


Abstract
In these studies, we characterized the influence of the novel benzodioxopiperazine serotonin (5-HT)1A ligand, S 15535, on the release of 5-HT in rat hippocampus and compared its potential anxiolytic properties with those of the 5-HT1A receptor partial agonist, buspirone, the 5-HT1A antagonist, WAY 100,635 and the benzodiazepine, diazepam (DZM). (Doses are in milligrams per kilogram s.c., unless otherwise specified.) S 15535 dose-dependently (0.3–3.0) reduced dialysate concentrations of 5-HT in the hippocampus of anesthetized rats. This action of S 15535 (3.0) was blocked by WAY 100,635 (0.3), (−)-penbutolol (2.0) and (−)-tertatolol (8.0), antagonists at 5-HT1A autoreceptors. In rats, fear-induced ultrasonic vocalizations (USVs) were dose-dependently abolished by S 15535 (0.16–2.5 s.c. and 0.63–10.0 p.o.), an action mimicked by buspirone (0.02–2.5) and DZM (0.16–10.0). Further, the action of S 15535 (0.63) was abolished by WAY 100,635 (0.16) and (−)-penbutolol (10.0), which were inactive alone. S 15535 dose-dependently (0.63–10.0 s.c. and 2.5–40.0 p.o.) blocked aggressive encounters in isolated mice; buspirone (0.16–10.0) and, at high doses, DZM (2.5–40.0) were also effective. WAY 100,635 (0.16), which was inactive alone, fully antagonized the antiaggressive actions of S 15535 (2.5). In an elevated plus-maze, neither S 15535 (0.0025–10.0), buspirone (0.0025–10.0) nor WAY 100,635 (0.00063–0.63) significantly increased open-arm entries, whereas they were increased by DZM (0.16–0.63). In the pigeon conflict test, S 15535 (0.04–0.16 i.m.) markedly increased punished responses and only slightly decreased unpunished responses, even at a 64-fold higher dose. In contrast, buspirone (0.16–2.5 i.m.) and DZM (0.04–2.5 i.m.) showed no or a less marked (4-fold) separation between doses increasing punished and decreasing unpunished responses. In the presence of the 5-HT1A antagonist, (−)-alprenolol (10.0 mg/kg i.m.), S 15535 did not increase punished responses. In a Geller conflict paradigm in rats, S 15535 dose-dependently (0.3–3.0) increased punished responses, and its action (1.0) was blocked by (−)-penbutolol (8.0). S 15535 (0.63–40.0 s.c. and 2.5–40.0 p.o.) exerted little influence on motor behavior. In conclusion, in line with its net inhibition of serotoninergic transmission by activation of 5-HT1A autoreceptors and blockade of postsynaptic 5-HT1A receptors, S 15535 expresses anxiolytic activity. In addition, it displays antiaggressive (and antidepressant, accompanying paper) properties. Further, S 15535 does not compromise motor behavior at doses over which it expresses its anxiolytic properties. Thus, S 15535 represents a promising candidate for the treatment of anxious states in man.
Serotoninergic pathways projecting from the median and dorsal raphe nuclei to the hippocampus, amygdala and other limbic structures play a major role in the control of mood (Coplan et al., 1995) and there is evidence that their overactivity contributes to anxious states (Barrett and Gleeson, 1991; Coplan et al., 1995; Lesch, 1991; Millan and Brocco, 1993). Correspondingly, a component of the anxiolytic action of BZPs can be attributed to a reinforcement of the inhibitory tone exerted by GABAergic neurons on these serotoninergic pathways (Lista et al., 1990; Pan and Williams, 1989). In analogy, the inhibition of ascending serotoninergic transmission via activation of inhibitory 5-HT1A autoreceptors localized on cell bodies by buspirone, for example, appears to underlie anxiolytic effects in both operant, conflict-based paradigms, such as the pigeon and rat conflict tests (Barrett and Gleeson, 1991; Cervo and Samanin, 1995a, b; Schefkeet al., 1989; Schreiber et al., 1995a), as well as in procedures based on unlearned behaviors; for example, fear-induced USVs in rats (De Vry et al., 1994;Sánchez, 1993; Schreiber and De Vry, 1993b). Anxiolytic properties of buspirone and other 5-HT1A receptor ligands have also been demonstrated in man (see Deakin, 1993; Lader, 1991). In addition to 5-HT1A autoreceptors, the possible significance of postsynaptic 5-HT1A sites in the modulation of anxious states should not be neglected. However, although stimulation of postsynaptic 5-HT1A receptors in the hippocampus has been associated with a decrease in anxiety (Carli et al., 1993; see Coplan et al., 1995; Jolas et al., 1995 for critical reviews), certain studies suggest that activation of postsynaptic 5-HT1A receptors in the hippocampus, amygdala and/or the periacqueductal grey may exacerbate anxious/aversive states (Andrews et al., 1994; Fletcher et al., 1996;Hodges et al., 1987; Jenck et al., 1989; Rodgers and Cole, 1994).
In addition to anxiety, a disturbance of serotoninergic transmission is implicated in various impulsive states including obsessive-compulsive disorders (Lesch et al., 1991; Miczek et al., 1995), alcohol abuse (Collins and Myers, 1987; Dillon et al., 1991; Sellers et al., 1992) and aggressive behavior (Blanchard et al., 1988; Mos et al., 1993; Sanchez and Hyttel, 1994; White et al., 1991). Further, 5-HT1A receptors have been particularly implicated in these disorders (see references above and Schreiber and De Vry, 1993b), although, with the exception of aggression (Ratey et al., 1991; Yudofsky et al., 1990), evidence that 5-HT1A agonists are of clinical utility in their treatment is limited (Bruno, 1989; Grady et al., 1993; Pato et al., 1991). Regarding the use of 5-HT1A ligands as anxiolytics, potential antiaggressive actions would be of particular interest in the light of the paradoxical aggression elicited by BZPs (Mos and Olivier, 1989).
The novel benzodioxopiperazine, S 15535, is a highly selective ligand at both rodent and cloned, human 5-HT1A receptors and behaves as an agonist and weak partial agonist/antagonist at pre- and postsynaptic 5-HT1A receptors, respectively (Millanet al., 1994). This distinctive combination of properties suggests that it may elicit anxiolytic actions in the absence of those disruptive motor, amnesic and endocrine effects which are elicited both by high-efficacy stimulation of postsynaptic 5-HT1Areceptors and by interactions at dopaminergic and adrenergic sites (Carli et al., 1995a, b; Millan et al., 1994;Steckler and Sahgal, 1995; Tricklebank, 1985, Van Wijngaarden et al., 1990). In addition, in light of the comments above, S 15535 might exert antiaggressive actions. As such, S 15535 would present a useful alternative to BZPs and buspirone in the management of anxious states. In the present studies, thus, we evaluated the influence of S 15535 on hippocampal 5-HT release and examined its potential anxiolytic actions by use of several paradigms sensitive to 5-HT1Areceptor ligands; that is, the rat and pigeon conflict tests (Barrett and Gleeson, 1991; Cervo and Samanin, 1995a, b), the elevated plus-maze (Handley et al., 1993) and fear-induced USV (De Vry et al., 1994) in rats. Putative antiaggressive properties were examined in isolated mice, a model responsive to 5-HT1Areceptor ligands (Sanchez and Hyttel, 1994). To control for potentially disruptive, motor actions, several measures of motor and locomotor behavior in mice and rats were used. Where active, we confirmed the involvement of 5-HT1A receptors in the actions of S 15535 by use of ligands that are antagonists at both pre- and postsynaptic 5-HT1A receptors; the arylalkylamines, (−)-penbutolol, (−)-tertatolol and (−)-alprenolol and the novel, highly selective arylpiperazine, WAY 100,635 (Fletcher et al., 1996; Hjorthet al., 1995; Hjorth and Sharp, 1993; Millan et al., 1994). The actions of S 15535 were compared with those of WAY 100,635, the (partial) agonist at 5-HT1A receptors, buspirone, and the BZP, DZM.
Materials and Methods
Laboratory conditions.
In all behavioral studies, with the exception of the dialysis and delayed non-matching-to-place experiments, room temperature was 21 ± 1°C and humidity, 60 ± 5%. There was a 12-h light-dark cycle, with lights on at 7:00 a.m..
Release of 5-HT in the hippocampus in vivo.
Male Sprague-Dawley rats (B&K Universal, Sollentuna, Sweden) weighing 270 to 350 g were used. They were adapted for 7 days before experimentation with free access to chow and water: temperature was 24 ± 2°C, humidity was 60 ± 5% and lights were on from 6:00 a.m. to 10:00 p.m.. As previously (Hjorthet al., 1995), chloral hydrate-anaesthetized rats (400 mg/kg i.p., plus supplementary dosing, about 80–100 mg/kg/h) were implanted with U-shaped dialysis probes with a total fiber length of 6 mm (= tip length, 3 mm). The tip was positioned in the ventral hippocampus (anteroposterior, −4.8; lateral, + 4.0; dorsoventral, −8.5 relative to bregma; Paxinos and Watson [1986]). Probes were perfused with artificial cerebrospinal fluid (composition in mM: NaCl,140; KCl, 3; CaCl2, 2.5; MgCl2, 1; Na2HPO4, 1.2; NaH2PO4, 0.27; glucose, 7.2; pH 7.4) containing citalopram (1 μM). Dialysates were analyzed for 5-HT by high-performance liquid chromatography and electrochemical detection, with the detector potential set at 590 to 610 mV. The composition of the mobile phase was: 126 mM NaH2PO4, 0.85 mM EDTA, 0.01 mM sodium octylsulfonate, 13% methanol, pH 4.0. The sensitivity of the assay was 1 fmol of 5-HT/20 μl of dialysate. Stable base-line levels of 5-HT were obtained 2 to 3 h after implantation. S 15535 or vehicle were injected and dialysates collected every 20 min. For antagonist studies, (−)-tertatolol (8.0 mg/kg s.c.), WAY 100,635 (0.36 mg/kg s.c.) or (−)-penbutolol (2.0 mg/kg s.c.) were injected 40 min before S 15535 (0.3 mg/kg s.c.). 5-HT levels were expressed as a function of control levels before injection of S 15535 (defined as 100%). A multiple analysis of variance (ANOVA) with the dose of S 15535 as the between-subject factor, and with sampling time as the repeated within-subject factor, was performed. For examination of antagonist actions, a multiple ANOVA with the dose of antagonist as the between-subject factor, and with sampling time as the repeated within-subject factor, was performed. Post hoc analyses were performed by the Dunnett’s test for comparison of individual values (P ≤ .05).
Rat Geller-Seifter conflict test.
Male Sprague Dawley rats (CD-COBS, Charles River, Calco, Italy) weighing 280 to 300 g, maintained at 85% of their free-feeding weight, were housed in sawdust-lined cages with free access to water. Animals were tested in operant chambers with two levers, and reinforcement consisted of 45-mg food pellets delivered to a magazine tray equidistant between the levers. As described previously, with use of a variable interval 20 s (VI, 20 s) schedule (Cervo and Samanin, 1995a, b), rats were trained to press for reinforcement. Thereafter, a multiple schedule for three 5-min components was established. Rewardperiods, signaled by illumination of a light in the ceiling of the chamber, during which lever responding was reinforced according to the above schedule; time-out periods signaled by absence of light, when no food was given and conflict periods signaled by illumination of three lights on the front panel, during which lever responding was reinforced according to the schedule but each reinforced response was punished by a foot-shock through the grid floor. The shock level, initially set at 0.1 mA for 0.5 s, was increased daily by 0.02 mA until responding during the conflict period was less than 10% of that of the reward period. The 30-min daily session consisted of two consecutive presentations of the multiple schedule. When responding had stabilized, drug studies were initiated. On Tuesdays and Wednesdays, rats received an injection of vehicle before behavioral testing (control sessions). On Thursdays, they received an injection of the test compound (drug session). S 15535 (s.c.), DZM (i.p.) or vehicle was injected 30 min before testing. In antagonist experiments, drugs or vehicle were administered 40 min before S 15535 (1.0 mg/kg s.c.) or its vehicle. Rats were used as their own controls and data are expressed as response rates during the various periods. For drug actions alone, response rates obtained after drug administration were compared with control values by Wilcoxon’s test. The effects of antagonistsversus S 15535 were evaluated by ANOVA, and the Student’st test was used to determine significance of differences to respective vehicle groups (P ≤ .05).
Pigeon conflict test.
White Carneaux pigeons (500–600 g) of either sex (Grozek, Lewarde, France) were housed singly in cages with unlimited access to water and crushed oyster shell grit, but controlled access to mixed grain to maintain body weight at approximately 80% of free-feeding values. As described previously (Brocco et al.,1990; Schreiber et al., 1995a), pigeons were trained to peck an illuminated (green or red) key for food. Every 30th response made during illumination of the green key produced access to food, whereas every 30th response made during illumination of the red key produced both food and electric shock to the groin. A 1-min time-out interval (no key light and no food reinforcement) separated each 3-min illumination period. Sessions were terminated after 5 cycles of alternating components and lasted 40 min in all. S 15535, buspirone, WAY 100,635, DZM or vehicle were injected i.m.(1 ml/kg) 5 min before the session. In antagonism studies, S 15535 and (−)-alprenolol or vehicle were simultaneously injected 60 min before testing. For each pigeon, response during unpunished (green) and punished (red) components of test sessions were expressed as a percentage of control responses during the previous saline session.
Elevated plus-maze in the rat.
Male Wistar rats (Iffa-Credo, L’Arbresle, France) weighing 220 to 240 g were housed in sawdust-lined cages with free access to rat chow and water for at least 1 week before the experimentation. The elevated plus-maze was made of white-mat, painted wood and consisted of two open (50 × 10 cm) and two enclosed arms of the same size with walls 40 cm high. The two open arms were opposite to each other. The maze was at a height of 50 cm and located in the center of the room. The procedure (see Millan and Brocco, 1993) was as follows. The day before the test, each animal was isolated in an individual polycarbonate cage. Thirty minutes after treatment with drugs or vehicle, rats were placed in the central square of the maze facing one of the enclosed arms. The number of entries and the time spent in open and enclosed arms were recorded directly over 5 min by an observer situated 2 m from the maze. An entry was counted only when the rat had its four limbs in one arm. Parameters analyzed were the total number of entries (into open and enclosed arms) and percentage entries and time spent in open arms. Drug effects were evaluated by ANOVA, followed by Dunnett’s test (P ≤ .05).
Fear-induced ultrasonic vocalizations in the rat.
Animals and laboratory conditions were the same as for the plus maze test. All experiments were performed in sound-proof chambers (modular test cage system, model EW-10SF, Coulbourn Instruments, Lehigh Valley, PA), equipped with an electrifiable grid and with a microphone in the center of the ceiling. Ultrasounds were transformed to the audible range with a bat-detector (model S-25, Buitenbedrijf, BBZ, Groningen, Holland) and the modified signals were led through a low-pass antialias filter to attenuate for frequencies above the cut-off of 25.5 kHz and to prevent the generation of spurious spectral material during acquisition and feedback. Subsequently, signals were led through a high-speed single-board analog and digital input/output system (250 kHz, model DT-28216, Data Transmission, Marlboro, MA) displayed by an active speaker system and on the screen of a computer controlling acquisition and display with Peak Time Spectrogram software (Engineering Design, Belmont, MA). The experimental procedure consisted of three different stages 24 h apart: training, selection and drug testing. On day 1 (training), rats were placed in the chambers and received six randomly distributed inescapable shocks (0.8 mA, 8 s) in a 7-min period. Electric stimuli were delivered by a automated shock source connected to a solid-state grid-floor scrambler (model ENV 412, Med Associates Inc, Georgia, VT) according to an intershock interval which varied between 30 and 90 s (training). On day 2 (selection), rats were placed in the chambers and a single shock delivered during a 2-min period. Rats were then returned to their home cages. Thirty minutes later, rats were returned to the operant chamber and the emission of USVs was measured in a 10-min session. Only rats emitting more than 150 s of ultrasonic calls were selected for drug testing. On day 3 (drug testing), rats were tested under identical conditions in the operant chamber as on day 2, but were injected with drug or vehicle at the end of the first 2-min period before being returned to home cages. Thirty minutes later, they were again placed in the chamber and USVs registered. The same rats were tested repeatedly in the operant chamber with use of a 2-day washout period. Animals were their own controls. Drugs or vehicle were injected 30 min pretesting. WAY 100,635 or vehicle were given s.c. 30 min before S 15535 or vehicle: that is, 60 min pretesting. Data were expressed as total duration of USVs. Dose effects were analyzed by ANOVA, followed by Dunnett’s test (P ≤ .05).
Aggressive behavior in isolated mice.
Male CD1 (ICR) BR mice (Charles River, Elbeuf, France) weighing 20 to 25 g at the beginning of the experiment were isolated in individual black-painted polycarbonate cages with a sawdust floor and maintained in the experimentation room during the entire study. After 1 month of isolation, selection of the pairs of mice to be used for drug studies was initiated: once or twice a week, one isolated mouse (“intruder”) was placed into the cage of another isolated mouse (“resident”) for 5 min. At the end of the trial, the “intruder” was isolated again in its home cage. If the mice fought during the trial, the same pair was used for the next scheduled trial; if not, each animal of the pair was confronted with a different mouse in the next trial. Usually within 3 weeks, pairs of aggressive mice entering the study could be defined. Mice remained isolated, with the exception of test days which were scheduled once a week. On the test day, the “intruder” mouse of the pair was placed into the cage of the “resident” mouse, for 3 min. The number of fights and total fight duration (s) were directly recorded by an observer blind as to treatment. Each mouse received the same treatment (drug or vehicle). Drugs or vehicle (s.c.) were administered to mice 30 min before testing. The effects of S 15535 were evaluated after s.c. or oral administration 30 min before testing. In antagonist experiments, WAY 100,635 (0.16 mg/kg, s.c.) or vehicle was administered 30 min before S 15535 (2.5 mg/kg s.c.). For drug actions alone, the total duration (s) and number of fights in drug-treated pairs of mice were compared with those in vehicle-treated pairs by ANOVA followed by Dunnett’s test (P ≤ .05). The effects of the antagonist upon the activity of S 15535 were evaluated by a multiple ANOVA followed by Dunnett’s test (P ≤ .05).
Tests of motor behavior.
These were performed on male NMRI mice (22–25 g) and male Wistar rats (220–280 g) (Iffa-Credo, L’Arbresle, France), maintained under standard laboratory conditions. For the rotarod test (Millan et al., 1994) the latency of mice to fall from a rotating bar was determined (accelerating rotation rate = 5–40 rpm over 300 s), with a cut-off time of 360 s. Spontaneous locomotor activity was measured in mice placed in individual white Plexiglas chambers (27 × 27 × 27 cm) equipped with two facing rows of four photocells, located 6 cm apart 2 cm above the floor. The cells were connected via an interface to a computer with software written by Osys/Orga System (Changéé France). The interruption of two adjacent beams was taken as a count of ambulation. The day before testing, mice were placed in individual cages. On the test day, they were administered drug or vehicle, then returned to their cages for 30 (s.c.) or 60 (p.o.) min. Thereafter, they were placed in the chambers and monitored for ambulation during 10 min. Data were analyzed by ANOVA, followed by Dunnett’s test. Spontaneous locomotor activity was measured as previously (Maurel-Rémy et al., 1995) viabeam interruption over 60 min in rats placed in clear Plexiglas cages. Drugs were given 30 (s.c.) or 60 (p.o.) min before testing. This procedure was also used for examination of drug effectsversus amphetamine-induced hyperlocomotion in rats, with administration of d-amphetamine (2.5 mg/kg i.p.) immediately before testing. In the test of stereotyped behavior, rats were transferred into individual, transparent, polycarbonate cages the day before testing. For the effects of drugs alone, drug or vehicle was injected 30 min before evaluation. Rats were observed for 10 s, every min of a 10-min period for presence (1) or absence (0) of locomotion, sniffing, rearing or gnawing. Each behavior was considered present if the animal spent at least 3 consecutive seconds performing the behavior. The maximal score for each behavior was determined. For the influence of drugs on methylphenidate-induced gnawing, drug or vehicle was administered 60 min, and methylphenidate (40.0 mg/kg i.p.) 30 min, before observation.
Drugs.
All drug doses are in terms of the base. Drugs were dissolved in sterile water, plus a few drops of lactic acid if necessary, and pH adjusted to as close to neutrality (>5.0) as possible. Drugs were, unless specified, injected subcutaneously (S.C.). Injection volumes were 1 ml/kg (rats) or 10 ml/kg (mice). In certain studies, S 15535 was administered orally (10 ml/kg p.o.) by gavage in a suspension of water plus a few drops of Tween 80. Drug sources, salts and structures were as follows: (−)-alprenolol, xylazine HCl and haloperidol (Sigma, Chesnes, France); buspirone HCl (Bristol Myers, Wallingford, CT); diazepam (Valium, 2 mg/10 ml ampullas) (Hoffman-La Roche, Basel, Switzerland); ICI 118,551, {[erythro-d, I-1-(7-methylindan-4-yloxy)-3 isopropyl aminobutan-2-ol]} HCl (Imperial Chemical Industries, England); (−)-metoprolol and (−)-penbutolol sulphate (Hoechst AG, Frankfurt, Germany); 8-OH-DPAT HBr and apomorphine HCl (R.B.I., Wayland, MA); UK 14,304 tartrate (Pfizer, Orsay, France); methylphenidate chlorhydrate (Ciba-Geigy, Rueil, France); d-amphetamine sulfate (Calaire Chimie, Calais, France); UK 14,304 tartrate (5-bromo-6-[2-imidazolin-2-yl-amino]-quinoxaline), S 15535 (4-(benzodioxan-5-yl)1-(indan-2-yl)piperazine, WAY 100,635 (N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl) cyclohexanecarboxamide 3HCl), 1-pyrimidinyl-piperazine and (−)-tertatolol were synthesized by Servier Chemists (Suresnes, France).
Results
Action of S 15535 on hippocampal release of 5-HT in the anesthetized rat.
As shown in figure 1, S 15535 dose-dependently decreased 5-HT levels in hippocampal dialysates of anesthetized rats. This action was abolished by the 5-HT1A autoreceptor antagonists, WAY 100,635, (−)-penbutolol and (−)-tertatolol (fig. 1), as well as by (−)-pindolol (8.0 mg/kg s.c., not shown). Administered alone, at these doses, they have previously been shown not to modify dialysate levels of 5-HT (Hjorth and Sharp, 1993; Hjorthet al., 1995) and, in the present study, before injection of S 15535, they did not significantly modify levels of 5-HT (fig. 1).

Influence of s.c. administration of S 15535 on 5-HT levels in dialysates of rat hippocampus. Serotonin levels are expressed relative to basal, preinjection values (= 100%). These were 45.8 ± 1.6 fmol/20 μl dialysate for vehicle-treated animals,n = 20. Data are means ± S.E.M.n = 4–6 per value. The upper left panel depicts the dose-response relationship for inhibition of 5-HT release by S 15535 alone and the other panels depict the inhibition of its actions by antagonists at 5-HT1A autoreceptors. For the dose-response: Effect of S 15535, F(5,17) = 25.7, P < .001; effect of time, F(5,85) = 39.2, P < .001 and interaction, F(25,85) = 3.4, P < .001. For antagonist activity, effect of WAY 100,635, F(1,7) = 182.5, P < .001; effect of (−)-penbutolol, F(1,8) = 73.1, P < .001 and effect of (−)-tertatolol,F(1,6) = 262.8, P < .001. Asterisks indicate significance of differences to corresponding vehicle values in the Dunnett’s test after ANOVA. * P ≤ .05.
Action of S 15535 in the Geller conflict test in rats.
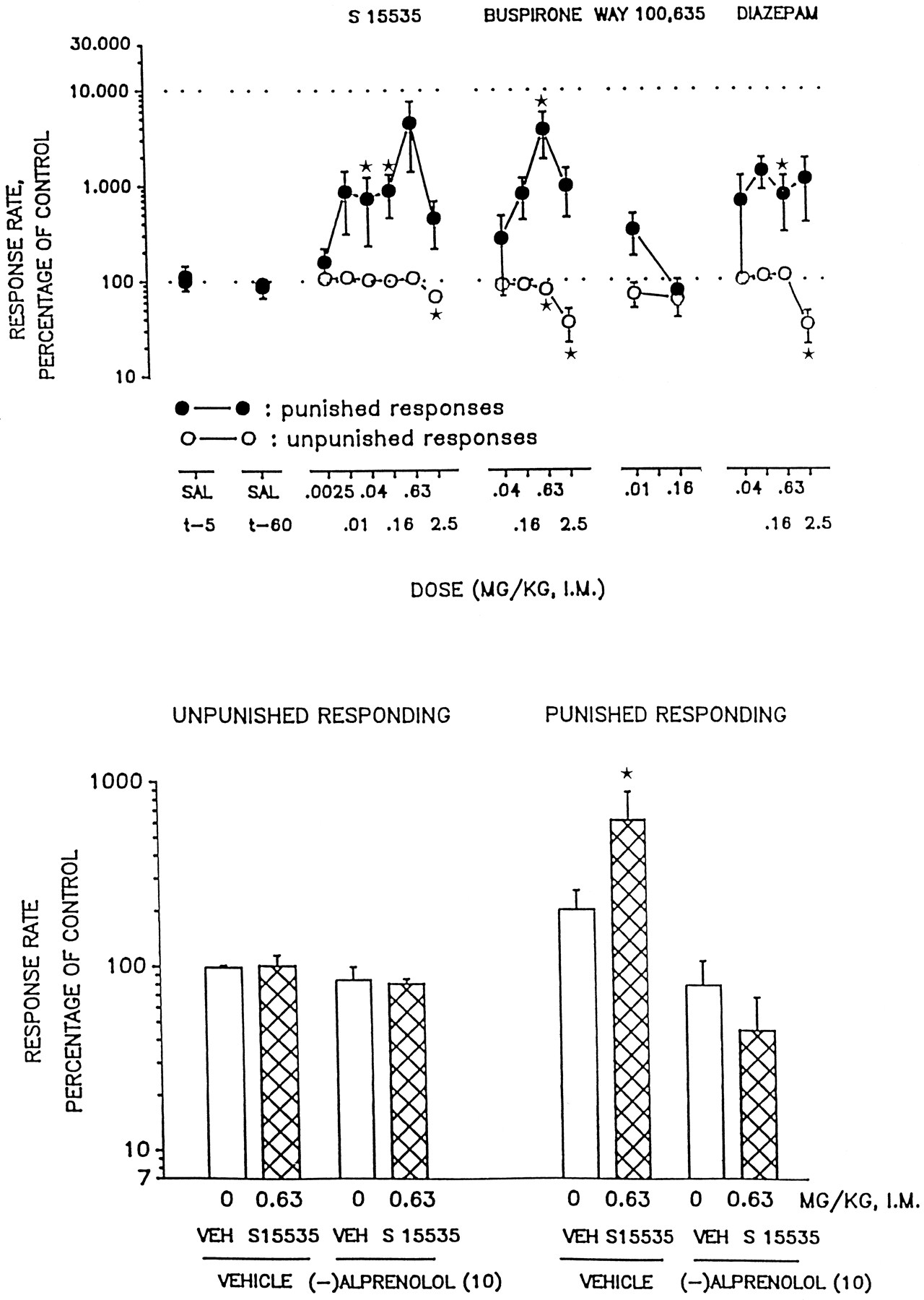
S 15535 elicited a dose-dependent increase in the number of responses emitted by rats in the presence of punishment (table 1). At the highest dose tested (3.0 mg/kg s.c), S 15535 induced a 6-fold increase over basal (vehicle-treated) response rates. Further, the action of S 15535 was significant even at the lowest dose examined (0.3 mg/kg s.c.), S 15535 also significantly enhanced time-out responses, but only at the two highest doses tested (1.0 and 3.0 mg/kg, s.c.) and with a maximal effect about half of that seen for punished responses (table1). In contrast, responses emitted during the control, unpunished (reward) periods were not significantly modified at any doses tested. DZM, at 1.25 and 2.5 mg/kg i.p., induced a marked increase in the conflict response rates without modifying the reward rates. However, like S 15535, DZM also significantly increased time-out responding at higher dose. As shown in table 2, the anxiolytic action of S 15535 was almost abolished in the presence of (−)-penbutolol. (−)-Penbutolol did not itself modify punished responses. In this experiment, there was no statistically significant influence of S 15535 on responses in the time-out period such that the possible antagonism of this action of S 15535 by (−)-penbutolol could not be evaluated. Nonpunished, reward responses were not modified by (−)-penbutolol (table 2). Although (−)-penbutolol possesses β-AR antagonist properties, in the presence of the combined application of a β1-AR antagonist ((−)-metaprolol) and a β2-AR antagonist (ICI 118,551), the anxiolytic action of S 15535 was maintained (table 2). Indeed, it was slightly enhanced, although this action may reflect the slight anxiolytic effects of (−)-metaprolol/ICI 118,551 themselves. S 15535 also did not modify time-out (or unpunished) responses in this experiment.
Actions of s.c. administration of S 15535 in the rat Geller-Seifter conflict test
Effect of the 5-HT1A receptor antagonist, (−)-penbutolol, as compared with the β-AR antagonists, (−)-metaprolol and ICI 118,551, on the anxiolytic activity of S 15535 in the rat Geller-Seifter conflict test
Action of S 15535 in the pigeon conflict test.
Figure2 shows that S 15535 elicited a significant increase in punished responses in the pigeon conflict test. As is characteristic for 5-HT1A receptor ligands in this procedure (Schreiberet al., 1995a), the dose response inflected at the highest dose (2.5 mg/kg i.m.) examined. Nevertheless, there was a dose-response relationship for the increase in punished responses across a lower range of doses (0.0025–0.63 mg/kg i.m.). At the highest dose tested, S 15535 significantly and slightly (10%) decreased nonpunished responses: that is, there was a 64-fold separation between the lowest effective doses increasing and decreasing punished as compared with nonpunished responses, respectively. Although WAY 100,635 was inactive, buspirone similarly elicited a marked and significant increase in punished responses, although there was no separation between the minimal effective dose in this respect and that for a (marked) decrease in nonpunished responses. The influence of S 15535 on punished responses was abolished in the presence of the 5-HT1A receptor antagonist, (−)-alprenolol, which did not, itself, modify either nonpunished or punished responses (fig. 2). (−)-Alprenolol was selected for this antagonist study to provide comparability with previous studies of 5-HT1A receptor ligands in pigeons in which we have used this ligand (Schreiberet al., 1995a). Further, the pigeon is liable to show unexpected toxic reactions for certain drugs, including serotoninergics, either alone or in combination. Nevertheless, it would be important to confirm this finding with a selective 5-HT1A antagonist. Finally as shown in figure 2, DZM elicited a significant increase in punished responses and, at a 4-fold higher dose, a significant decrease in unpunished responses.

Influence of i.m. administration of S 15535 in the pigeon conflict test. In the upper panel, the dose-response relationship for S 15535 (n = 9–12 per value) as compared with buspirone (n = 9), WAY 100,635 (n = 8) and DZM (n = 7 or 8) is depicted and, in the lower panel, the blockade of the actions of S 15535 by (−)-alprenolol (n = 5– 9) is shown. Data are means ± S.E.M. Response rates are expressed as a percentage of those obtained on vehicle treatment (animals used as their own controls). Average control (vehicle treatment) values were 1834.0 ± 60.3 (unpunished) and 11.8 ± 1.5 (punished) responses. Asterisks indicate significance of differences (* P ≤ .05) to control values in the permutation test for paired replicates. The test was two-tailed for unpunished and one-tailed for punished responses.
Action of S 15535 in the plus-maze test in rats.
Administered over a broad range of doses (0.002–10.0 mg/kg s.c.), S 15535 did not significantly modify the percentage entries into the open arms of an elevated plus-maze, although at high doses it reduced both percentage time in the open-arms and total entries (fig. 3). Buspirone presented a similar pattern of data with a significant reduction in open arm entries (number and time), and a marked decrease in total entries, which was significant at doses of 2.5 to 40.0 mg/kg, s.c. WAY 100,635 (0.0063–0.63 mg/kg s.c.) did not significantly modify the behavior of rats in this paradigm (fig. 3). In distinction, DZM presented a biphasic dose-response curve for the number and time of open-arm entries, with a significant increase in these at intermediate doses (0.16–2.5 mg/kg s.c.) (fig. 3). The curve inflected at higher doses, at which a significant reduction in total entries was also seen (10.0 mg/kg s.c.)

Influence of s.c. administration of S 15535 in the elevated plus-maze test in rats. Data are means ± S.E.M. per value. For upper panel (% entries into open arms): S 15535 (n = 10 per value), F(7,80) = 0.3, P > .05; buspirone (n = 7 to 11),F(8,64) = 2.6, P < .05, DZM (n= 6 to 10), F(6,51) = 7.2, P < .001 and WAY 100,635 (n = 7 or 8), F(5,42) = 1.2, P > .05. Middle panel (% time in open arms), S 15535,F(7,80) = 3.5, P < .01; buspirone,F(8,64) = 4.1, P < .001, DZM,F(6,51) = 7.7, P < .001 and WAY 100,635,F(5,42) = 0.6, P > .05. Lower panel (total entries), S 15535, F(7,80) = 5.0, P < .05, buspirone, F(8,68) = 15.4, P < .001, DZM,F(6,53) = 6.7, P < .001 and WAY 100,635,F(5,42) = 1.1, P > .05. Asterisks indicate significance of differences to control values in Dunnett’s test (* P ≤ .05).
Action of S 15535 in the ultrasonic vocalization test in rats.
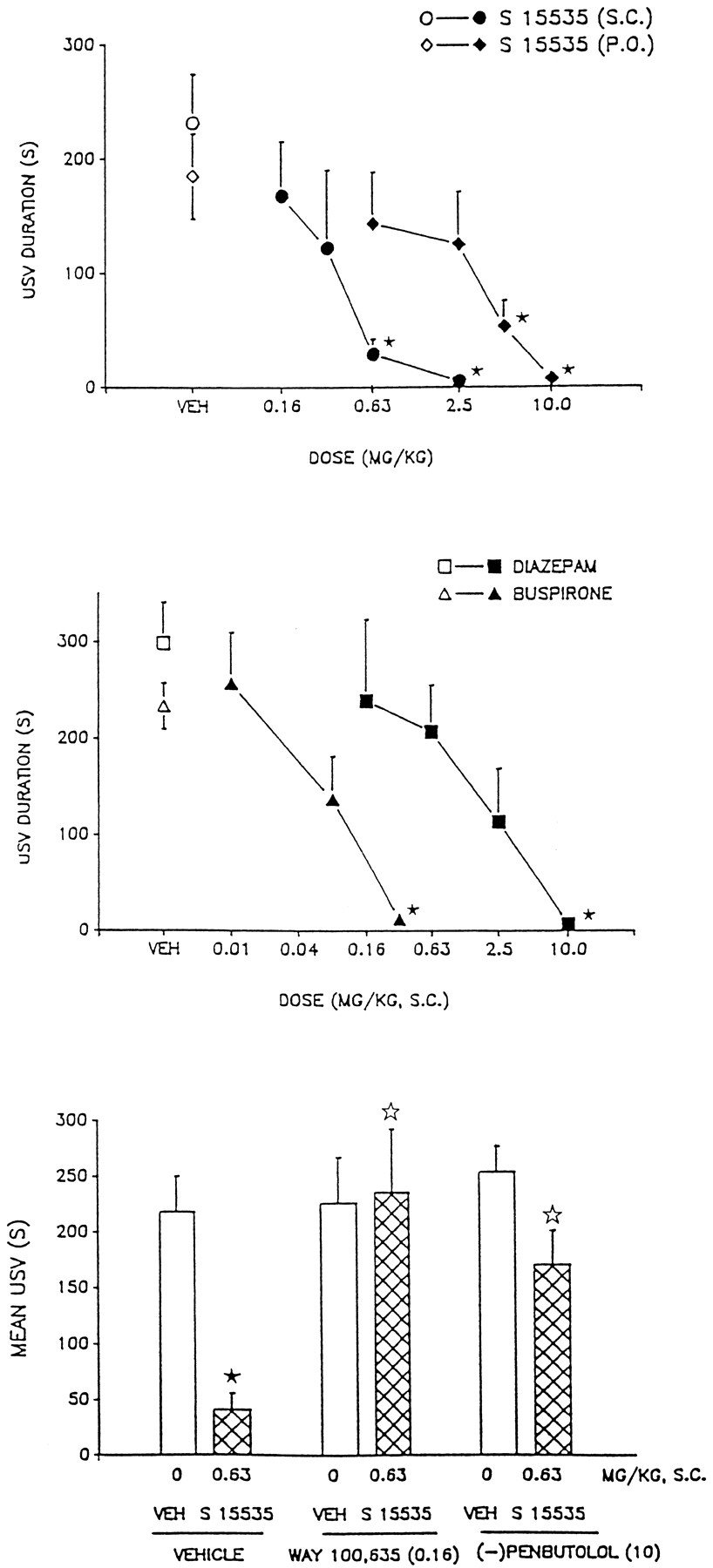
Figure 4 shows that S 15535, administered either s.c. or p.o., dose-dependently and completely blocked fear-associated USVs in rats. Similarly, buspirone and DZM dose-dependently blocked USVs under these conditions, although the dose-response curve of the latter was shallow and high doses were required. The 5-HT1A receptor antagonists, WAY 100,635 and (−)-penbutolol, did not themselves modify USVs and, in their presence, the action of S 15535 was abolished (fig.4).

Influence of s.c. administration of S 15535 upon fear-induced ultrasonic vocalizations in rats. Upper and middle panels, dose-response relationships; lower panel, antagonism of the actions of S 15535 (0.63) by the 5-HT1A autoreceptor antagonists, (−)-penbutolol (10) and WAY 100,635 (0.16). Data (duration of vocalization in seconds) are means ± S.E.M.n = 5 per value. (upper and middle panels) absolute control values were 231.6 ± 42.9, 184.7 ± 37.6, 233.4 ± 24.2 and 298.4 ± 42.3 for S 15535 s.c. (n= 4–6 per value), S 15535 p.o. (n = 5–8), buspirone (n = 5) and diazepam (n = 4–8), respectively. ANOVA as follows: S 15535 s.c., F(3,15) = 5.5, P < .01; S 15535 p.o.F(4,32) = 2.74, P < .05; buspirone,F(3,25) = 8.9, P < .001 and DZM,F(4,32) = 4.9, P < .01. Asterisks indicate significance of differences to control values in Dunnett’s test after ANOVA (* P ≤ .05). Lower panel, control (vehicle/vehicle) values were 218.5 ± 31.9 s, and data were pooled for presentation from two separate experiments. ANOVA as follows: effect of WAY 100,635,F(1,28) = 7.4, P < .01; effect of S 15535,F(1,28) = 11.2, P < .01 and interaction,F(3,2) = 6.9, P < .05. Effect of (−)-penbutolol, F(1,28) = 3.2, P > .05; effect of S 15535 = 20.0, P < .001 and interaction,F(3,25) = 5.7, P < .05. Closed asterisks indicate difference to vehicle/vehicle values, and open asterisks indicate significance of differences to vehicle/S 15535 values, with Student’st test (* P ≤ .05).
Action of S 15535 upon aggressive behavior in isolated mice.
As depicted in figure 5, upon both s.c. and p.o. application, S 15535 dose-dependently and markedly inhibited aggressive encounters in isolated mice as concerns both the number and duration of attacks emitted by the dominant towards a familiar submissive conspecific. This action was mimicked by both s.c. administration of buspirone and DZM, although the latter was active only at high doses and did not completely block aggressive behavior as indicated by the duration of attacks (fig. 5). Figure 6 shows that WAY 100,635 failed to modify aggressive behavior and, in its presence, the anti-aggressive actions of S 15535 were abolished, in line with their mediation by 5-HT1A receptors.
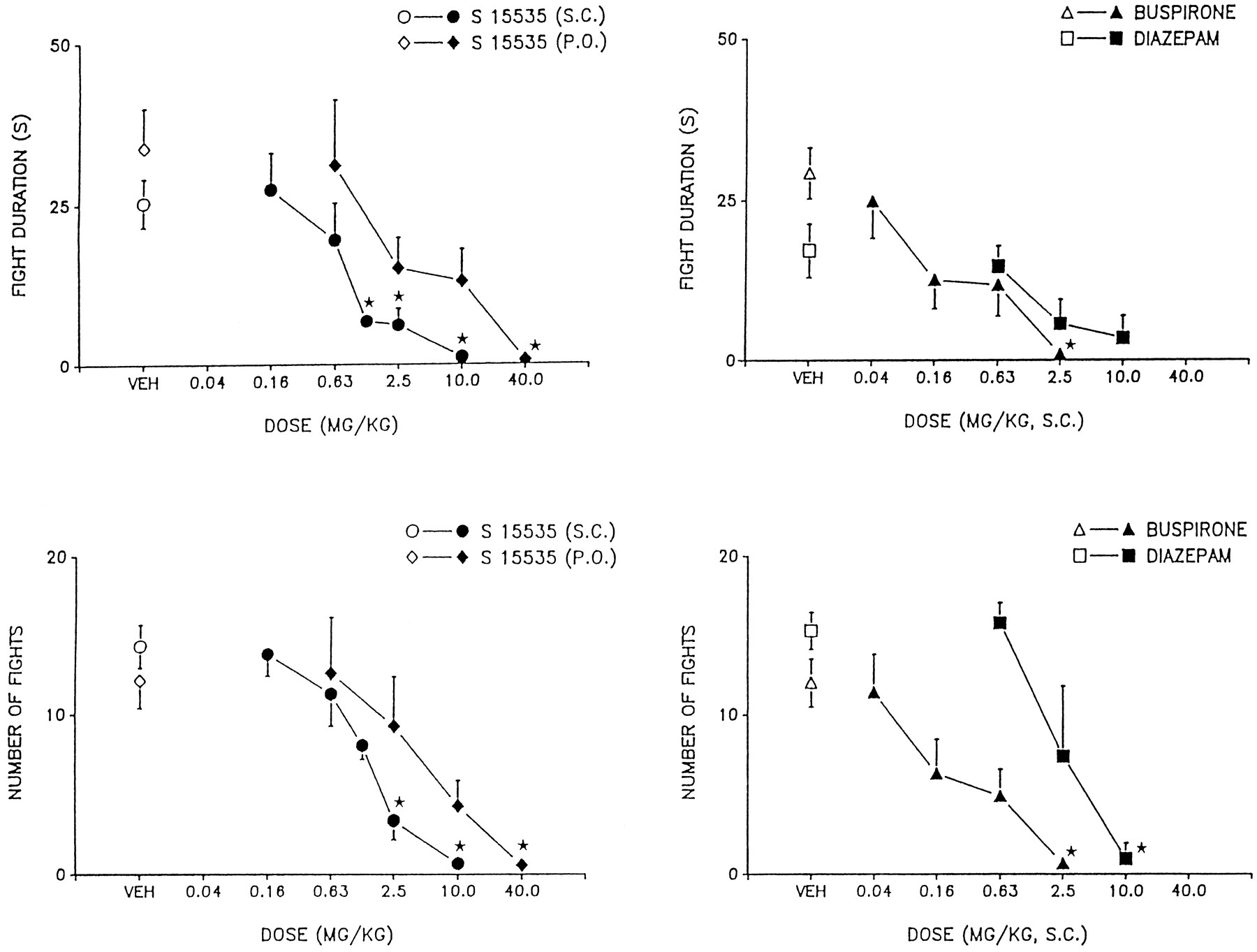
Influence of s.c. and p.o. administration of S 15535 on aggressive behavior in isolated mice. Data are means ± S.E.M. The effects of S 15535 s.c. (n = 5–10 per value), S 15535 p.o. (n = 7–15), buspirone (n = 4–8) and DZM (n = 6–7) on the duration of fighting in seconds and the number of attacks by the dominant on the submissive mouse, are shown in the upper and lower panels, respectively. (upper panel) S 15535 s.c.,F(5,51) = 5.4, P < .001; S 15535 p.o.,F(4,59) = 2.3, P < .05; buspirone,F(4,35) = 5.0, P < .01 and DZM,F(3,17) = 3.0, P > .05. (lower panel) S 15535 s.c., F(5,51) = 11.1, P < .001; S 15535 p.o.,F(4,59) = 4.1, P < .01; buspirone,F(4,35) = 5.4, P < .01 and DZM,F(3,17) = 9.0, P < .001. Asterisks indicate the significance of differences to vehicle values in Dunnett’s test after ANOVA (* P ≤.05).

Blockade of the antiaggressive actions of S 15535 by the 5-HT1A autoreceptor antagonist, WAY 100,635, in mice. Data are means ± SEM. n = 7 per value. Doses are in milligrams per kilogram s.c. The upper panel shows the duration of fighting in seconds, and the lower panel shows the number of attacks by the dominant on the submissive mouse. (upper panel) effect of WAY 100,635, F(1,24) = 1.4, P > .05; effect of S 15535, F(1,24) = 12.5, P < .01 and interaction, F(1,24) = 7.4, P < .01. (lower panel) effect of WAY 100,635, F(1,24) = 12.9, P < .01; effect of S 15535, F(1,24) = 9.3, P < .01 and interaction, F(1,24) = 4.7, P < .05. Closed asterisks indicate the significance of differences to vehicle/vehicle values, and open asterisks indicate significance of differences to vehicle/S 15535 values, in Student’s t test (* P ≤ .05).
Action of S 15535 upon motor and locomotor behavior.
In contrast to the dopaminergic agonist, apomorphine and the dopamine releaser, methylphenidate (scores of 10 at 10.0 mg/kg s.c.), S 15535 and buspirone did not elicit stereotyped behavior in rats, even at high doses administered s.c. (score = 0 at 10.0) or p.o. (score = 0 at 40.0). Further, as reported previously (Millan et al.,1994), S 15535 does not interfere with the stereotyped gnawing provoked by methylphenidate (40.0 mg/kg i.p.): vehicle + methylphenidate, score = 10.00 ± 0.0, n = 10; S 15535 (40.0 mg/kg, s.c.), score = 9.2 ± 0.3, n = 5, not significant. In contrast, buspirone completely blocked the action of methylphenidate with an inhibitory dose50(ID50) (95% confidence limits [C.L.]) of 0.64 (0.32–1.28) mg/kg s.c. Table 3 shows that S 15535 diminished spontaneous locomotor behavior only at a very high dose (40.0 mg/kg s.c.) relative to its efficacy range in therapeutic models. Further, on oral administration, it was inactive. S 15535 did not significantly decrease the stimulation of locomotor behavior elicited by amphetamine (1.25 mg/kg s.c.); vehicle + amphetamine = 289 ± 31 counts, n = 8 versus S 15535 (10.0 mg/kg s.c.) + amphetamine = 240 ± 36 counts,n = 12, not significant. In contrast, buspirone completely blocked the action of amphetamine with an ID50(95% C.L.) of 0.47 (0.23–0.94) mg/kg, s.c. In rats and mice, as shown in figure 7, in contrast to DZM and the α2-AR agonists, xylazine and UK 14,304, S 15535 did not elicit sedation (loss of righting reflex) in rats. Buspirone was similarly inactive but, reflecting the α2-AR antagonist properties of its metabolite, 1-pyrimidinyl-piperazine, it dose-dependently blocked the action of xylazine with an effective dose50 (ED50) (95% C.L.) of 0.5 (0.05–5.0) mg/kg s.c. The action of xylazine was not, in contrast, modified by S 15535 (0% inhibition at a dose of 40.0 mg/kg s.c., not shown). In the rotarod procedure (fig. 7), in contrast to buspirone and the other reference ligands, S 15535 elicited only a mild decrease in latencies upon either the s.c. or the p.o. route. Finally, in each of the above-described procedures, WAY 100,635 was virtually inactive (<20% effect) over a dose range of 0.0025 to 0.63 mg/kg s.c. (not shown).
Relative lack of modification of locomotor activity by S 15535 in comparison with dopaminergic agents in rats and mice3-a

Effect of s.c. and p.o. administration of S 15535 in models of potential sedative properties. In the upper panel, the induction of a loss of righting reflex in rats (n = 4–9 per value) is shown and, in the lower panel, the induction of ataxia in the rotarod procedure (n = 5–14 per value) is depicted. (upper panel) Data are percentage of animals showing a loss of reflex. ED50 values (95% C.L.) were 0.32 (0.03–3.98) and 6.1 (4.0–9.2) for UK 14,304 and xylazine, respectively; and asterisks indicate significance of differences to vehicle in Fisher’s Exact Probability Test. (lower panel) Data are means ± S.E.M. of animals showing latency to fall in seconds. Asterisks indicate significance of differences to vehicle values by Log-rank analysis (* P ≤ .05).
Discussion
Modulation of hippocampal release of 5-HT by S 15535.
In analogy to buspirone and other agonists at 5-HT1Aautoreceptors (Gobert et al., 1995; Hjorth and Sharp, 1993;Hjorth et al., 1995), S 15535 reduced hippocampal dialysate levels of 5-HT. This observation is in line with its ability to reduce 5-HT release in frontal cortex (Millan et al., 1997, accompanying paper), hippocampal synthesis of 5-HT (Gobert et al., 1995; Millan et al., 1994) and the firing rate of raphe-localized serotoninergic neurons (Millan et al., 1994). The 5-HT1A autoreceptor antagonists, WAY 100,635, (−)-penbutolol and (−)-tertatolol, which do not modify 5-HT release in the hippocampus (Hjorth and Sharp, 1993; Hjorth et al., 1995), abolished the influence of S 15535 (fig. 1), in line with their ability to block the inhibitory influence of S 15535 on 5-HT release in frontal cortex and electrical discharge of raphe-localized serotoninergic cell bodies (Millan et al., 1994; Lejeuneet al., 1996; Millan et al., 1997, accompanying paper). These data show that the inhibition of serotoninergic projections by S 15535 reflects its agonist properties at 5-HT1A autoreceptors and provide a mechanistic basis for its anxiolytic properties (Coplan et al., 1995).
Actions of S 15535 in tests of potential anxiolytic activity.
The increase in punished responses elicited by S 15535 in a rat conflict test (Cervo and Samanin, 1995b) indicates that it expresses robust anxiolytic activity over a broad dose range. Although this action was selective for conflict as compared with reward periods, the response rate during the time-out period was also increased. It might be argued that S 15535 may reduce discrimination of the schedule. However, this seems unlikely inasmuch as S 15535 does not disrupt acquisition and performance of complex behavioral tasks including mnesic paradigms (Jaffard, R. and Samanin, R., unpublished observations) and drug-discrimination models (Schreiber et al., 1995b). Moreover, DZM itself yielded a similar pattern of data to S 15535 in that it also increased time-out responses, and similar effects have been reported previously with benzodiazepines (Hodges et al., 1987; Tye et al., 1979). S 15535 also does not possess stimulant-like properties as indicated by both behavioral (herein) and biochemical (Millan et al., 1997, accompanying paper) studies. Indeed, amphetamine increases time-out responses without increasing conflict responses (Tye et al.,1979). As concerns buspirone, although time-out responses are little affected, we have also not been able to obtain reliable increases in the conflict behavior with this drug (Samanin, R. et al., unpublished observations). Irrespective of the underlying reasons, it is of note that the influence of S 15535 upon punished and time-out responses could be dissociated, inasmuch as the latter change was more variable, less marked and only seen at high doses. Overall, it is likely that the increase in time-out responses with S 15535 reflects its disinhibitory rather than suppressive influence on behavior. Indeed, the relative absence of motor-suppressive actions of S 15535 is exemplified by its lack of marked influence in a battery of tests for the detection changes in motor behavior (table 3, fig. 7). These findings suggest that S 15535 exhibits anxiolytic actions at doses which were below those high doses that modify motor behavior. Correspondingly, in the pigeon conflict procedure, at doses of 0.04 and 0.16 mg/kg i.m., S 15535 increased punished responses, whereas even a 64-fold higher dose only slightly decreased unpunished responses. In contrast, DZM and buspirone showed little separation (fig. 2). Although a low dose (0.04 mg/kg) of S 15535 tended to increase time spent in the open arm of a plus-maze, statistical significance was not obtained. Further, whereas DZM was active at modest doses, neither buspirone nor WAY 100,635 expressed anxiolytic properties in this procedure. In fact, for S 15535, buspirone and DZM, biphasic curves were seen, a phenomenon frequently encountered (Handley et al., 1993; Millan and Brocco, 1993). Further, the influence of drugs on locomotor activity may modify their effects in this model (Dawson and Tricklebank, 1995). Correspondingly, the decrease in open arm presence seen with S 15535 and, more markedly, with buspirone, is unlikely to reflect “anxiogenic” actions (Dawson and Tricklebank, 1995; Handleyet al., 1993; Thiébot et al., 1988). Indeed, the dose-response curve for DZM inflected at high doses because of the onset of its motor-sedative actions. The USV procedure in fearful rats is sensitive to 5-HT1A receptor agonists (Sanchez, 1993; Schreiber and De Vry, 1993a) and S 15535 was active in this paradigm. Although WAY 100,635 was inactive, DZM and buspirone reduced USVs, albeit with little separation to sedative doses (table4). Molewijk et al. (1995) suggested that the activity of drugs in paradigms of aversive stimulation-induced USV may be related to antipanic actions, and a perturbation of central serotoninergic (5-HT1A) mechanisms may be involved in such disorders (Coplan et al., 1992; Lesch et al., 1991). However, no experimental models of panic attacks are currently recognized and the clinical utility of 5-HT1A ligands for treating panic attacks is not established (Coplan et al., 1992; Deakin, 1993; Lader, 1991). Thus, the actions of S 15535, buspirone and DZM in this USV model likely reflect their anxiolytic properties (Schreiber and De Vry, 1993a).
Summary of drug actions in tests of potential anxiolytic and antiaggressive activity as compared with motor disruption
Mechanism(s) underlying the anxiolytic actions of S 15535.
The anxiolytic actions of S 15535 likely reflect an action at 5-HT1A receptors. First, S 15535 is a highly selective 5-HT1A ligand and expresses its anxiolytic actions over a moderate dose-range (Millan et al., 1994). Second, the potency of S 15535 in the pigeon conflict test correlated to its affinity at 5-HT1A receptors (Schreiber, R., unpublished observations; Schreiber et al., 1995a). Third, S 15535 did not increase punished responding in the presence of the 5-HT1A receptor antagonists, (−)-alprenolol or (−)-penbutolol, whereas the β1- and β2-AR antagonists, (−)-metaprolol and ICI 118,551, respectively, little modified the anxiolytic actions of S 15535. Fourth, both (−)-penbutolol and WAY 100,635, which is devoid of β-AR blocking properties, abolished the action of S 15535 in the USV procedure. Hyperactive serotoninergic transmission may underlie anxious states, and microinjection and lesion studies with other 5-HT1Aagonists favor a predominant role of 5-HT1A autoreceptors in their anxiolytic actions (Picazo et al., 1995; seeBarrett and Gleeson, 1991; Coplan et al., 1995; Schreiber and De Vry, 1993b). Indeed, if blockade of postsynaptic 5-HT1A sites played a major role in the effects of S 15535, WAY 100,635, (−)-alprenolol and (−)-penbutolol should mimic rather than block its actions. These were not active, however. In fact, some studies indicate that activation of postsynaptic 5-HT1Areceptors elicits anxiolytic actions (see Barrett and Gleeson, 1991;Coplan et al., 1995; Millan et al., 1992a;Schreiber and De Vry, 1993a), but their role is complex and may depend on the precise level of serotoninergic tone. In fact, stimulation of 5-HT1A receptors in the hippocampus, amygdala or periaqueductal grey may increase anxiety under certain conditions (Andrews et al., 1994; Hodges et al., 1987; Jencket al., 1989). Moreover, anxiolytic actions of WAY 100,635 (and WAY 100,135) have been seen in certain models (Bickerdike et al., 1995; Fletcher et al., 1996; Rodgers and Cole, 1994). Thus, the antagonist/weak partial agonist properties of S 15535 at postsynaptic sites may complement its agonist actions at 5-HT1A autoreceptors in mediating its anxiolytic properties.
Action of S 15535 on aggressive behavior in mice.
Like other 5-HT1A ligands (Mos et al., 1993; Sanchez and Hyttel, 1994; White et al., 1991), S 15535 inhibited aggression in isolated mice. Although buspirone was active, it showed no separation between doses for antiaggressive versusmotor-disruptive actions (table 4). Nevertheless, buspirone is effective in the clinical treatment of aggression, and both serotoninergic systems and 5-HT1A receptors are implicated in aggressive states (Blanchard et al., 1988;Moeller et al., 1996; Ratey et al., 1991;Yudofsky et al., 1990). It has been hypothesized that decreasing serotoninergic activity may encourage aggressive behavior but the situation may be more complex (Miczek et al., 1995). Thus, transgenic mice possessing (10-fold) elevated levels of cerebral serotonin show increased aggressivity (Cases et al., 1995). Further, whether the antiaggressive actions of 5-HT1Areceptor agonists in rodents reflects stimulation of post- or presynaptic 5-HT1A receptors remains unclear (Miczeket al., 1995; Mos et al., 1993; Schreiber and De Vry, 1993a; Sjibesma et al., 1991). In fact, the present observation that WAY 100,635 blocks the action of S 15535 supports a role of 5-HT1A autoreceptors in its antiaggressive actions. Aggressive behavior may reflect a loss of impulse control and is related to such states as obsessive-compulsive disorders and alcohol abuse (Lesch et al., 1991; Sellers et al., 1992). There is evidence for a role of serotoninergic systems in the pathology of these disorders and activation of 5-HT1A autoreceptors may counter processes underlying excessive intake of alcohol and other drugs (Bruno, 1989; Schreiber et al., 1993; Sellers et al., 1992). However, clinical data concerning 5-HT1Areceptor ligands in the treatment of obsessive-compulsive disorders are inconclusive (Grady et al., 1993; Pato et al., 1991) and the relative relationship between the anxiolytic and anti-impulsive properties of 5-HT1A ligands remains under discussion (Millan et al., 1992a; Schreiber and De Vry, 1993a).
Comparison of the profile of S 15535 to that of buspirone.
S 15535 may possess certain advantages over buspirone and related drugs in the management of anxiety (table 4). First, S 15535 shows superior selectivity for 5-HT1A versus dopamine D2 receptors and lower efficacy at post-synaptic 5-HT1A receptors. Correspondingly, S 15535 does not elicit a “5-HT syndrome” and little modifies motor behavior (table 4,Millan et al., 1992b,1994). Second, the onset of anxiolytic effect of buspirone is slower than for BZPs; this may be due to its metabolism to the α2-AR antagonist, 1-pyrimidinyl-piperazine, because blockade of α2-ARs exacerbates anxious states (Charney and Redmond, 1983; Goudie and Leathley, 1991; Lader, 1991). Unlike buspirone, S 15535 is devoid of α2-AR antagonist properties and cannot be metabolized to 1-pyrimidinyl-piperazine. The anxiety which ensues upon termination of BZP treatment may be caused by an overactivation of serotoninergic pathways (File and Andrews, 1994; Goudie and Leathley, 1991), and S 15535 could be of utility in the control of such states. Third, buspirone and other 5-HT1A agonists drugs induce insomnia as a side effect and reduce REM sleep time in insomniacs (Gillinet al., 1994; Mendelson et al., 1990). This action, which reflects both their dopaminergic (D2antagonist) properties and activation of postsynaptic 5-HT1A receptors (Sanford et al., 1994; Tissieret al., 1993) decreases their utility in patients with sleep difficulties, contributes to difficulties of replacing BZP therapy and may also underlie the delay in achieving therapeutic efficacy (Mendelson et al., 1990). Inasmuch as S 15535 has low efficacy at postsynaptic 5-HT1A sites and does not show D2 antagonist properties, it should not disrupt sleep. Indeed, the postsynaptic antagonist, (−)-alprenolol, does not modify sleep patterns in rat (Bjorvatn et al., 1992).
Comparison of the profile of S 15535 with that of DZM.
S 15535 may also be favorably compared with DZM and other BZPs inasmuch as, first, it expresses its anxiolytic properties at doses not markedly modifying motor function (table 4). Second, BZPs present a major dependence/abuse potential (Barrett and Gleeson, 1991; Lader, 1991) whereas S 15535 is not self-administered in the rat (E. Mocaër, personal communication), and clinical experience with 5-HT1A ligands suggests that they do not have a major abuse potential (see Evans et al., 1994; Lader, 1991). Third, in contrast to BZPs (Barbee et al., 1991; Lister, 1991), S 15535 does not appear to exert a negative influence on mnesic function (unpublished observation). Fourth, co-morbid anxious and depressive states, which may reflect excessive output from the serotoninergic DRN (Deakin, 1994), are being increasingly diagnosed (Gammans et al., 1992). S 15535 inhibits DRN activity (Millan et al., 1994) and possesses antidepressant properties (Millanet al., 1997, accompanying paper). This suggests potential advantages in the treatment of anxiety associated with depression. Finally, one problem encountered clinically with BZPs is that of “paradoxical” aggression (see Mos and Olivier, 1993), and S 15535 displays potential antiaggressive properties.
Summary and conclusions.
To summarize, S 15535 expresses marked anxiolytic activity in several experimental procedures, in line with its inhibition of serotoninergic transmission via an agonist and antagonist interaction at pre- and postsynaptic 5-HT1A receptors, respectively. In addition, possiblyvia 5-HT1A autoreceptors, S 15535 may exert antiaggressive properties. These actions, as well as the antidepressant properties described in the preceding paper, are expressed at doses below those eliciting behavioral disruption (table 4). In conclusion, reflecting its selectivity and dual pattern of high/low efficacy at pre- and postsynaptic 5-HT1A receptors, respectively, S 15535 may display advantages to currently available drugs in the management of anxious states.
Acknowledgments
The skillful technical assistance of Gerd Leonsson in thein vivo microdialysis studies is gratefully acknowledged. These dialysis studies were supported, in part, by grants from Svenska Lundbeck-Stiftelsen in the Sw. MRC (#07486), to S.H.
Footnotes
-
Send reprint requests to: Dr. Mark J. Millan, Institut de Recherches Servier. Centre de Recherches de Croissy, Psychopharmacology Department. 125, Chemin de Ronde, 78290, Croissy-sur-Seine, Paris, France.
-
↵1 Present address: Troponwerke, Institut for Neurobiology, Berliner Straáe 156, D-51063 Köln, Germany.
- Abbreviations:
- AR
- adrenergic receptor
- BZP
- benzodiazepine
- DZM
- diazepam
- USV
- ultrasonic vocalization
- 5-HT
- serotonin
- Received March 19, 1996.
- Accepted March 10, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}