated with a reduction in the ability of SB 207499 to produce one of the gastrointestinal side effects observed with rolipram, increased gastric acid secretion.
Methods
Aminopyrine accumulation rabbit isolated gastric glands.
Gastric glands were isolated from rabbits of either sex as described previously (Barnette et al., 1995a) according to the procedures originally outlined by Berglindh and Obrink (1976) and Sack and Spenney (1982). To measure acid secretion, gastric glands were incubated with 14C-aminopyrine (1.0 nmol/ml; 0.1 μCi/nmol), a range of concentrations of SB 207499 or (R)-rolipram and a threshold concentration of histamine (0.3–1.0 μM) at 37°C in a horizontal shaker (110 cycles/min) for 20 min according to the procedures of Sack and Spenney (1982). Samples were then centrifuged, and radioactivity in aliquots of the supernatant fraction and pellet was determined. Aminopyrine accumulation ratios (RAP) were calculated as described by Sack and Spenney (1982). The data were expressed as a percentage of a response produced by a maximal concentration of histamine (100 μM). EC50 values were determined by linear interpolation using the maximum response.
cAMP accumulation in U937 cells.
U937 cells, obtained from American Type Culture Collection (Rockville, MD), were grown in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum in plastic flasks (175 cm2) in a humidified atmosphere of 95% air/5% CO2 at 37°C. The culture medium was replaced every 3 to 4 days. Cells were harvested when they reached a concentration of ≈1 × 106 cells/ml. At this time, they were isolated from the medium by centrifugation (500 × g for 5 min) and washed once with Krebs-Ringer buffer of the following composition (in mM): NaCl 118, KCl 4.6, NaHCO3 24.9, KH2PO4 1.0, CaCl2 1.0, MgCl2 1.0, glucose 11.1 and HEPES 5.0, pH 7.5. The harvested cells were resuspended in Krebs-Ringer buffer supplemented with bovine serum albumin (0.2 mg/ml) at a concentration of ≈20 × 106 cells/ml.
Cells (1–2 × 106) were incubated at 37°C in a shaking water bath with SB 207499 or rolipram for 1 min before the addition of 0.1 μM PGE2 (total volume of 200 μl). The incubation proceeded for an additional 4 min and was stopped by the addition of 0.1 ml of HClO4 (17.5%), neutralized with 0.15 ml of K2CO3 (1.0 M) and diluted to 1 ml with sodium acetate buffer. Samples were centrifuged at 3000 × g for 10 min. Aliquots of the supernatant fraction were assayed for cAMP content by radioimmunoassay using commercially available kits obtained from New England Nuclear Research Prodcuts (Boston, MA). Results were expressed as pmol/1 × 106 cells and were corrected for cAMP content in the presence of PGE2 alone.
LPS-induced TNF-α formation in monocytes.
Peripheral blood monocytes were purified from freshly obtained buffy coats or plasmaphoresis residues of blood obtained from normal donors according to published procedures (Barnette et al., 1996b; Collataet al., 1984). Cells were incubated for 45 min to 1 hr in the absence or presence of a range of SB 207499 concentrations before the addition of LPS (0.1 μg/ml). Culture supernatants were removed from the monocytes after a 14- to 16-hr incubation at 37°C/5% CO2 and centrifuged at 1000 × gto remove cell debris. TNF-α levels of these samples were either determined immediately by ELISA, or the culture supernatants were stored at −70°C until assayed.
TNF-α was measured using a sandwich ELISA (Winston et al., 1987) with a murine monoclonal anti-human TNF-α antibody as the capture antibody and a polyclonal rabbit anti-human TNF-α as the second antibody. For detection, a peroxidase-conjugated goat anti-rabbit antibody was added followed by the addition of 1 mg/ml of orthophenylenediamine with 0.1% urea peroxide. TNF-α content was calculated from a standard curve generated with recombinant human TNF-α produced in Escherichia coli. Monoclonal antibodies to human TNF-α were prepared from spleens of BALB/c mice immunized with human TNF-α by a modification of the method of Kohler and Milstein, (1975). Polyclonal rabbit anti-human TNF-α antibodies were prepared by repeated immunization of a New Zealand White rabbit with recombinant human TNF-α emulsified in complete Freunds’ adjuvant.
MPO release from human neutrophils.
Human neutrophils were isolated from heparinized blood by gradient centrifugation using Ficoll (Histopaque 1077) followed by dextran sedimentation to remove the erythrocytes. Any remaining erythrocytes were lysed with water for 30 sec, and isotonicity was restored using 10× Dulbecco’s phosphate-buffered saline (without Ca++ or Mg++). Neutrophils were isolated by centrifugation and were washed one additional time with 1× Dulbecco’s phosphate-buffered saline before determination of cell number and viability (trypan blue dye exclusion). Cell number was adjusted to 0.75 to 1.5 × 106 cells/ml depending on the individual donor.
To assess MPO release, an index of degranulation, an aliquot (0.1 ml) of the above cell suspension was incubated in Earle’s balanced salt solution containing 20 mM HEPES buffer (pH 7.4) and 0.1% gelatin in the presence of 5 μg/ml cytochalasin B for 5 min at 37°C in a shaking water bath. Cells were pretreated for additional 5 min with a range of SB 207499 or (R)-rolipram concentrations along with PGE2 (3–10 nM) before the addition of fMLP (30 nM) for 30 min. The reaction was terminated by placing the samples on ice followed by centrifugation. The supernatant fraction was removed and stored frozen (−30°C) until assay for MPO activity.
MPO activity was determined using o-dianisidine as substrate and horseradish peroxidase as a standard (Barnette et al., 1996b). Data were expressed as percentage of control (amount of MPO released in the presence of PGE2 alone). Because the maximum inhibition produced by PDE4 inhibitors was ≈30%, IC15 values were calculated by linear interpolation using the responses obtained from at least three experiments.
Histamine release from human basophils.
Venous blood, collected from normal and atopic volunteers who had given informed consent, was collected in tubes containing EDTA, diluted with normal saline and fractionated over Percoll (density, 1.080 g/ml). The mononuclear cell layer containing 1% to 5% basophils was collected and washed once with saline-EDTA and twice with PAG buffer of the following composition: 25 mM piperazine-N,N′-bis(2-ethanesulfonic acid), 110 mM NaCl, 5 mM KCl, 0.003% human serum albumin and 0.1% glucose. PAG buffer supplemented with 1 mM CaCl2and 1 mM MgCl2 was used during the release experiments.
Basophils were incubated for 10 min in the presence of SB 207499 or rolipram before the addition of anti-IgE (0.1 μg/ml). The incubation continued for an additional 40 min at 37°C, after which the cells were pelleted by centrifugation (1000 × g for 3 min). Histamine released into the medium was determined by the automated fluorometric method of Siraganian (1974). Total histamine content was determined by lysing aliquots of cells with 1.6% HClO4. Cells incubated in buffer alone served as a measure of spontaneous histamine release.
Antigen-induced proliferation and cytokine mRNA production from PBMCs.
Fifty milliliters of whole blood from allergic volunteers was drawn into a heparinized 60-ml syringe. The blood was diluted 1:1 with serum-free RPMI 1640 medium supplemented with 1% penicillin/streptomycin. This mixture was overlaid onto 10 ml Ficoll-Paque in 50-ml centrifuge tubes. Samples were centrifuged at 800 × g for 30 min at room temperature. PBMCs were harvested from the interface and washed twice in serum-free media. The cells were then resuspended in RPMI 1640 medium containing 5% human AB serum and aliquoted into 96-well flat-bottom plates at a density of 2 × 105 cells/well. The cells were incubated at 37°C with 5% CO2. Using this procedure, 50 ml of whole blood typically yielded 50 to 80 × 106 cells. Platelet contamination of these cell populations was negligible. Viability as determined by trypan blue exclusion was uniformly ≥99%.
To determine the effects of SB 207499 on PBMC proliferation, SB 207499 or (R)-rolipram was preincubated with the cells for 2 hr before the addition of house dust mite antigen (0.1–2.5 μg/ml). Cells were cultured for 96 hr before the addition of [3H]thymidine (1 μCi/well), and the culture was continued for an additional 24 hr before harvest. Cells were harvested using a multiwell filtration apparatus, and the amount of radioactivity was determined by liquid scintillation spectrometry. Samples were run in triplicate. Results are expressed as a percentage of the control [3H]thymidine incorporation.
The ability of SB 207499 to inhibit antigen-induced cytokine mRNA expression was determined by incubating PBMCs in the absence and presence of a range of SB 207499 concentrations and ragweed antigen for 14 hr. The expression of cytokine mRNA was determined by semiquantitative RT-PCR as described previously (Huang et al., 1994).
Materials.
[3H]Thymidine and the cAMP radioimmunoassay kits were obtained from New England Nuclear Research Products. 14C-Aminopyrine was purchased from Amersham Life Sciences (Arlington Heights, IL). Histamine, fMLP, PGE2, o-dianisidine, cytochalasin B, Histopaque 1077, HEPES, gelatin, endotoxin (E. coli 055:B5), human A/B serum and Earle’s balanced salt solution were obtained from Sigma Chemical (St. Louis, MO). Buffer salts, glucose and hydrogen peroxide were purchased from J. T. Baker Chemical (Phillipsburg, NJ) Horseradish peroxidase was obtained from Boehringer-Mannheim Biochemica (Mannheim, Germany). RPMI 1640 medium was obtained from either MA Bioproduct (Waldersville, MD) or Life Technologies (Gaithersburg, MD). Ficoll-Paque was acquired from Pharmacia (Piscataway, NJ). Antibiotics and Dulbecco’s phosphate-buffered saline were purchased from Life Technologies. Freeze-dried dust mites (D. pteronyssinus) were obtained from Greer Laboratories (Lenoir, NC), and a defatted, lyophilized preparation of ragweed was obtained from Dr. D. Marsh of Johns Hopkins Allergy and Asthma Center (Baltimore, MD). (R)-Rolipram and SB 207499 were synthesized by the department of Medicinal Chemistry, SmithKline Beecham Pharmaceuticals (King of Prussia, PA).
Results.
To determine the ability of SB 207499 ability to inhibit PDE4 in intact cells, we examined its capability to increase cAMP content of U937 cells and compared its action with that of (R)-rolipram. Both SB 207499 and (R)-rolipram produced a concentration-dependent increase in cAMP content in U937 cells (fig. 1). Thus, even though SB 207499 is charged at physiological pH, this does not appear to prevent its access into cells.

Elevation of cAMP content in U937 cells. Cells (1–2 × 106) cells were incubated with either (R)-rolipram (○) or SB 207499 (•) at 37°C for 1 min before the addition of PGE2 (0.1 μM). The incubation continued for an additional 4 min before being terminated. cAMP content of the samples was determined by radioimmunoassay using commercially available kits. The results are corrected for the increase in cAMP produced by PGE2 alone. Values are mean ± S.E.M. of 5 or 6 experiments.
SB 207499 was identified as a second-generation PDE4 inhibitor with reduced activity for HPDE4. To determine whether the decreased activity of SB 207499 against HPDE4 affected its anti-inflammatory activity, we compared its activity with that of (R)-rolipram in a variety of cellular assays. SB 207499 produced a concentration-dependent suppression of several immune and inflammatory cell functions similar to that observed with (R)-rolipram, the prototype PDE4 inhibitor (fig. 2). In isolated human monocytes, SB 207499 and (R)-rolipram were equipotent at suppressing LPS-induced TNF-α formation [−log (IC50) = 7.0 ± 0.1 for SB 207499vs. + 7.2 ± 0.1 for (R)-rolipram;n = 4–7]. Both SB 207499 and (R)-rolipram produced a modest inhibition of fMLP-induced degranulation of human neutrophils. As observed previously with human monocytes, SB 207499 and (R)-rolipram were equipotent [−log (IC50) = 7.1 ± 0.2 vs. 6.4 ± 0.5 for (R)-rolipram; n = 3–12] at suppressing neutrophil activation.

A, Suppression of LPS-induced TNF-α secretion by peripheral human blood monocytes. Monocytes (1 × 106cells/ml) isolated from buffy coats or plasmaphoresis residues obtained from normal donors were preincubated for 45 min with (R)-rolipram (○) or SB 207499 (•) before the addition of LPS (0.1 μg/ml). After 14 to 16 hr, TNF-α levels were determined in the supernatant fractions by ELISA. Results are expressed as a percentage of the control TNF-α production and are the mean ± S.E.M. of 4 to 7 experiments. B, Inhibition of fMLP-induced degranulation of human neutrophils. Purified neutrophils were incubated for 30 min with fMLP (30 nM) and PGE2 (3 or 10 nM) in the absence or presence of increasing concentrations of (R)-rolipram (○) or SB 207499 (•). After removal of the cells by centrifugation, MPO activity was measured in the supernatant fraction. Data are expressed as a percentage of the control release and are the mean ± S.E.M. of 3 to 12 experiments. C, Suppression of antigen-induced T cell proliferation. Peripheral blood mononuclear cells (2 × 105 cells/well) were preincubated for 2 hr with rolipram (○) or SB 207499 (•) in 96-well plates before the addition of house dust mite antigen (0.1–2.5 μg/ml). Cells were cultured for 96 hr before incorporated [3H]thymidine was measured. The results are expressed as a percentage of the control proliferation (18 ± 9.3-fold increase) and are the mean ± S.E.M. of 3 to 7 experiments. D, Inhibition of anti-IgE-induced histamine release from isolated human basophils. Basophils were purified by elutriation from normal donors undergoing hemapheresis. Cells were incubated for 10 min with various concentrations of (R)-rolipram (○) or SB 207499 (•) before the addition of anti-IgE (0.1 μg/ml). The reaction continued for an additional 40 min before histamine release into the medium was determined. The results are expressed as percentage inhibition of the maximum amount of histamine released and are the mean ± S.E.M. of 5 experiments.
To examine the effects of SB 207499 on T cell function, we compared its activity with that of (R)-rolipram with regard to inhibition of antigen-induced proliferation. Again, SB 207499 had comparable activity to (R)-rolipram [−log (IC50) = 6.5 ± 0.3 vs. 6.4 ± 0.3 for (R)-rolipram; n = 4–7]. In contrast to our previous observations with monocytes, T cells and neutrophils, SB 207499 was less potent than (R)-rolipram at suppressing antigen-induced histamine from human basophils [−log (IC50) = 6.6 ± 0.3 vs. 8.0 for (R)-rolipram; n = 5].
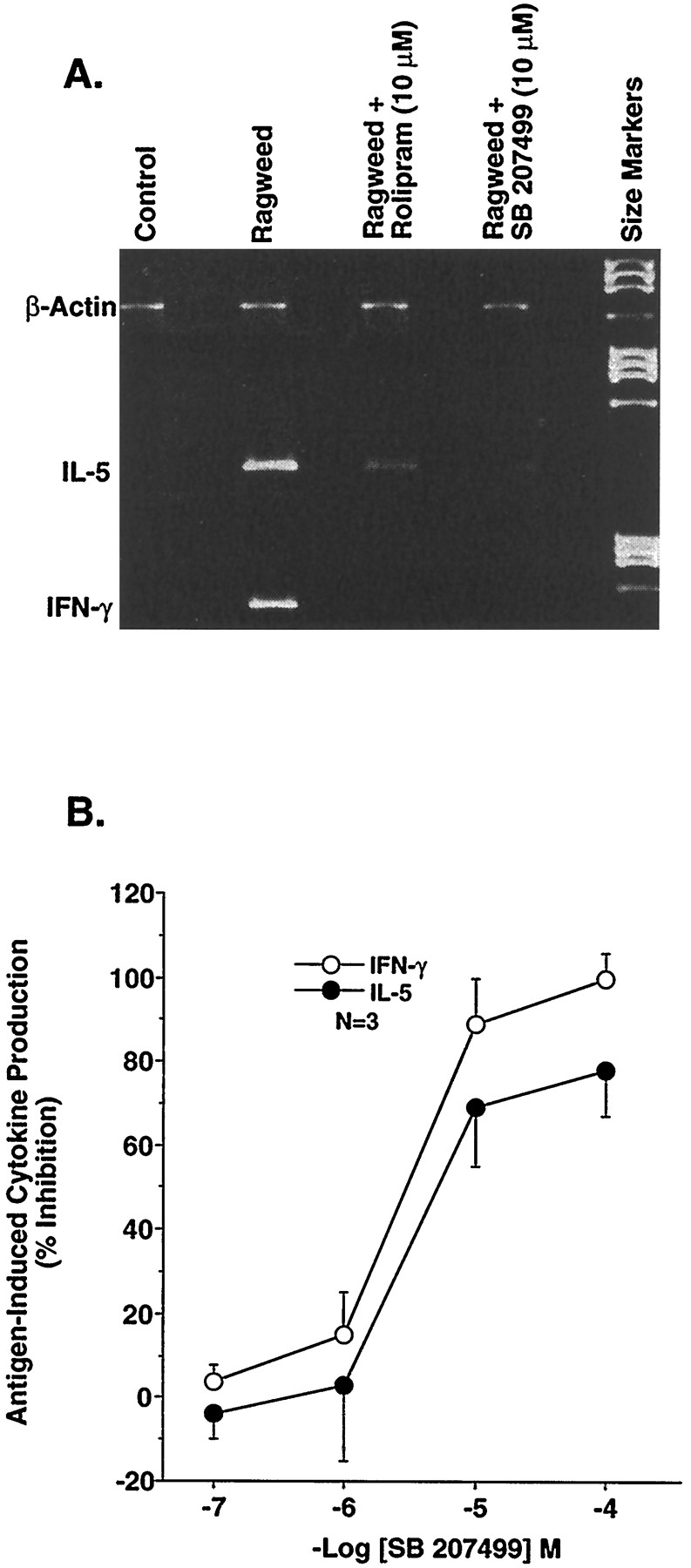
In recent years, it has been established that cytokines, especially T cell-derived cytokines, play an important role in the pathophysiology of many inflammatory diseases. To explore the effects of PDE4 inhibitors, in general, and SB 207499, in particular, on antigen-driven T cell cytokine production, we examined the ability of SB 207499 to inhibit antigen-induced production of IFN-γ and IL-5. SB 207499 produced a concentration-dependent reduction in ragweed-induced stimulation of both IFN-γ and IL-5 (fig.3). Furthermore, SB 207499 inhibited the production of both cytokines with the same potency. As summarized in table 1, the equivalent potency of SB 207499 and (R)-rolipram at suppressing the activation of inflammatory cells supports the observations that many of the anti-inflammatory effects of PDE4 inhibitors are associated with the ability of compounds to inhibit LPDE4 rather than HPDE4 (Barnetteet al., 1996a).

A, Suppression of ragweed antigen-induced cytokine production in PBMCs by (R)-rolipram or SB 207499. Representative example of the ability of (R)-rolipram (10 μM) or SB 207499 (10 μM) to inhibit ragweed-induced IL-5 and IFN-γ mRNA in PBMCs from a single allergic individual. Cytokine mRNA production in PBMCs was measured 14 hr after the addition of antigen by RT-PCR. B, Concentration-dependent inhibition of IL-5 or IFN-γ by SB 207499. Cytokine production was determined as described above. The results are expressed as percentage of inhibition and are the mean ± S.E.M. of 3 experiments.
Comparison of the ability of SB 207499 to suppress immune and inflammatory cell function with rolipram
In contrast to the equivalent actions of SB 207499 on immune and inflammatory cell function, SB 207499 was >100-fold less potent than (R)-rolipram as an acid secretagogue in isolated rabbit parietal glands [−log (IC50) = 6.1 ± 0.11 vs. 8.3 ± 0.2 for (R)-rolipram; n = 3–5; fig. 4]. These results confirm our earlier observations that the ability of PDE4 inhibitors to enhance gastric acid secretion is associated with their ability to inhibit HPDE4 activity (Barnette et al., 1995a)

Enhancement of acid secretion in isolated rabbit gastric glands. Gastric acid secretion was measured by the accumulation of 14C-aminopyrine in rabbit parietal glands. Glands were incubated with 14C-aminopyrine and a threshold concentration of histamine (0.3–1.0 μM) in the absence and presence of either (R)-rolipram (○) or SB 207499 (•) for 20 min at 37°C. The results are expressed as percentage of the maximum response obtained for each compound and are the mean ± S.E.M. of 3 to 5 experiments.
Discussion
The present results demonstrate that SB 207499 with its potent anti-inflammatory activity and its markedly reduced acid secretagogue activity, has the potential to produce significant anti-inflammatory actions in humans without eliciting gastrointestinal side effects secondary to excess acid secretion.
The potential usefulness of selective PDE4 inhibitors as novel antiasthmatic and anti-inflammatory agents has been demonstrated in scores of in vitro and in vivo models (Barnetteet al., 1996a; Torphy and Undem, 1991). The encouraging therapeutic profile of these agents has been tempered by the side effect profile of the compounds identified to date. Thus, the challenge to drug discovery remains the identification of compounds that maintain the anti-inflammatory effects of the first-generation compounds (e.g., rolipram or denbufylline) but with a reduced potential to elicit class-associated side effects of these earlier compounds, including nausea, vomiting and acid secretion.
Many of the side effects of first-generation compounds appear to be related to inhibition of HPDE4, a distinct conformer of the enzyme that is enriched in the central nervous system and parietal glands (Barnetteet al., 1996a). SB 207499 was selected as a second-generation PDE4 inhibitor based on its equivalent potency to (R)-rolipram (Ki ≈100 nM) for inhibiting LPDE4, the conformer of PDE4 that predominates in selected inflammatory cells, and its reduced potency as an inhibitor of HPDE4 (Barnette et al., 1994; Christensen et al., in press).
A unique structural feature of SB 207499 is the presence of a carboxylic acid moiety that confers a net negative charge on the molecule. This characteristic has the potential of limiting the ability of compounds to cross cell membranes. Thus, before comparing the therapeutic activity of SB 207499 with rolipram, which is not charged at physiological pH, we compared the ability of both compounds to enhance cAMP content. In U937 cells, PDE4 constitutes the major cAMP-metabolizing activity (DiSanto and Heaslip, 1993; Torphy et al., 1992b). Thus, this monocytic cell serves as an excellent model system to examine the ability of PDE4 inhibitors to gain access to the internal milieu. (R)-Rolipram and SB 207499 produced a concentration-dependent elevation in intracellular cAMP content in U937 cells. These results indicate that although SB 207499 is negatively charged at physiological pH, this does not interfere with its ability to enter into cells and alter PDE4 activity.
SB 207499 produced a concentration-dependent inhibition of TNF-α formation in isolated human monocytes similar to that observed with other selective PDE4 inhibitors (Barnette et al., 1996b;Semmler et al., 1993; Souness et al., 1996;Verghese et al., 1995). SB 207499 and (R)-rolipram were equipotent at suppressing this cytokine. The equivalent potencies of (R)-rolipram and SB 207499 are consistent with the hypothesis that the suppressive effects of PDE4 inhibitors in isolated human monocytes are associated with inhibition of LPDE4 activity, not HPDE4 (Barnette et al., 1996b;Souness et al., 1996).
SB 207499 and (R)-rolipram produced a concentration-dependent and equipotent suppression of fMLP-induced neutrophil degranulation. As demonstrated previously (Barnette et al., 1996b; Nielson et al., 1990), the maximal inhibition of degranulation produced by either (R)-rolipram or SB 207499 was ≈30%. Furthermore, the equivalent potency of both compounds was somewhat surprising because suppression of fMLP-induced MPO release tends to be associated with inhibition of HPDE4 rather than LPDE4 (Barnette et al., 1996b). Accordingly, one would have expected that (R)-rolipram would be more potent than SB 207499 because its affinity for HPDE4 is 20- to 40-fold higher than that of SB 207499. Perhaps, the lack of potency of (R)-rolipram reflects the inability of this compound to gain access to the appropriate compartment within the neutrophil, or because SB 207499 is more potent than rolipram at inhibiting PDE4D (Torphyet al., 1997), the apparent equivalent potency of SB 207499 and rolipram may reflect the importance of PDE4D subtype in regulating this response. Nevertheless, the neutrophil modulatory actions of SB 207499 further enhance its therapeutic profile.
Because T cells are important immune cells in the initiation and maintenance of inflammatory reactions, we determined the ability of both rolipram and SB 207499 to suppress several functional responses of T cell activation. Previous work by Essayan et al. (1994,1995) demonstrated that rolipram inhibited both T cell proliferation and cytokine production in response to ragweed antigen. In the present study, we also observed that SB 207499 produced a concentration-dependent inhibition of house dust mite antigen-induced T cell proliferation. Again, SB 207499 was equipotent with (R)-rolipram. Identical results were obtained when a different antigen, ragweed, was used (data not shown). Cytokine production was inhibited by SB 207499 with a potency similar to that observed for rolipram (Essayan et al., 1995). Interestingly, the inhibition of IL-5 production produced by the PDE4 inhibitors contrasts with their activity in murine immune cells. In mouse T cells, agents that elevate cAMP seem to enhance IL-5 production (Betz and Fox, 1991), whereas agents such as rolipram inhibit IL-4 and IL-5 production in human T cells (Crocker et al., 1996; Essayan et al., 1995).
Basophils have been implicated in the pathophysiology of asthma, especially in the late phase (Peachell et al., 1992), and PDE inhibitors, particularly PDE4 inhibitors, have been demonstrated to inhibit their activation (Peachell et al., 1992). In contrast to our observations with other immune/inflammatory cells, (R)-rolipram was more potent than SB 207499 at suppressing the IgE-mediated release of histamine. These preliminary findings suggest that the HPDE4 is more important in regulating the activation state of basophils.
We (Barnette et al., 1995a) and others (Puurunen et al., 1978) have demonstrated that PDE4 inhibitors are potent acid secretagogues. This pharmacological action is correlated with the ability of PDE4 inhibitors to inhibit HPDE4 rather than LPDE4 (Barnetteet al., 1995a). Because SB 207499 is markedly less potent than (R)-rolipram for this high-affinity site, it was predicted that SB 207499 would have less acid secretagogue activity. Comparison of the dose-response relationship for both compounds confirms that SB 207499 is indeed markedly less potent an acid secretagogue than (R)-rolipram. Obviously, acid secretagogue activity could be viewed as inherently detrimental, but perhaps of equal importance is that the ability of PDE4 inhibitors to produce nausea and vomiting, which although believed to be primarily a central nervous system action (Heaslip and Evans, 1995), can be exacerbated by local irritant activity, such as acid secretion (Heaslip and Evans, 1995).
In summary, these results demonstrate that SB 207499, a second-generation PDE4 inhibitor, maintains the potency of rolipram as an anti-inflammatory agent in vitro but is markedly less active in producing one potential side effect, acid secretion. The molecular basis for the advantageous profile of SB 207499 can be explained at least in part by its decreased potency against HPDE4 (Barnette et al., 1996a; Torphy et al., 1993). Importantly, other factors, such as its negative charge, which may limit its access to certain tissues such as the central nervous system or gastric glands, and PDE4D subtype selectivity also may be playing a role. Regardless, the results of this study raise the possibility that SB 207499 will provide significant anti-inflammatory activity in vivo while producing less side effects than first-generation PDE4 inhibitors. This proposal is currently being evaluated in the clinic.
Footnotes
-
Send reprint requests to: Mary S. Barnette, Ph.D., Assistant Director, Department of Pulmonary Pharmacology, SmithKline Beecham Pharmaceuticals, 709 Swedeland Road, King of Prussia, PA 19406-0939. E-mail:Mary_S_Barnette{at}sbphrd.com
- Abbreviations:
- AP
- aminopyrine
- fMLP
- formyl methionine leucine phenylalanine
- HPDE4
- phosphodiesterase 4 conformer that binds rolipram with high affinity (previously termed “high affinity rolipram-binding site”)
- IL-4
- interleukin 4
- IL-5
- interleukin 5
- IFN-γ
- interferon-γ
- LTC4
- leukotriene C4
- LPDE4
- phosphodiesterase 4 conformer that binds rolipram with low affinity
- LPS
- lipopolysacchride
- MPO
- myeloperoxidase
- PBMC
- peripheral blood mononuclear cell
- PDE
- phosphodiesterase
- RT
- reverse transcription
- ELISA
- enzyme-linked immunosorbent assay
- PCR
- polymerase chain reaction
- TNF–α
- tumor necrosis factor-α
- PGE
- prostaglandin E
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- Received March 12, 1997.
- Accepted September 15, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}